
A Population-Based Human In Vitro Approach to Quantify Inter-Individual Variability in Responses to Chemical Mixtures

 , ,
, ,  ,
, 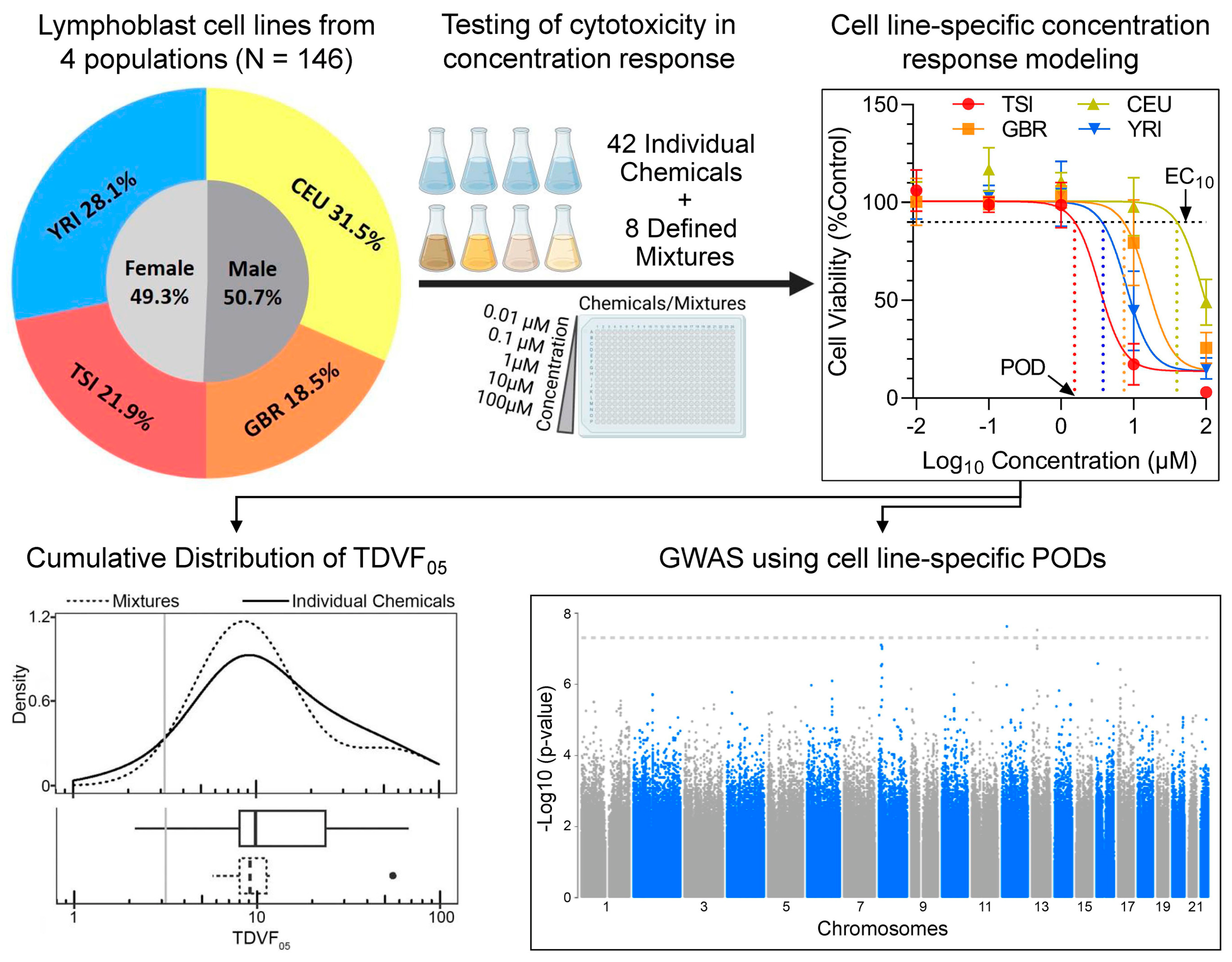{kind=link}
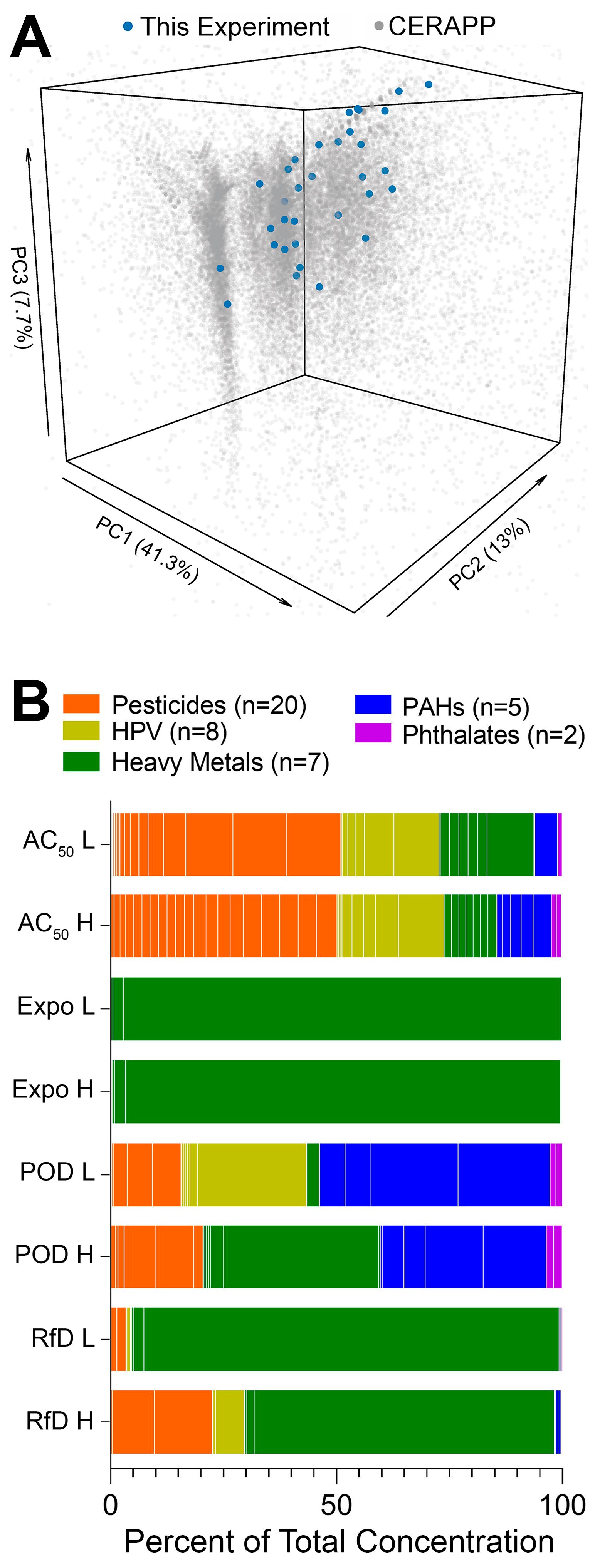{kind=link}
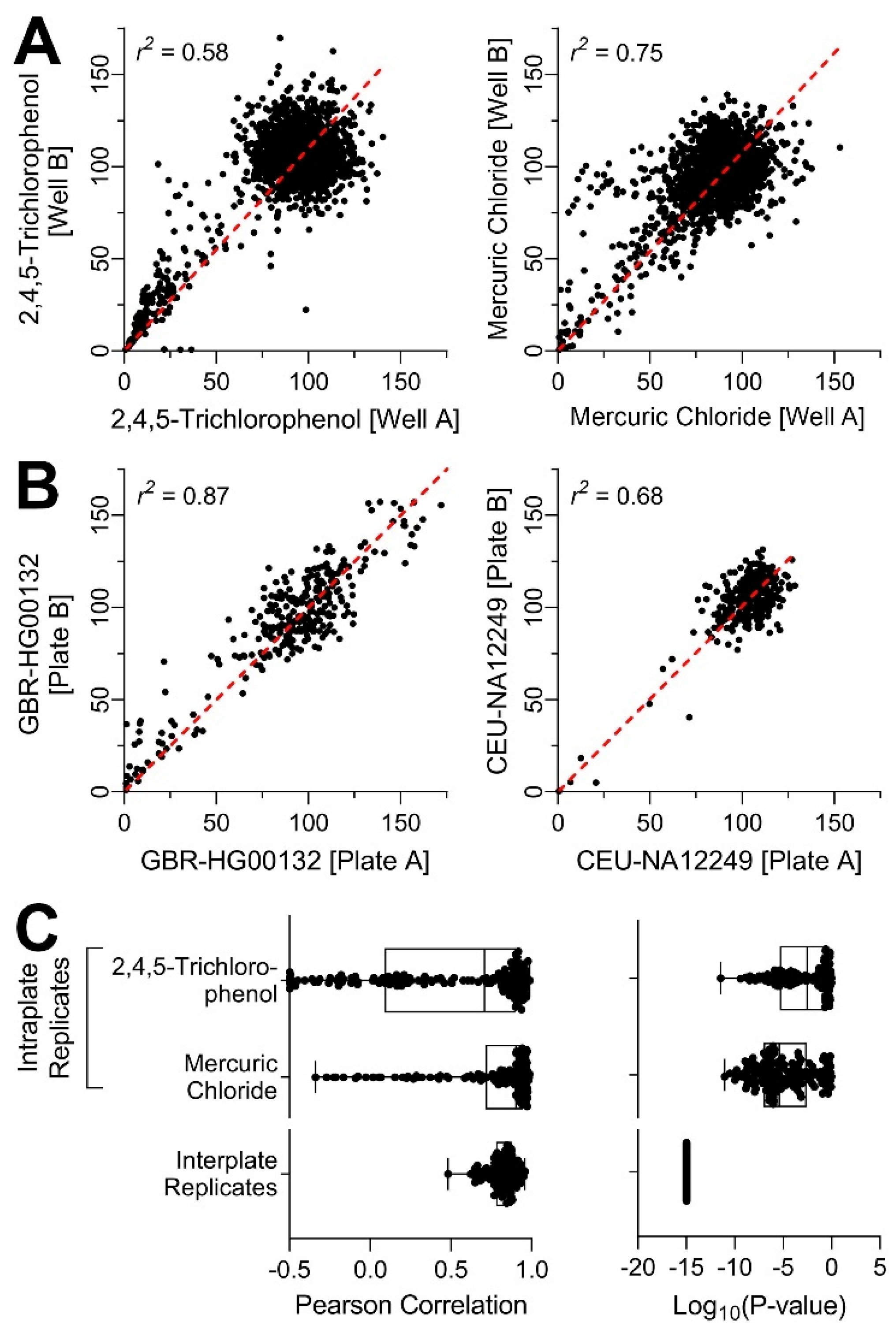{kind=link}
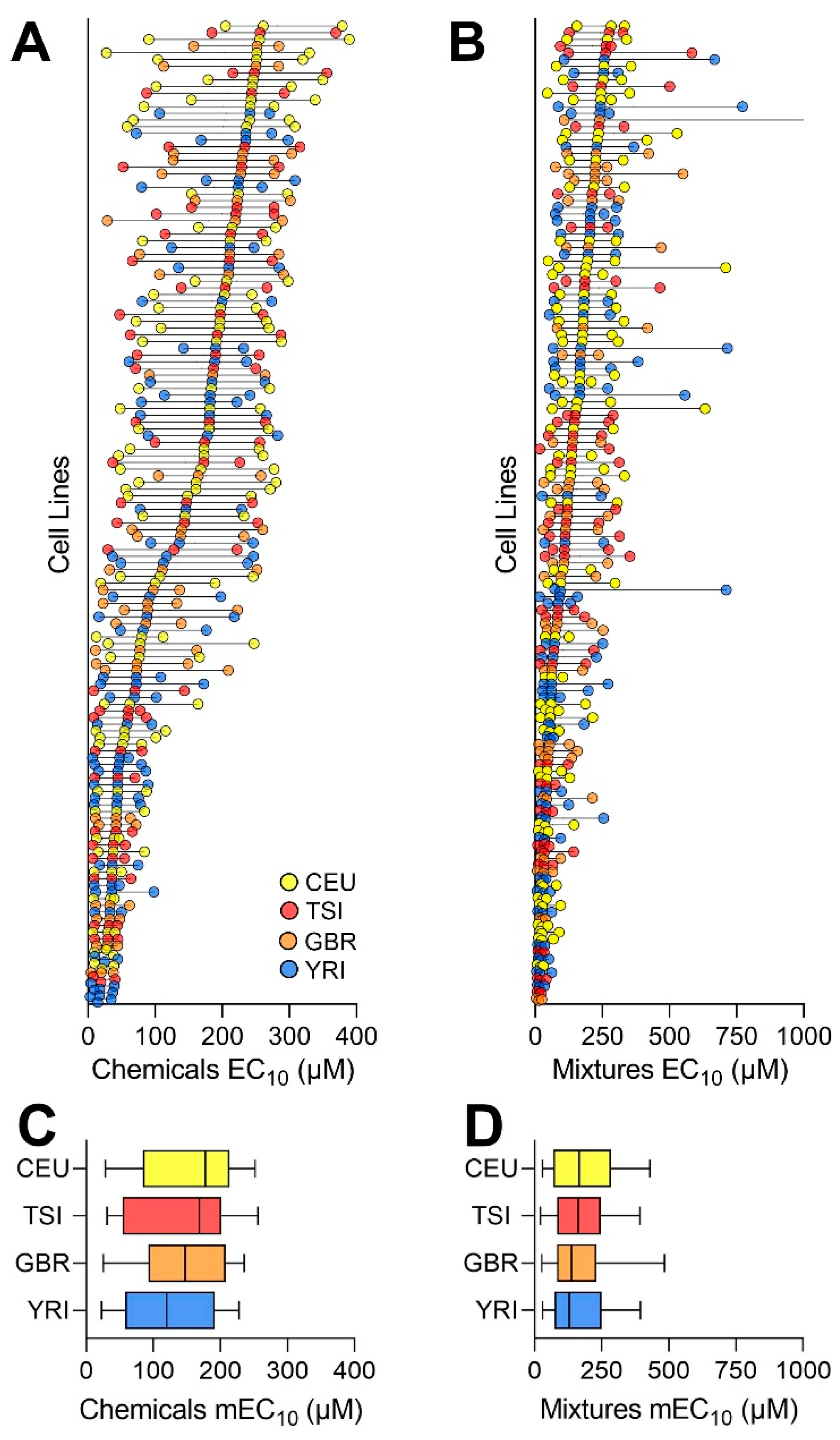{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
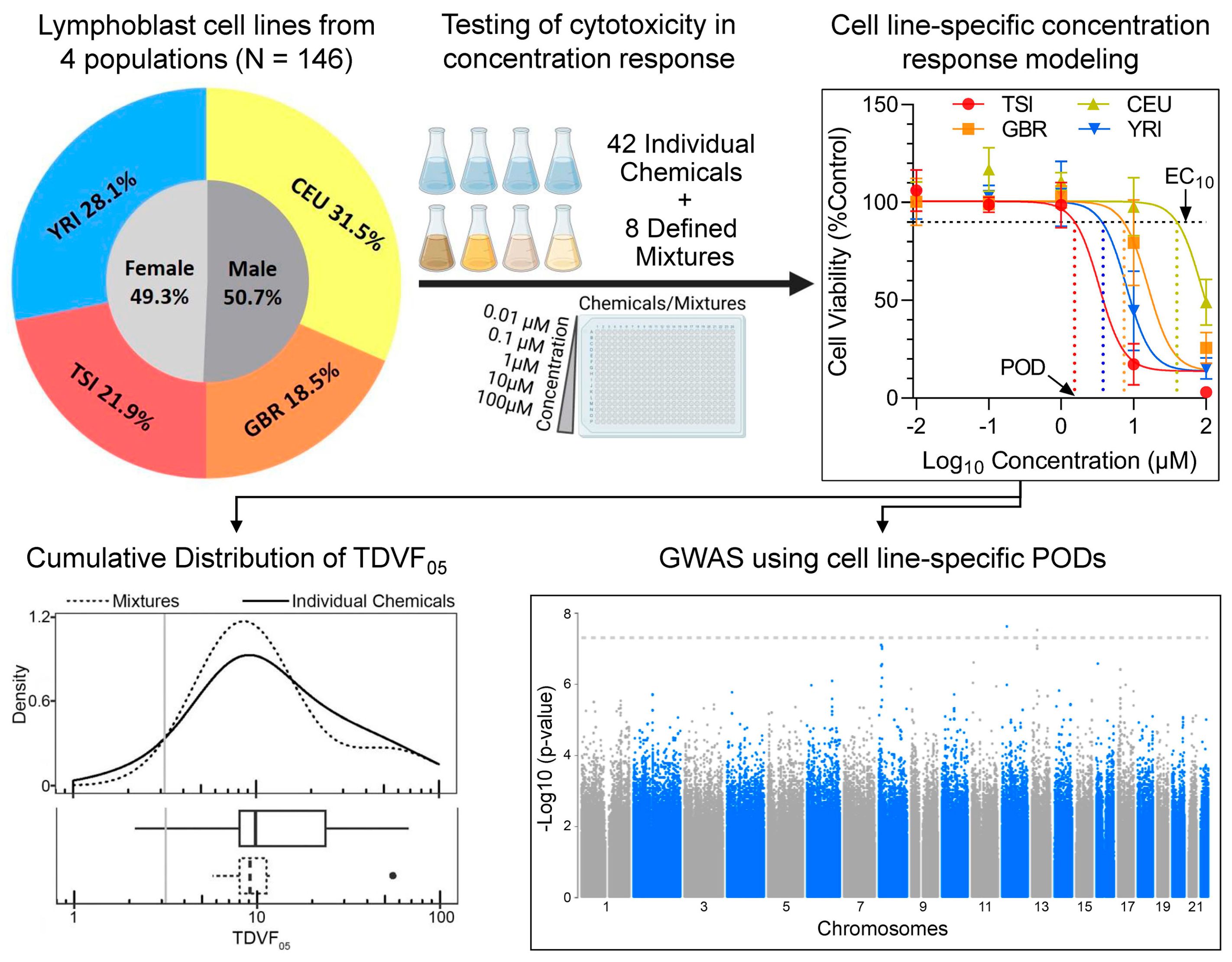
2.1. Chemicals and Biologicals
2.2. Cell Lines
2.3. Cell Culture
2.4. Chemicals and Mixtures
2.5. Cell Viability Screening
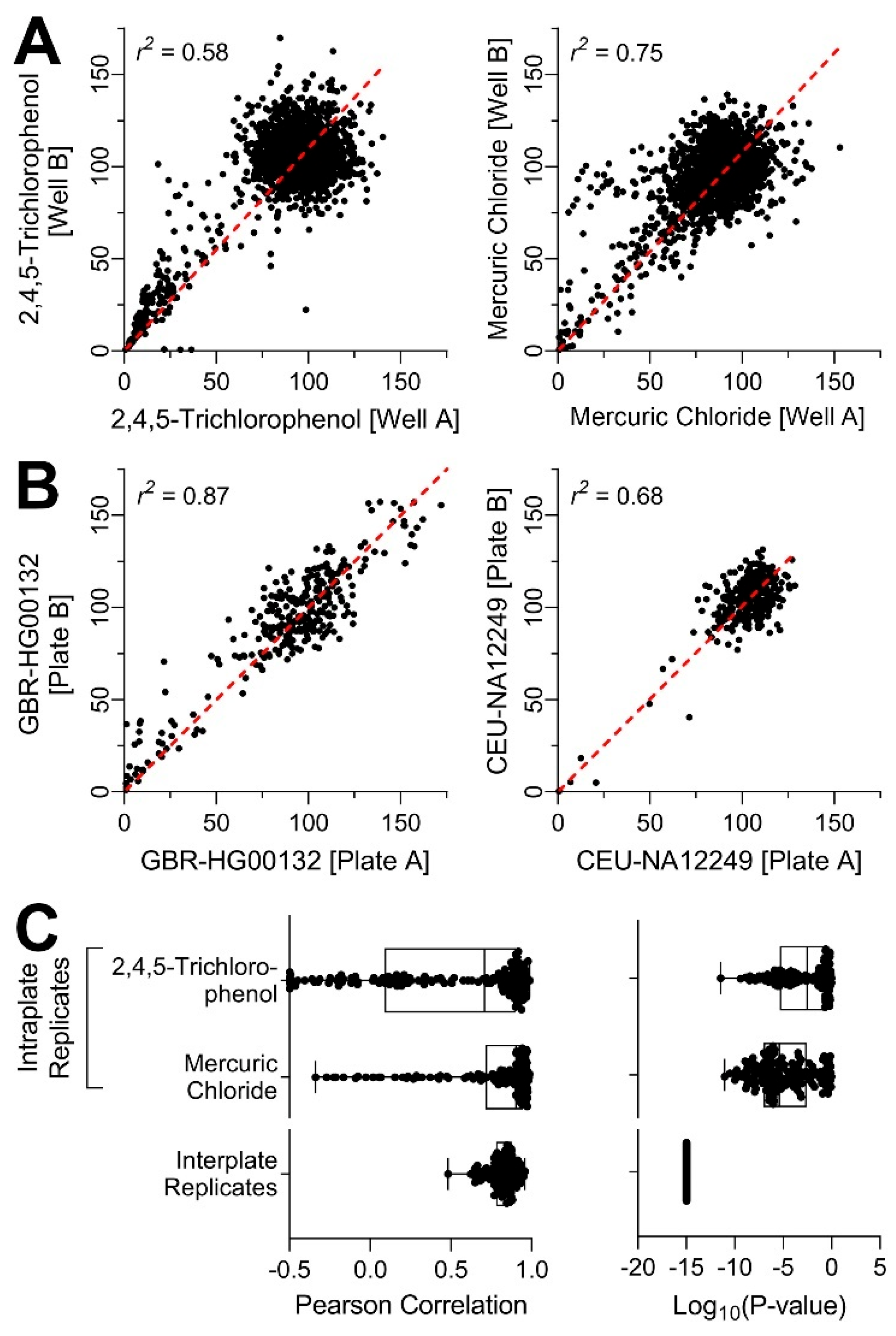
2.6. Assessment of Experimental Reproducibility
2.7. Derivation of Chemical and Mixture-Specific Concentration–Response Profiles
2.8. Derivation of Chemical and Mixture-Specific Point of Departure (POD) and Toxicodynamic Variability Factor (TDVF05)
2.9. Genome-Wide Association Mapping
3. Results
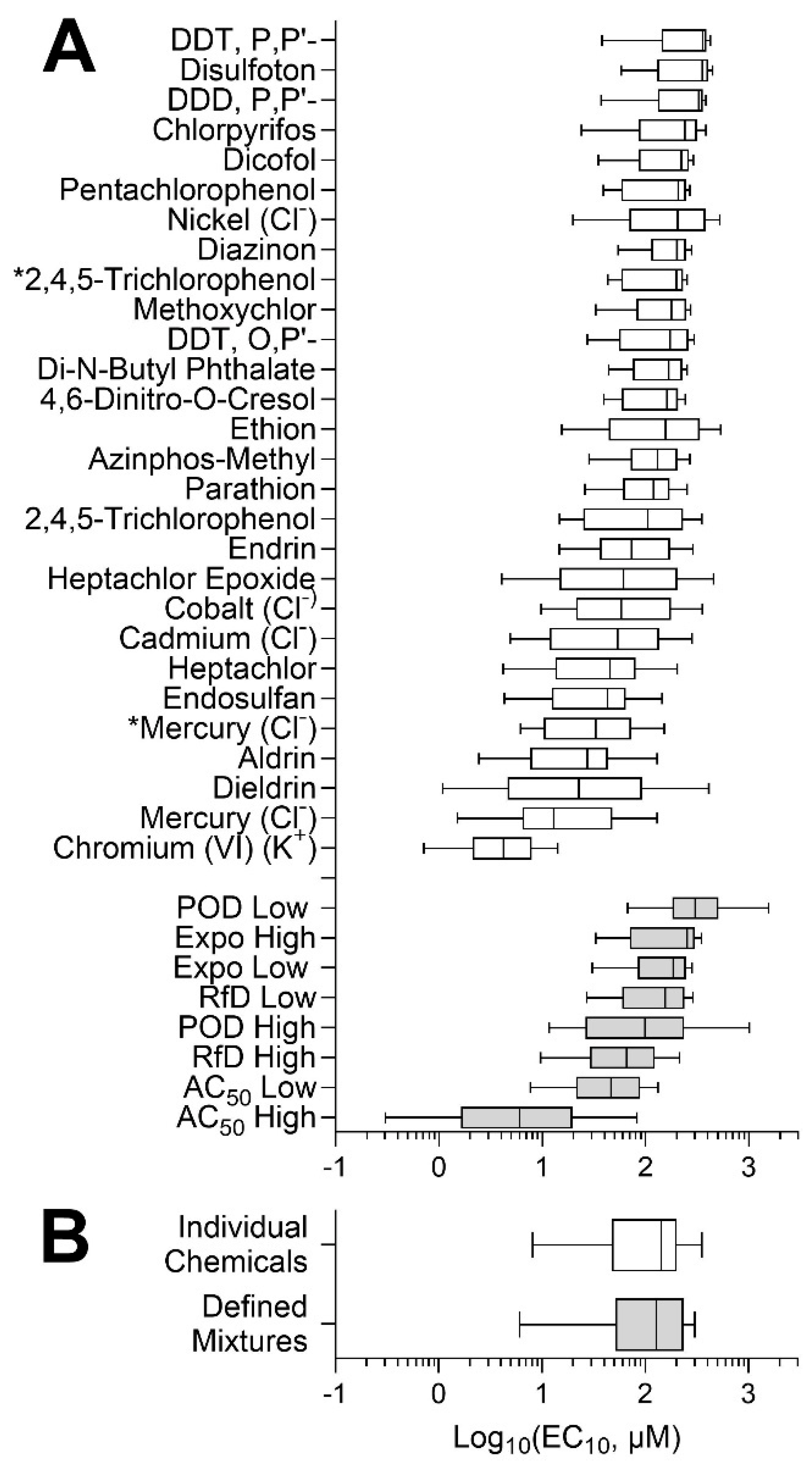
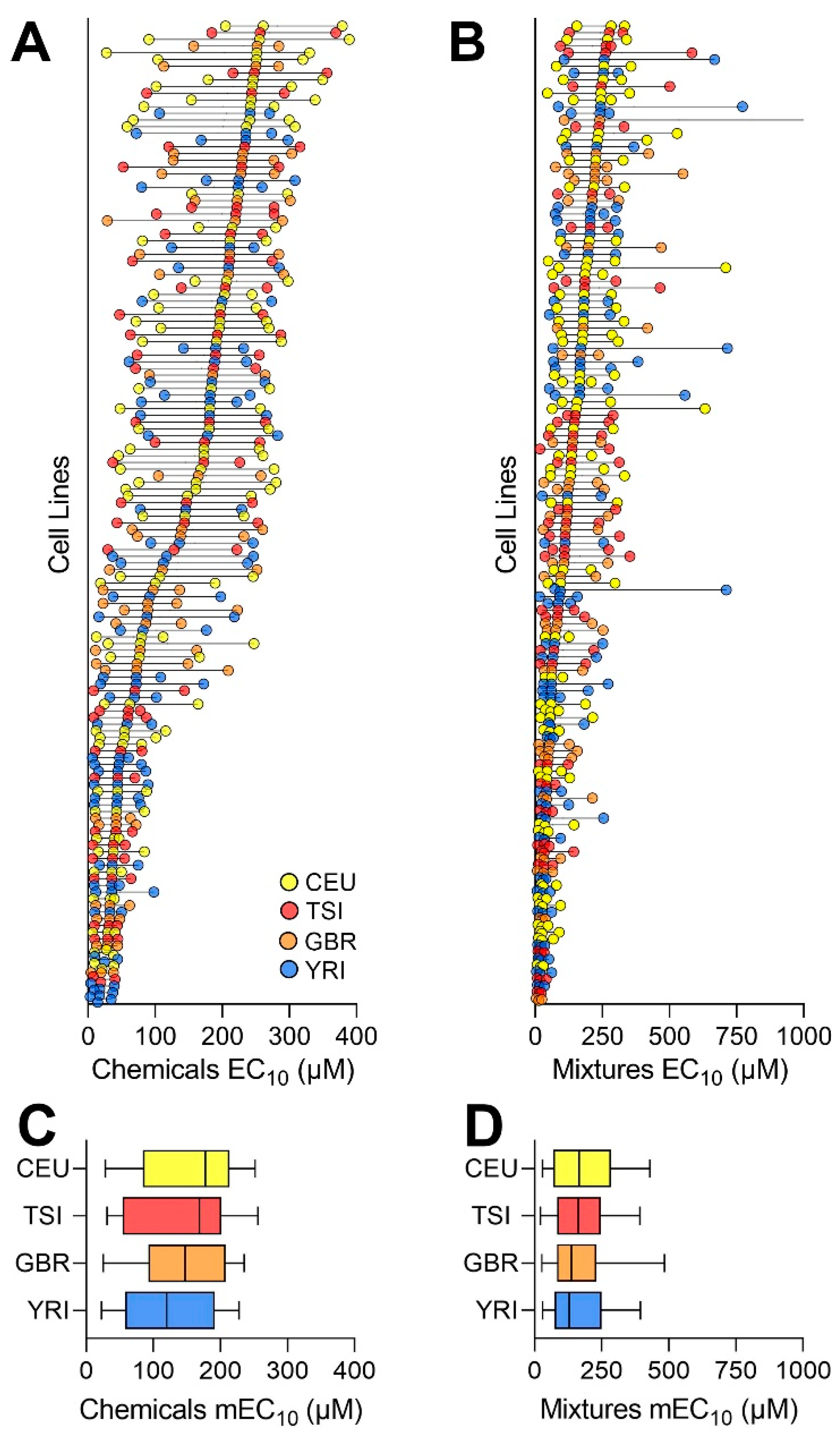
3.1. Population Variability Screening of Chemicals and Mixtures in Human Lymphoblasts
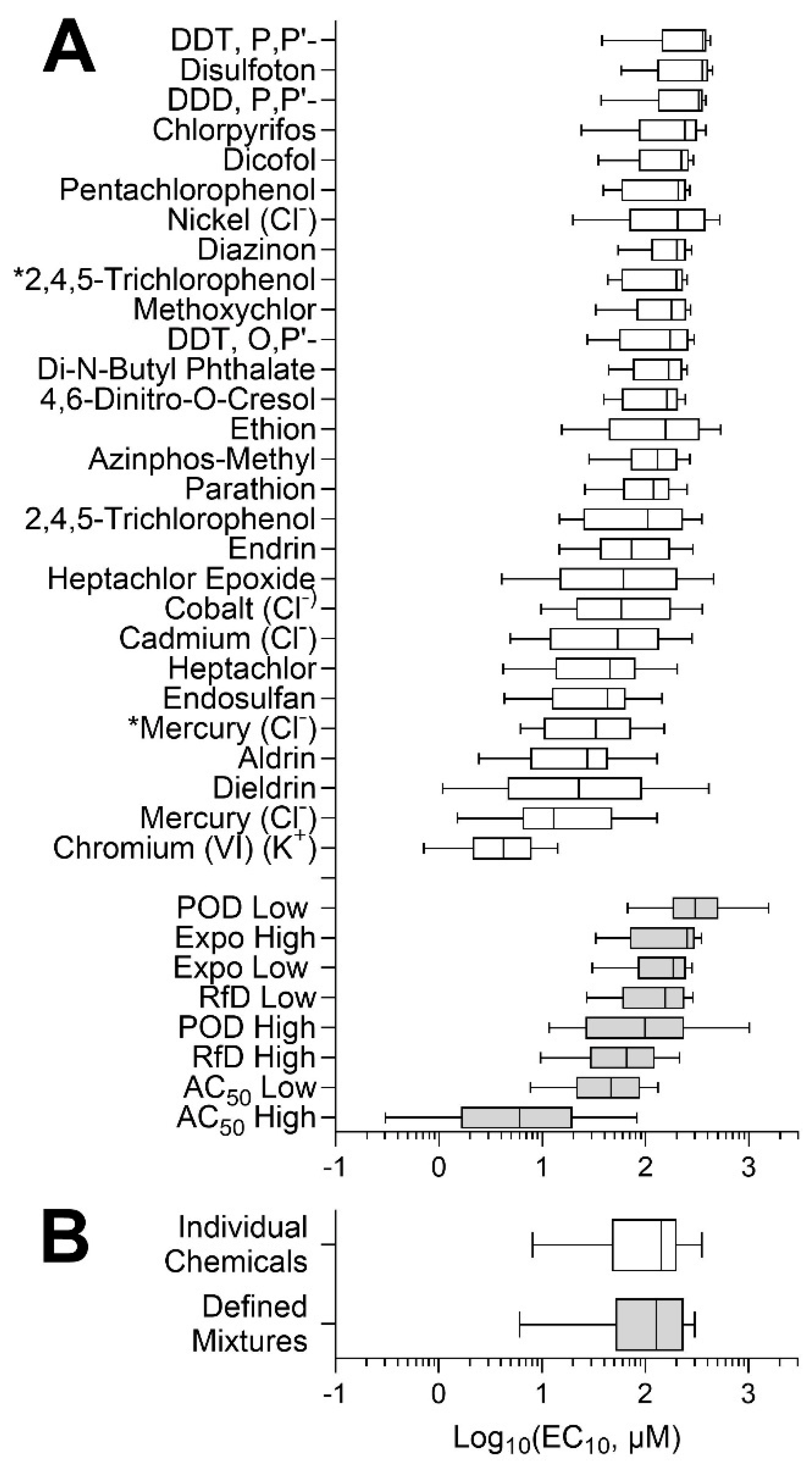
3.2. Concentration–Response Analyses of the Effects of Chemicals and Mixtures
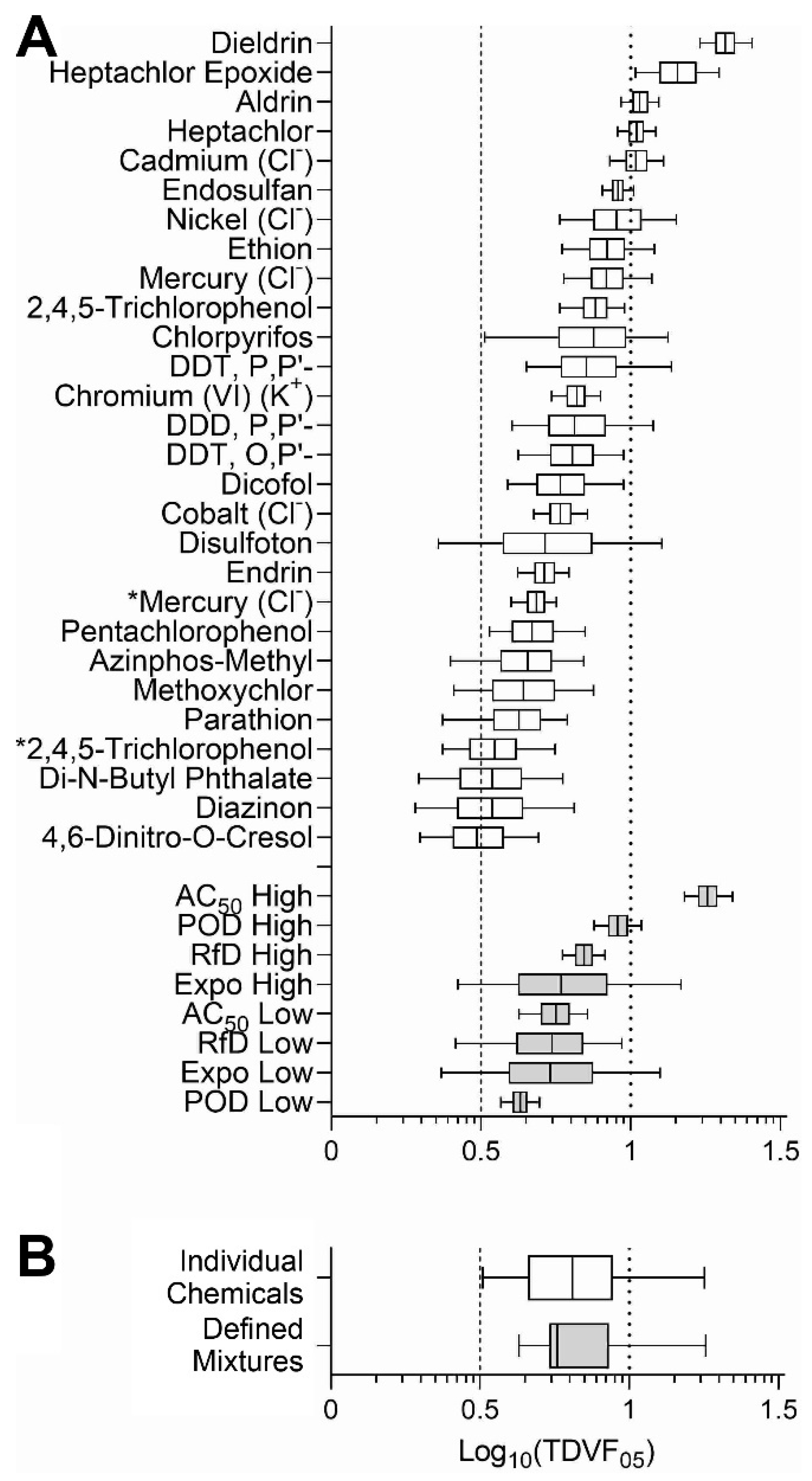
3.3. Analysis of the Inter-Individual Variability in Effects of Chemicals and Mixtures
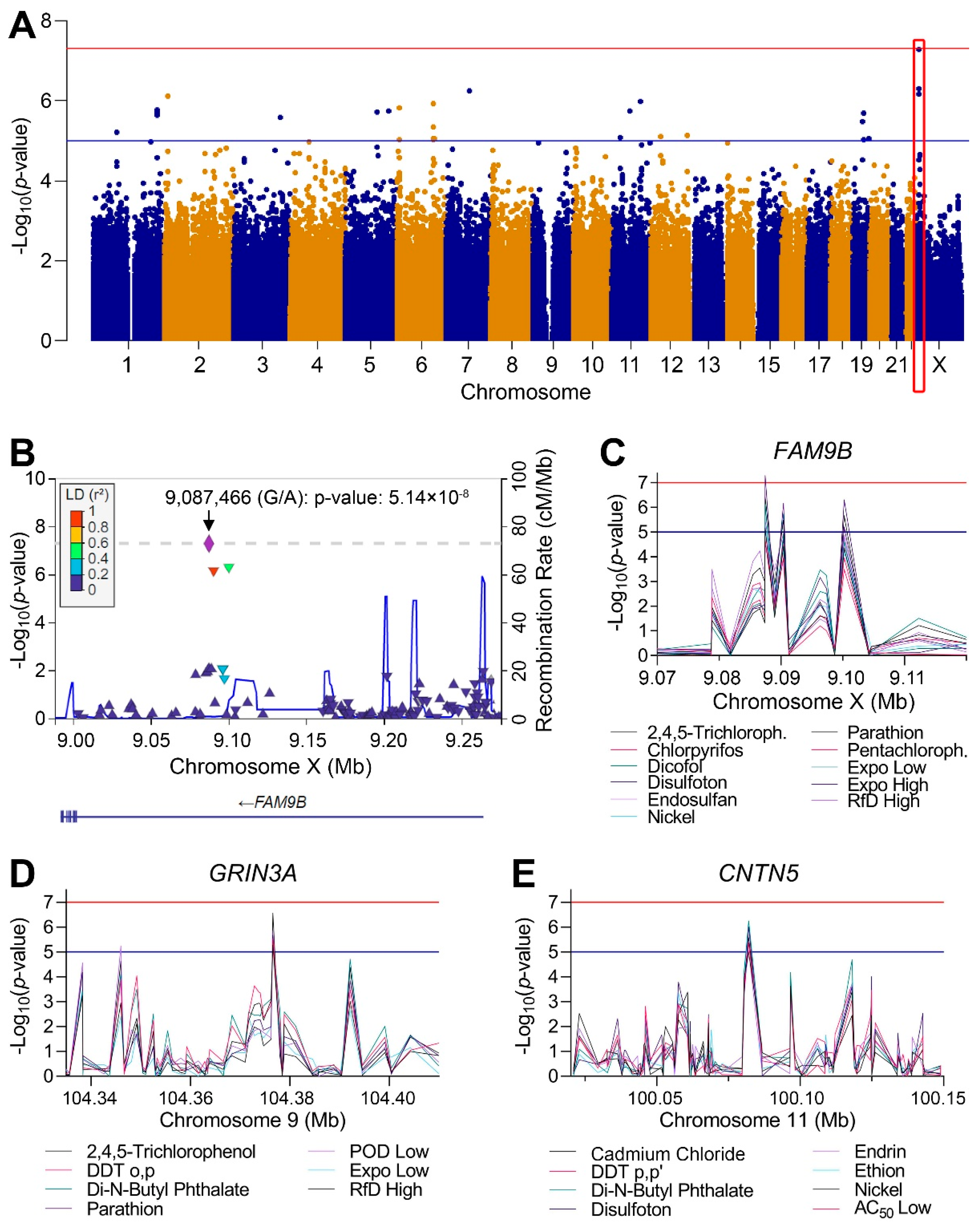
3.4. Genome-Wide Association Study (GWAS) Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kumar, V.; Boobis, A.R.; Moretto, A. Test and risk assessment strategies for combined exposure to multiple chemicals. Food Chem. Toxicol. 2020, 144, 111607. [Google Scholar] [CrossRef] [PubMed]
- Sexton, K.; Hattis, D. Assessing cumulative health risks from exposure to environmental mixtures—Three fundamental questions. Environ. Health Perspect. 2007, 115, 825–832. [Google Scholar] [CrossRef] [PubMed]
- OECD. Considerations for Assessing the Risks of Combined Exposure to Multiple Chemicals, Series on Testing and Assessment No. 296, Evironment, Health and Safety Division, Environment Directorate; Organisation for Economic Co-Operation and Development: Paris, France, 2018. [Google Scholar]
- Kienzler, A.; Bopp, S.K.; van der Linden, S.; Berggren, E.; Worth, A. Regulatory assessment of chemical mixtures: Requirements, current approaches and future perspectives. Regul. Toxicol. Pharmacol. 2016, 80, 321–334. [Google Scholar] [CrossRef]
- U.S. Environmental Protection Agency. Framework for Cumulative Risk Assessment; U.S. Environmental Protection Agency: Washington, DC, USA, 2003.
- U.S. Environmental Protection Agency. Cumulative Assessments of Risk from Pesticides; Office of Pesticide Programs, U.S. Environmental Protection Agency: Washington, DC, USA, 2016.
- Efsa Scientific Committee; More, S.J.; Bampidis, V.; Benford, D.; Bragard, C.; Hernandez-Jerez, A.; Bennekou, S.H.; Halldorsson, T.I.; Koutsoumanis, K.P.; Lambre, C.; et al. Guidance document on scientific criteria for grouping chemicals into assessment groups for human risk assessment of combined exposure to multiple chemicals. EFSA J. 2021, 19, e07033. [Google Scholar] [PubMed]
- National Research Council. Phthalates and Cumulative Risk Assessment: The Tasks Ahead; National Academies Press: Washington, DC, USA, 2008. [Google Scholar]
- U.S. Environmental Protection Agency. Concepts, Methods, and Data Sources for Cumulative Health Risk Assessment of Multiple Chemicals, Exposure and Effects: A Resource Document; United States Environmental Protection Agency: Washington, DC, USA, 2008.
- European Centre for Ecotoxicology and Toxicology of Chemicals. Workshop on Combined Exposure to Chemicals; European Centre for Ecotoxicology and Toxicology of Chemicals: Berlin, Germany, 2011. [Google Scholar]
- OECD. Guidance Document on Aqueous-Phase Aquatic Toxicity Testing of Difficult Test Chemicals; OECD: Paris, France, 2019. [Google Scholar]
- Luo, Y.S.; Chen, Z.; Hsieh, N.H.; Lin, T.E. Chemical and biological assessments of environmental mixtures: A review of current trends, advances, and future perspectives. J. Hazard. Mater. 2022, 432, 128658. [Google Scholar] [CrossRef]
- Zeise, L.; Bois, F.Y.; Chiu, W.A.; Hattis, D.; Rusyn, I.; Guyton, K.Z. Addressing human variability in next-generation human health risk assessments of environmental chemicals. Environ. Health Perspect. 2013, 121, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, C.F.; Christoph, G.R.; Julien, E.; Kelley, J.M.; Kronenberg, J.; McCarthy, J.; Reiss, R. Assessing the risks of exposures to multiple chemicals with a common mechanism of toxicity: How to cumulate? Regul. Toxicol. Pharmacol. 2000, 31, 30–43. [Google Scholar] [CrossRef]
- Rusyn, I.; Chiu, W.A. Decision-making with new approach methodologies: Time to replace default uncertainty factors with data. Toxicol. Sci. 2022, kfac033. Available online: https://pubmed.ncbi.nlm.nih.gov/35404442/ (accessed on 1 June 2022). [CrossRef]
- Rusyn, I.; Chiu, W.A.; Wright, F.A. Model systems and organisms for addressing inter- and intra-species variability in risk assessment. Regul. Toxicol. Pharmacol. 2022, 132, 105197. [Google Scholar] [CrossRef]
- Wheeler, H.E.; Dolan, M.E. Lymphoblastoid cell lines in pharmacogenomic discovery and clinical translation. Pharmacogenomics 2012, 13, 55–70. [Google Scholar] [CrossRef] [Green Version]
- Fermini, B.; Coyne, S.T.; Coyne, K.P. Clinical trials in a dish: A perspective on the coming revolution in drug development. Slas Discov. 2018, 23, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Abdo, N.; Xia, M.; Brown, C.C.; Kosyk, O.; Huang, R.; Sakamuru, S.; Zhou, Y.H.; Jack, J.R.; Gallins, P.; Xia, K.; et al. Population-based in vitro hazard and concentration-response assessment of chemicals: The 1000 genomes high-throughput screening study. Environ. Health Perspect. 2015, 123, 458–466. [Google Scholar] [CrossRef] [Green Version]
- Eduati, F.; Mangravite, L.M.; Wang, T.; Tang, H.; Bare, J.C.; Huang, R.; Norman, T.; Kellen, M.; Menden, M.P.; Yang, J.; et al. Prediction of human population responses to toxic compounds by a collaborative competition. Nat. Biotechnol. 2015, 33, 933–940. [Google Scholar] [CrossRef] [Green Version]
- Burnett, S.D.; Blanchette, A.D.; Chiu, W.A.; Rusyn, I. Cardiotoxicity hazard and risk characterization of toxcast chemicals using human induced pluripotent stem cell-derived cardiomyocytes from multiple donors. Chem. Res. Toxicol. 2021, 34, 2110–2124. [Google Scholar] [CrossRef]
- Abdo, N.; Wetmore, B.A.; Chappell, G.A.; Shea, D.; Wright, F.A.; Rusyn, I. In vitro screening for population variability in toxicity of pesticide-containing mixtures. Environ. Int. 2015, 85, 147–155. [Google Scholar] [CrossRef] [Green Version]
- 1000 Genomes Project Consortium; Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar]
- Chen, Z.; Liu, Y.; Wright, F.A.; Chiu, W.A.; Rusyn, I. Rapid hazard characterization of environmental chemicals using a compendium of human cell lines from different organs. ALTEX 2020, 37, 623–638. [Google Scholar] [CrossRef]
- Lock, E.F.; Abdo, N.; Huang, R.; Xia, M.; Kosyk, O.; O’Shea, S.H.; Zhou, Y.H.; Sedykh, A.; Tropsha, A.; Austin, C.P.; et al. Quantitative high-throughput screening for chemical toxicity in a population-based in vitro model. Toxicol. Sci. 2012, 126, 578–588. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, N.H.; Chen, Z.; Rusyn, I.; Chiu, W.A. Risk characterization and probabilistic concentration-response modeling of complex environmental mixtures using new approach methodologies (nams) data from organotypic in vitro human stem cell assays. Environ. Health Perspect. 2021, 129, 17004. [Google Scholar] [CrossRef]
- Wignall, J.A.; Shapiro, A.J.; Wright, F.A.; Woodruff, T.J.; Chiu, W.A.; Guyton, K.Z.; Rusyn, I. Standardizing benchmark dose calculations to improve science-based decisions in human health assessments. Environ. Health Perspect. 2014, 122, 499–505. [Google Scholar] [CrossRef] [Green Version]
- Wambaugh, J.F.; Setzer, R.W.; Reif, D.M.; Gangwal, S.; Mitchell-Blackwood, J.; Arnot, J.A.; Joliet, O.; Frame, A.; Rabinowitz, J.; Knudsen, T.B.; et al. High-throughput models for exposure-based chemical prioritization in the expocast project. Environ. Sci. Technol. 2013, 47, 8479–8488. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.J.; Grulke, C.M.; Edwards, J.; McEachran, A.D.; Mansouri, K.; Baker, N.C.; Patlewicz, G.; Shah, I.; Wambaugh, J.F.; Judson, R.S.; et al. The comptox chemistry dashboard: A community data resource for environmental chemistry. J. Cheminformatics 2017, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Chiu, W.A.; Wright, F.A.; Rusyn, I. A tiered, bayesian approach to estimating of population variability for regulatory decision-making. ALTEX 2017, 34, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, A.D.; Burnett, S.D.; Grimm, F.A.; Rusyn, I.; Chiu, W.A. A bayesian method for population-wide cardiotoxicity hazard and risk characterization using an in vitro human model. Toxicol. Sci. 2020, 178, 391–403. [Google Scholar] [CrossRef]
- Gelman, A.; Goodrich, B.; Gabry, J.; Vehtari, A. R-squared for bayesian regression models. Am. Stat. 2019, 73, 307–309. [Google Scholar] [CrossRef]
- Gelman, A.; Rubin, D.B. Inference from iterative simulation using multiple sequences. Stat. Sci. 1992, 7, 457–472. [Google Scholar] [CrossRef]
- US EPA. Benchmark Dose Technical Guidance; Risk Assessment Forum; US EPA: Washington, DC, USA, 2012.
- WHO/IPCS. Chemical-Specific Adjustment Factors for Interspecies Differences in Human Variability: Guidance Document for Use of Data in Dose/Concentration-Response Assessment; World Health Organization: Geneva, Switzerland, 2005. [Google Scholar]
- Dudbridge, F.; Gusnanto, A. Estimation of significance thresholds for genomewide association scans. Genet. Epidemiol. 2008, 32, 227–234. [Google Scholar] [CrossRef]
- Price, A.L.; Zaitlen, N.A.; Reich, D.; Patterson, N. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 2010, 11, 459–463. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zaitlen, N.A.; Goddard, M.E.; Visscher, P.M.; Price, A.L. Advantages and pitfalls in the application of mixed-model association methods. Nat. Genet. 2014, 46, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Mansouri, K.; Abdelaziz, A.; Rybacka, A.; Roncaglioni, A.; Tropsha, A.; Varnek, A.; Zakharov, A.; Worth, A.; Richard, A.M.; Grulke, C.M.; et al. Cerapp: Collaborative estrogen receptor activity prediction project. Environ. Health Perspect. 2016, 124, 1023–1033. [Google Scholar] [CrossRef]
- Guha, R. Chemical informatics functionality in R. J. Stat. Softw. 2007, 18, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Hattis, D.; Lynch, M.K. Empirically observed distributions of pharmacokinetic and pharmacodynamic variability in humans-implications for the derivation of single point component uncertainty factors providing equivalent protection as existing rfds. In Toxicokinetics in Risk Assessment; Lipscomb, J.C., Ohanian, E.V., Eds.; Informa Healthcare, USA, Inc.: NewYork, NY, USA, 2007; pp. 69–93. [Google Scholar]
- Devlin, B.; Roeder, K. Genomic control for association studies. Biometrics 1999, 55, 997–1004. [Google Scholar] [CrossRef]
- Chiu, W.A.; Rusyn, I. Advancing chemical risk assessment decision-making with population variability data: Challenges and opportunities. Mamm. Genome 2018, 29, 182–189. [Google Scholar] [CrossRef]
- National Research Council. Science and Decisions: Advancing Risk Assessment; National Academies Press: Washington, DC, USA, 2009. [Google Scholar]
- EFSA Scientific Committee; More, S.J.; Bampidis, V.; Benford, D.; Bennekou, S.H.; Bragard, C.; Halldorsson, T.I.; Hernández-Jerez, A.F.; Koutsoumanis, K.; Naegeli, H. Guidance on harmonised methodologies for human health, animal health and ecological risk assessment of combined exposure to multiple chemicals. EFSA J. 2019, 17, e05634. [Google Scholar]
- Dorne, J.L.; Amzal, B.; Bois, F.; Crepet, A.; Tressou, J.; Verger, P. Population effects and variability. Methods Mol. Biol. 2012, 929, 521–581. [Google Scholar]
- Bois, F.Y.; Hsieh, N.H.; Gao, W.; Chiu, W.A.; Reisfeld, B. Well-tempered mcmc simulations for population pharmacokinetic models. J. Pharmacokinet. Pharmacodyn. 2020, 47, 543–559. [Google Scholar] [CrossRef]
- Kostewicz, E.S.; Aarons, L.; Bergstrand, M.; Bolger, M.B.; Galetin, A.; Hatley, O.; Jamei, M.; Lloyd, R.; Pepin, X.; Rostami-Hodjegan, A.; et al. Pbpk models for the prediction of in vivo performance of oral dosage forms. Eur. J. Pharm. Sci. 2014, 57, 300–321. [Google Scholar] [CrossRef]
- Jamei, M.; Marciniak, S.; Edwards, D.; Wragg, K.; Feng, K.; Barnett, A.; Rostami-Hodjegan, A. The simcyp population based simulator: Architecture, implementation, and quality assurance. Silico Pharm. 2013, 1, 9. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, J.C.; Evans, M.V.; Krishnan, K. Cutting edge pbpk models and analyses: Providing the basis for future modeling efforts and bridges to emerging toxicology paradigms. J. Toxicol. 2012, 2012, 852384. [Google Scholar] [CrossRef]
- Quignot, N.; Wiecek, W.; Amzal, B.; Dorne, J.L. The yin-yang of cyp3a4: A bayesian meta-analysis to quantify inhibition and induction of cyp3a4 metabolism in humans and refine uncertainty factors for mixture risk assessment. Arch. Toxicol. 2019, 93, 107–119. [Google Scholar] [CrossRef]
- US EPA. New Approach Methods Work Plan: Reducing Use of Animals in Chemical Testing; US Environmental Protection Agency: Washington, DC, USA, 2020.
- US EPA. Strategic Plan to Promote the Development and Implementation of Alternative Test Methods within the TSCA Program; Office of Chemical Safety and Pollution Prevention, US Environmental Protection Agency: Washington, DC, USA, 2018.
- ECHA. The Use of Alternatives to Testing on Animals for the Reach Regulation: Fourth Report (2020) under Article 117(3) of the Reach Regulation; European Chemicals Agency: Helsinki, Finland, 2020.
- ECHA. Read-Across Assessment Framework (RAAF)-Considerations on Multi-Constituent Substances and UVCBs; European Chemical Agency: Helsinki, Finland, 2017.
- Rusyn, I.; Daston, G.P. Computational toxicology: Realizing the promise of the toxicity testing in the 21st century. Environ. Health Perspect. 2010, 118, 1047–1050. [Google Scholar] [CrossRef] [PubMed]
- International HapMap Consortium. A haplotype map of the human genome. Nature 2005, 437, 1299–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altshuler, D.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Collins, F.S.; De la Vega, F.M.; Donnelly, P.; Egholm, M.; et al. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar]
- Burnett, S.D.; Karmakar, M.; Murphy, W.J.; Chiu, W.A.; Rusyn, I. A new approach method for characterizing inter-species toxicodynamic variability. J. Toxicol. Environ. Health Part A 2021, 84, 1020–1039. [Google Scholar] [CrossRef] [PubMed]
- Drakvik, E.; Altenburger, R.; Aoki, Y.; Backhaus, T.; Bahadori, T.; Barouki, R.; Brack, W.; Cronin, M.T.D.; Demeneix, B.; Hougaard Bennekou, S.; et al. Statement on advancing the assessment of chemical mixtures and their risks for human health and the environment. Environ. Int. 2020, 134, 105267. [Google Scholar] [CrossRef]
- Carnesecchi, E.; Svendsen, C.; Lasagni, S.; Grech, A.; Quignot, N.; Amzal, B.; Toma, C.; Tosi, S.; Rortais, A.; Cortinas-Abrahantes, J.; et al. Investigating combined toxicity of binary mixtures in bees: Meta-analysis of laboratory tests, modelling, mechanistic basis and implications for risk assessment. Environ. Int. 2019, 133, 105256. [Google Scholar] [CrossRef]
- Howard, G.J.; Schlezinger, J.J.; Hahn, M.E.; Webster, T.F. Generalized concentration addition predicts joint effects of aryl hydrocarbon receptor agonists with partial agonists and competitive antagonists. Environ. Health Perspect. 2010, 118, 666–672. [Google Scholar] [CrossRef] [Green Version]
- Hadrup, N.; Taxvig, C.; Pedersen, M.; Nellemann, C.; Hass, U.; Vinggaard, A.M. Concentration addition, independent action and generalized concentration addition models for mixture effect prediction of sex hormone synthesis in vitro. PLoS ONE 2013, 8, e70490. [Google Scholar] [CrossRef] [Green Version]
- WHO/IPCS. Guidance Document on Evaluating and Expressing Uncertainty in Hazard Characterization, 2nd ed.; World Health Organization & International Programme on Chemical Safety: Geneva, Switzerland, 2018; p. 159. [Google Scholar]
- Zhuang, X.J.; Feng, X.; Tang, W.H.; Zhu, J.L.; Li, M.; Li, J.S.; Zheng, X.Y.; Li, R.; Liu, P.; Qiao, J. Fam9b serves as a novel meiosis-related protein localized in meiotic chromosome cores and is associated with human gametogenesis. PLoS ONE 2021, 16, e0257248. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, T.; Li, X.; Yu, F.; Guo, Y. Comprehensive analysis of coexpressed long noncoding rnas and genes in breast cancer. J. Obstet. Gynaecol. Res. 2019, 45, 428–437. [Google Scholar] [CrossRef]
- Fan, X.; Zhu, Y.; Wang, N.; Zhang, B.; Zhang, C.; Wang, Y. Therapeutic dose of hydroxyurea-induced synaptic abnormalities on the mouse spermatocyte. Front. Physiol. 2021, 12, 666339. [Google Scholar] [CrossRef]
- Yoshihara, Y.; Kawasaki, M.; Tamada, A.; Nagata, S.; Kagamiyama, H.; Mori, K. Overlapping and differential expression of big-2, big-1, tag-1, and f3: Four members of an axon-associated cell adhesion molecule subgroup of the immunoglobulin superfamily. J. Neurobiol. 1995, 28, 51–69. [Google Scholar] [CrossRef]
- Haas, D.W.; Abdelwahab, M.T.; van Beek, S.W.; Baker, P.; Maartens, G.; Bradford, Y.; Ritchie, M.D.; Wasserman, S.; Meintjes, G.; Beeri, K.; et al. Pharmacogenetics of between-individual variability in plasma clearance of bedaquiline and clofazimine in South Africa. J. Infect. Dis. 2022, jiac024. Available online: https://pubmed.ncbi.nlm.nih.gov/35091749/ (accessed on 1 June 2022). [CrossRef]
- Kehoe, L.A.; Bernardinelli, Y.; Muller, D. Glun3a: An nmda receptor subunit with exquisite properties and functions. Neural Plast. 2013, 2013, 145387. [Google Scholar] [CrossRef] [Green Version]
- Slotkin, T.A.; Seidler, F.J. Comparative developmental neurotoxicity of organophosphates in vivo: Transcriptional responses of pathways for brain cell development, cell signaling, cytotoxicity and neurotransmitter systems. Brain Res. Bull. 2007, 72, 232–274. [Google Scholar] [CrossRef] [Green Version]
- Park, S.J.; Ciccone, S.L.; Beck, B.D.; Hwang, B.; Freie, B.; Clapp, D.W.; Lee, S.H. Oxidative stress/damage induces multimerization and interaction of fanconi anemia proteins. J. Biol. Chem. 2004, 279, 30053–30059. [Google Scholar] [CrossRef] [Green Version]
- Castella, M.; Pujol, R.; Callen, E.; Trujillo, J.P.; Casado, J.A.; Gille, H.; Lach, F.P.; Auerbach, A.D.; Schindler, D.; Benitez, J.; et al. Origin, functional role, and clinical impact of fanconi anemia fanca mutations. Blood 2011, 117, 3759–3769. [Google Scholar] [CrossRef]
- Macko, P.; Palosaari, T.; Whelan, M. Extrapolating from acute to chronic toxicity in vitro. Toxicol. In Vitro 2021, 76, 105206. [Google Scholar] [CrossRef]
- Deisenroth, C.; DeGroot, D.E.; Zurlinden, T.; Eicher, A.; McCord, J.; Lee, M.Y.; Carmichael, P.; Thomas, R.S. The alginate immobilization of metabolic enzymes platform retrofits an estrogen receptor transactivation assay with metabolic competence. Toxicol. Sci. 2020, 178, 281–301. [Google Scholar] [CrossRef]
- Wetmore, B.A. Quantitative in vitro-to-in vivo extrapolation in a high-throughput environment. Toxicology 2015, 332, 94–101. [Google Scholar] [CrossRef]
- U.S. EPA. Guidelines for the health risk assessment of chemical mixtures. Fed. Regist. 1986, 51, 34014–34025. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ford, L.C.; Jang, S.; Chen, Z.; Zhou, Y.-H.; Gallins, P.J.; Wright, F.A.; Chiu, W.A.; Rusyn, I. A Population-Based Human In Vitro Approach to Quantify Inter-Individual Variability in Responses to Chemical Mixtures. Toxics 2022, 10, 441. https://doi.org/10.3390/toxics10080441
Ford LC, Jang S, Chen Z, Zhou Y-H, Gallins PJ, Wright FA, Chiu WA, Rusyn I. A Population-Based Human In Vitro Approach to Quantify Inter-Individual Variability in Responses to Chemical Mixtures. Toxics. 2022; 10(8):441. https://doi.org/10.3390/toxics10080441
Chicago/Turabian StyleFord, Lucie C., Suji Jang, Zunwei Chen, Yi-Hui Zhou, Paul J. Gallins, Fred A. Wright, Weihsueh A. Chiu, and Ivan Rusyn. 2022. "A Population-Based Human In Vitro Approach to Quantify Inter-Individual Variability in Responses to Chemical Mixtures" Toxics 10, no. 8: 441. https://doi.org/10.3390/toxics10080441
APA StyleFord, L. C., Jang, S., Chen, Z., Zhou, Y.-H., Gallins, P. J., Wright, F. A., Chiu, W. A., & Rusyn, I. (2022). A Population-Based Human In Vitro Approach to Quantify Inter-Individual Variability in Responses to Chemical Mixtures. Toxics, 10(8), 441. https://doi.org/10.3390/toxics10080441