
HiPSC-Derived Hepatocyte-like Cells Can Be Used as a Model for Transcriptomics-Based Study of Chemical Toxicity

 , , ,
, , ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
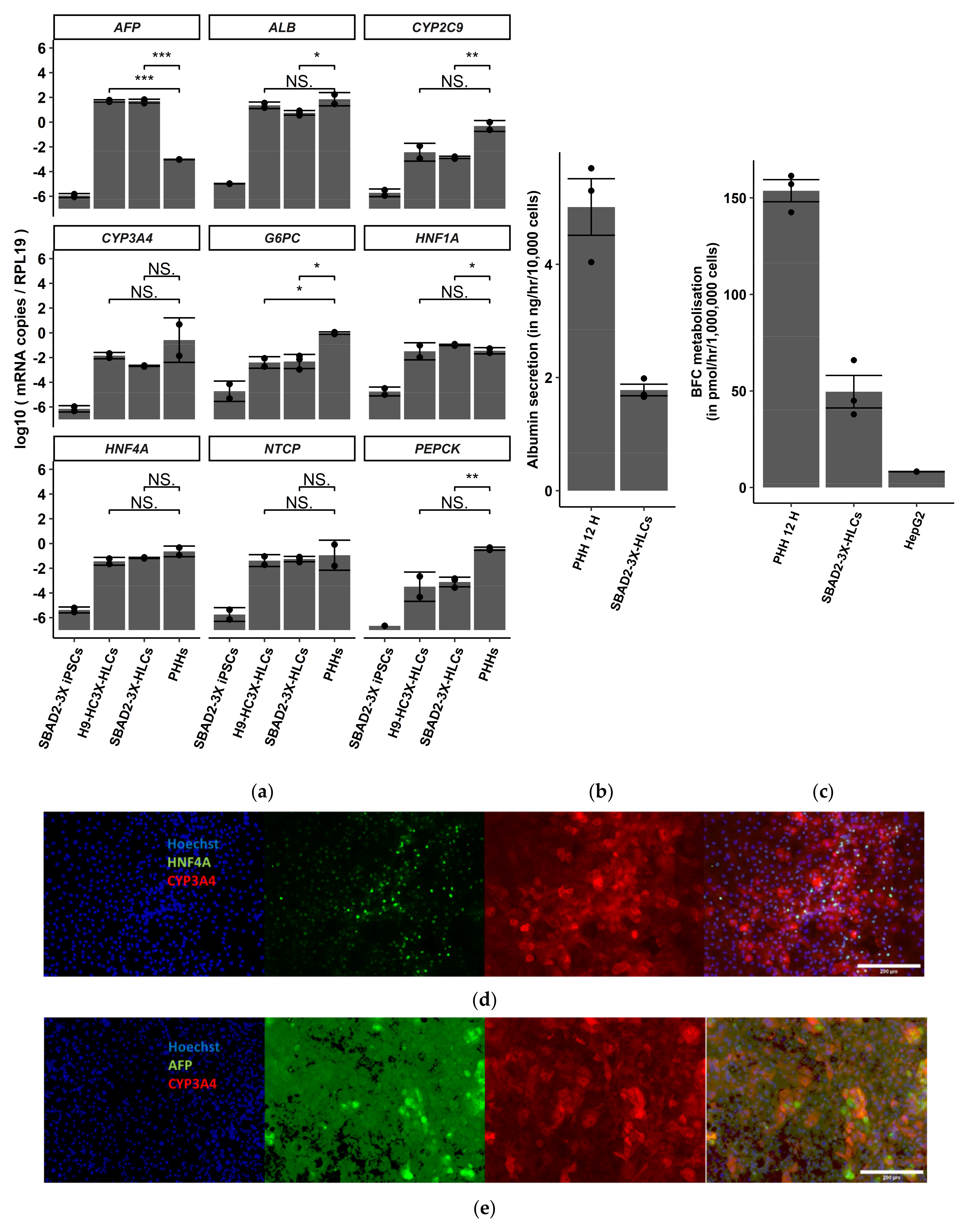
3.1. SBAD2-3x-AAGLY-HLCs Express Hepatocyte Markers, Produce Albumin and Have CYP3A4 Activity
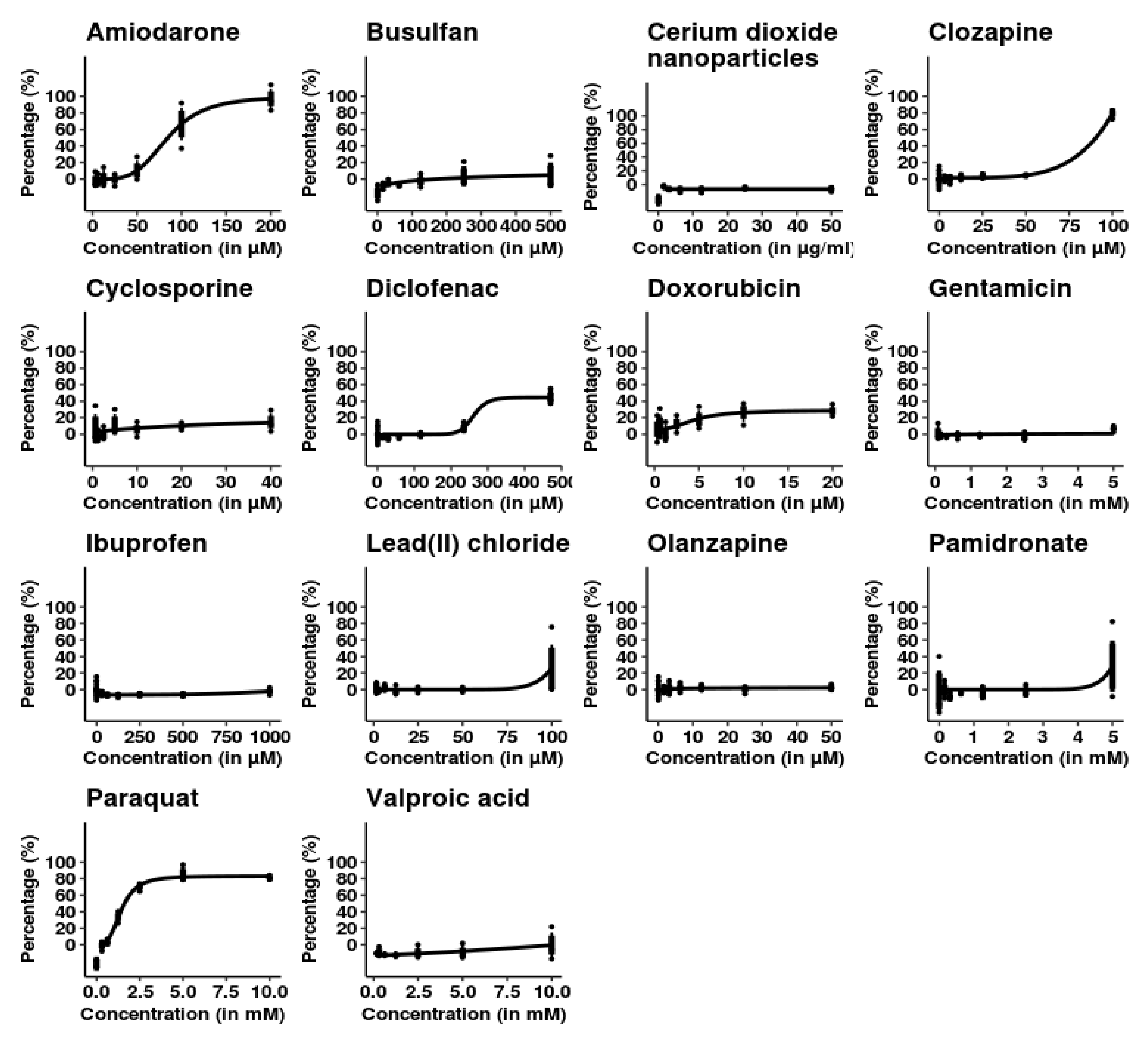
3.2. SBAD2-3x-AAGLY HLCs Cells Accurately Classify Chemicals Causing Acute Hepatocellular Injury
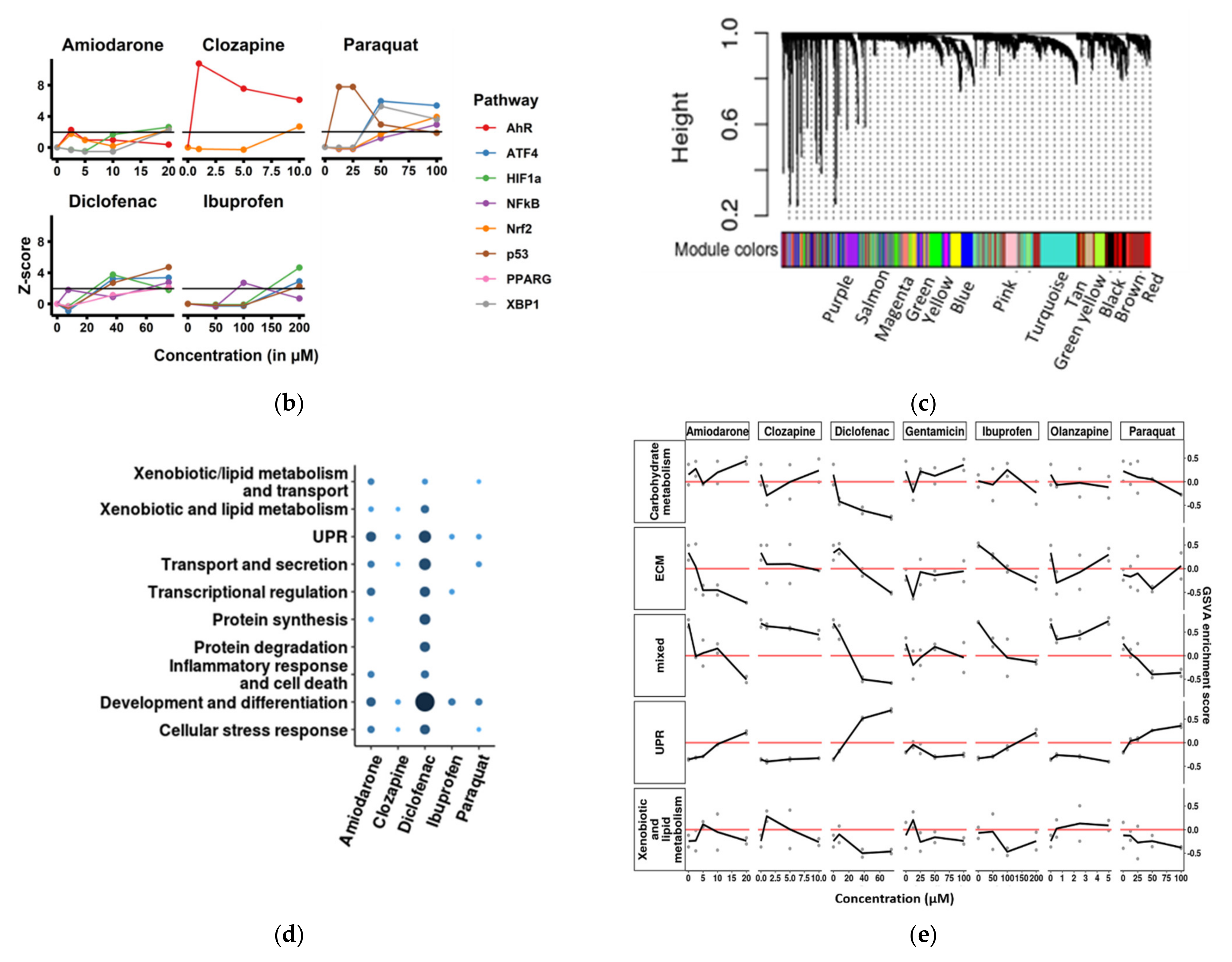
3.3. Cellular Stress Pathway Genes Are Highly Differentially Expressed in SBAD2-3x-AAGLY HLCs upon Chemical Treatment
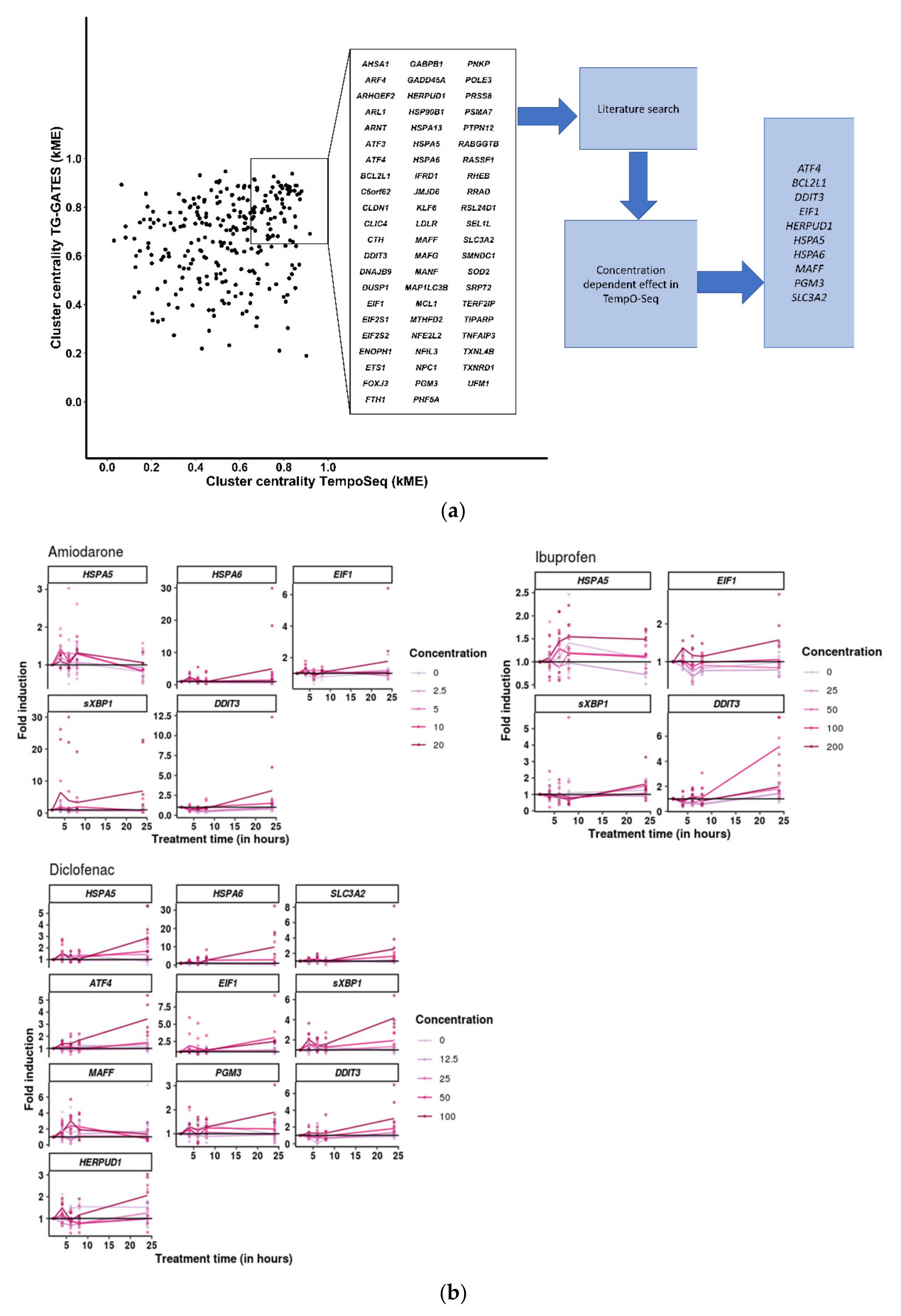
3.4. Cellular Stress Genes Show Differential Expression at Different Time-Points after Chemical Treatment
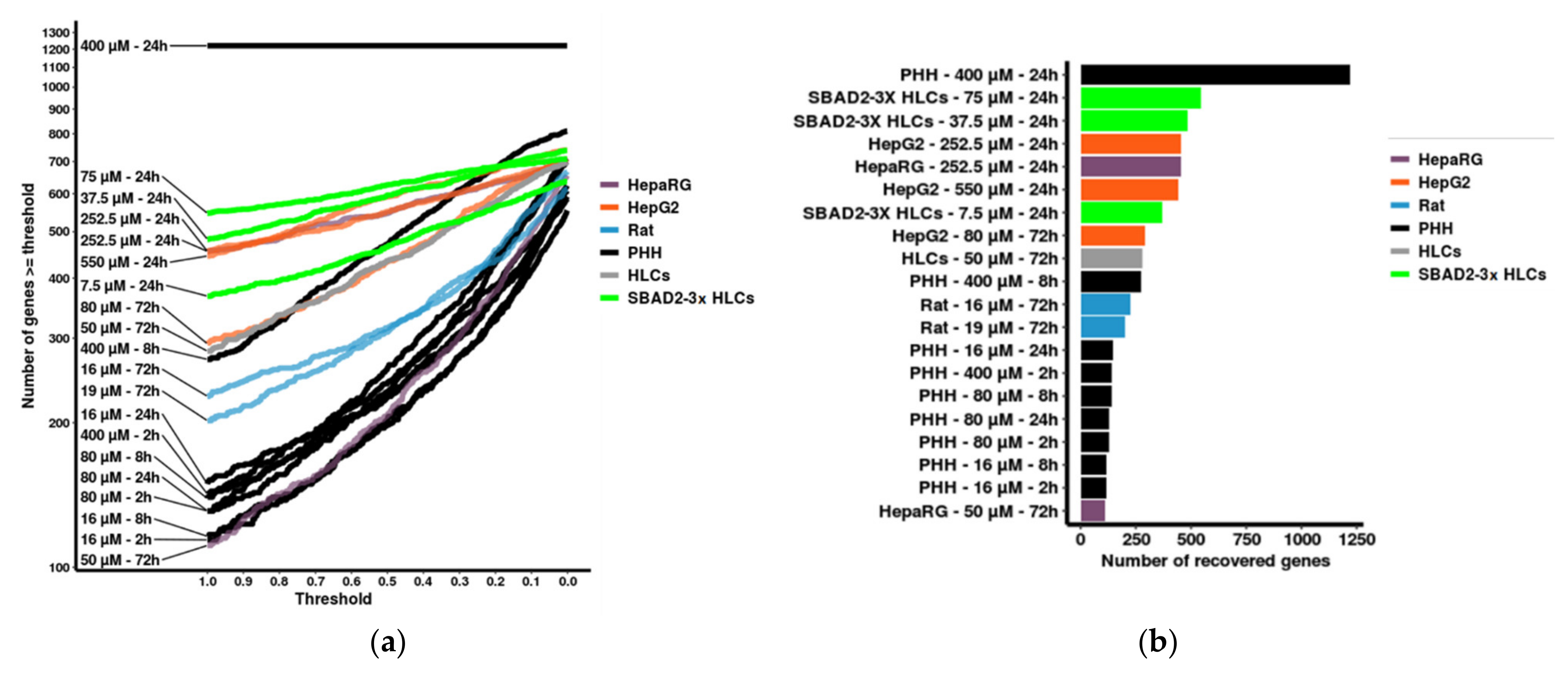
3.5. Benchmarking
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dresser, M.J.; Gray, A.T.; Giacomini, K.M. Kinetic and Selectivity Differences between Rodent, Rabbit, and Human Organic Cation Transporters (OCT1). J. Pharmacol. Exp. Ther. 2000, 292, 1146–1152. [Google Scholar]
- Fashe, M.M.; Juvonen, R.O.; Petsalo, A.; Räsänen, J.; Pasanen, M. Species-Specific Differences in the in Vitro Metabolism of Lasiocarpine. Chem. Res. Toxicol. 2015, 28, 2034–2044. [Google Scholar] [CrossRef] [PubMed]
- Li, A.P. Accurate prediction of human drug toxicity: A major challenge in drug development. Chem. Interact. 2004, 150, 3–7. [Google Scholar] [CrossRef]
- Garside, H.; Marcoe, K.F.; Chesnut-Speelman, J.; Foster, A.J.; Muthas, D.; Kenna, G.; Warrior, U.; Bowes, J.; Baumgartner, J. Evaluation of the use of imaging parameters for the detection of compound-induced hepatotoxicity in 384-well cultures of HepG2 cells and cryopreserved primary human hepatocytes. Toxicol. Vitro 2014, 28, 171–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanebratt, K.P.; Andersson, T.B. Evaluation of HepaRG Cells as an in Vitro Model for Human Drug Metabolism Studies. Drug Metab. Dispos. 2008, 36, 1444–1452. [Google Scholar] [CrossRef] [Green Version]
- Liguori, M.J.; Blomme, E.A.; Waring, J.F. Trovafloxacin-Induced Gene Expression Changes in Liver-Derived in Vitro Systems: Comparison of Primary Human Hepatocytes to HepG2 Cells. Drug Metab. Dispos. 2008, 36, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.C.; Lauschke, V.M.; Vorrink, S.U.; Palmgren, H.; Duffin, R.; Andersson, T.B.; Ingelman-Sundberg, M. Transcriptional, Functional, and Mechanistic Comparisons of Stem Cell–Derived Hepatocytes, HepaRG Cells, and Three-Dimensional Human Hepatocyte Spheroids as Predictive In Vitro Systems for Drug-Induced Liver Injury. Drug Metab. Dispos. 2017, 45, 419–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingelman-Sundberg, M.; Sim, S.C.; Gomez, A.; Rodriguez-Antona, C. Influence of cytochrome P450 polymorphisms on drug therapies: Pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol. Ther. 2007, 116, 496–526. [Google Scholar] [CrossRef]
- Gómez-Lechón, M.J.; Tolosa, L.; Conde, I.; Donato, M.T. Competency of different cell models to predict human hepatotoxic drugs. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1553–1568. [Google Scholar] [CrossRef]
- Donato, M.T.; Jover, R.; Gómez-Lechón, M. Hepatic Cell Lines for Drug Hepatotoxicity Testing: Limitations and Strategies to Upgrade their Metabolic Competence by Gene Engineering. Curr. Drug Metab. 2013, 14, 946–968. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, N.; Chen, S.; Cai, J.; Guo, Y.; Song, Z.; Che, J.; Liu, C.; Wu, C.; Ding, M.; Deng, H. Derivation and Characterization of Hepatic Progenitor Cells from Human Embryonic Stem Cells. PLoS ONE 2009, 4, e6468. [Google Scholar] [CrossRef] [Green Version]
- Hannan, N.; Segeritz, C.-P.; Touboul, T.; Vallier, L. Production of hepatocyte-like cells from human pluripotent stem cells. Nat. Protoc. 2013, 8, 430–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Duan, Y.; Tschudy-Seney, B.; Roll, G.; Behbahan, I.S.; Ahuja, T.P.; Tolstikov, V.; Wang, C.; McGee, J.; Khoobyari, S.; et al. Highly Efficient Differentiation of Functional Hepatocytes from Human Induced Pluripotent Stem Cells. Stem Cells Transl. Med. 2013, 2, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.; Kumar, M.; Tricot, T.; Elia, I.; Ordovas, L.; Jacobs, F.; One, J.; De Smedt, J.; Eelen, G.; Bird, M.; et al. Amino acid levels determine metabolism and CYP450 function of hepatocytes and hepatoma cell lines. Nat. Commun. 2020, 11, 1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hof, W.V.D.; Coonen, M.; Van Herwijnen, M.; Brauers, K.; Wodzig, W.K.W.H.; Van Delft, J.H.M.; Kleinjans, J.C.S. Classification of Hepatotoxicants Using HepG2 Cells: A Proof of Principle Study. Chem. Res. Toxicol. 2013, 27, 433–442. [Google Scholar] [CrossRef]
- Wink, S.; Hiemstra, S.; Herpers, B.; Van De Water, B. High-content imaging-based BAC-GFP toxicity pathway reporters to assess chemical adversity liabilities. Arch. Toxicol. 2016, 91, 1367–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wink, S.; Hiemstra, S.W.; Huppelschoten, S.; Klip, J.E.; Van De Water, B. Dynamic imaging of adaptive stress response pathway activation for prediction of drug induced liver injury. Arch. Toxicol. 2018, 92, 1797–1814. [Google Scholar] [CrossRef] [Green Version]
- Mueller, S.O.; Guillouzo, A.; Hewitt, P.G.; Richert, L. Drug biokinetic and toxicity assessments in rat and human primary hepatocytes and HepaRG cells within the EU-funded Predict-IV project. Toxicol. Vitro 2015, 30, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.Q.; Smith, P.C. Glucuronidation and Covalent Protein Binding of Benoxaprofen and Flunoxaprofen in Sandwich-Cultured Rat and Human Hepatocytes. Drug Metab. Dispos. 2009, 37, 2314–2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordovás, L.; Boon, R.; Pistoni, M.; Chen, Y.; Wolfs, E.; Guo, W.; Sambathkumar, R.; Bobis-Wozowicz, S.; Helsen, N.; Vanhove, J.; et al. Efficient Recombinase-Mediated Cassette Exchange in hPSCs to Study the Hepatocyte Lineage Reveals AAVS1 Locus-Mediated Transgene Inhibition. Stem Cell Rep. 2015, 5, 918–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, M.T.; Jimenez, N.; Castell, J.V.; Gómez-Lechón, M.J. Fluorescence-based assays for screening nine cytochrome p450 (p450) activities in intact cells expressing individual human p450 enzymes. Drug Metab. Dispos. 2004, 32, 699–706. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.J.; Henstock, P.V.; Dunn, M.C.; Smith, A.R.; Chabot, J.R.; De Graaf, D. Cellular Imaging Predictions of Clinical Drug-Induced Liver Injury. Toxicol. Sci. 2008, 105, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.; Huang, Y.-W.; Zhou, X.-D.; Ma, Y. Toxicity of Cerium Oxide Nanoparticles in Human Lung Cancer Cells. Int. J. Toxicol. 2006, 25, 451–457. [Google Scholar] [CrossRef]
- García-Rubio, L.; García-Abad, A.M.; Soler, F.; Míguez, M.P. Cytotoxicity of paraquat in freshly isolated rat hepatocytes: Effects of L-carnitine. BioFactors 1998, 8, 59–64. [Google Scholar] [CrossRef]
- Bramlage, P.; Goldis, A. Bioequivalence study of three ibuprofen formulations after single dose administration in healthy volunteers. BMC Pharmacol. 2008, 8, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barpe, D.R.; Rosa, D.D.; Froehlich, P.E. Pharmacokinetic evaluation of doxorubicin plasma levels in normal and overweight patients with breast cancer and simulation of dose adjustment by different indexes of body mass. Eur. J. Pharm. Sci. 2010, 41, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Tchounwou, P.B.; Yedjou, C.G.; Foxx, D.N.; Ishaque, A.B.; Shen, E. Lead-induced cytotoxicity and transcriptional activation of stress genes in human liver carcinoma (HepG2) cells. Mol. Cell. Biochem. 2004, 255, 161–170. [Google Scholar] [CrossRef]
- Regec, A.L.; Trifillis, A.L.; Trump, B.F. The Effect of Gentamicin on Human Renal Proximal Tubular Cells. Toxicol. Pathol. 1986, 14, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Rha, J.H.; Jang, I.J.; Lee, K.H.; Chong, W.S.; Shin, S.G.; Lee, N.; Myung, H.J. Pharmacokinetic comparison of two valproic acid formulations: A plain and a controlled release enteric-coated tablets. J. Korean Med Sci. 1993, 8, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Mav, D.; Shah, R.R.; Howard, B.E.; Auerbach, S.S.; Bushel, P.R.; Collins, J.B.; Gerhold, D.; Judson, R.S.; Karmaus, A.; Maull, E.A.; et al. A hybrid gene selection approach to create the S1500+ targeted gene sets for use in high-throughput transcriptomics. PLoS ONE 2018, 13, e0191105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limonciel, A.; Ates, G.; Carta, G.; Wilmes, A.; Watzele, M.; Shepard, P.J.; VanSteenhouse, H.C.; Seligmann, B.; Yeakley, J.M.; Van De Water, B.; et al. Comparison of base-line and chemical-induced transcriptomic responses in HepaRG and RPTEC/TERT1 cells using TempO-Seq. Arch. Toxicol. 2018, 92, 2517–2531. [Google Scholar] [CrossRef] [Green Version]
- Wellens, S.; Dehouck, L.; Chandrasekaran, V.; Singh, P.; Loiola, R.A.; Sevin, E.; Exner, T.; Jennings, P.; Gosselet, F.; Culot, M. Evaluation of a human iPSC-derived BBB model for repeated dose toxicity testing with cyclosporine A as model compound. Toxicol. Vitro 2021, 73, 105112. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.; Chandrasekaran, V.; Hardy, B.; Wilmes, A.; Jennings, P.; Exner, T.E. Temporal transcriptomic alterations of cadmium exposed human iPSC-derived renal proximal tubule-like cells. Toxicol. Vitro 2021, 76, 105229. [Google Scholar] [CrossRef]
- Kutmon, M.; van Iersel, M.; Bohler, A.; Kelder, T.; Nunes, N.; Pico, A.; Evelo, C.T. PathVisio 3: An Extendable Pathway Analysis Toolbox. PLoS Comput. Biol. 2015, 11, e1004085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, Y.; Nakatsu, N.; Yamashita, T.; Ono, A.; Ohno, Y.; Urushidani, T.; Yamada, H. Open TG-GATEs: A large-scale toxicogenomics database. Nucleic Acids Res. 2014, 43, D921–D927. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutsumi, S.; Gotoh, T.; Tomisato, W.; Mima, S.; Hoshino, T.; Hwang, H.-J.; Takenaka, H.; Tsuchiya, T.; Mori, M.; Mizushima, T. Endoplasmic reticulum stress response is involved in nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death Differ. 2004, 11, 1009–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, W.W.-L.; Puthalakath, H. Bcl-2 family proteins: The sentinels of the mitochondrial apoptosis pathway. IUBMB Life 2008, 60, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Hori, O.; Ichinoda, F.; Yamaguchi, A.; Tamatani, T.; Taniguchi, M.; Koyama, Y.; Katayama, T.; Tohyama, M.; Stern, D.M.; Ozawa, K.; et al. Role of Herp in the endoplasmic reticulum stress response. Genes Cells 2004, 9, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Katsuoka, F.; Motohashi, H.; Ishii, T.; Aburatani, H.; Engel, J.D.; Yamamoto, M. Genetic Evidence that Small Maf Proteins Are Essential for the Activation of Antioxidant Response Element-Dependent Genes. Mol. Cell. Biol. 2005, 25, 8044–8051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Li, X.; Li, C.; Zhang, Z.; Gao, X.; Jia, Z.; Chen, H.; Jia, Q.; Zhao, X.; Liu, J.; et al. SLC3A2 is a novel endoplasmic reticulum stress-related signaling protein that regulates the unfolded protein response and apoptosis. PLoS ONE 2018, 13, e0208993. [Google Scholar] [CrossRef] [PubMed]
- Ashida, H.; Kanazawa, K.; Danno, G.-I. Hepatic Phosphoglucomutase Activity as a Marker of Oxidative Stress Induced by Pro-oxidative Drugs. Biosci. Biotechnol. Biochem. 1994, 58, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Van Schadewijk, A.; Wout, E.F.A.V.; Stolk, J.; Hiemstra, P. A quantitative method for detection of spliced X-box binding protein-1 (XBP1) mRNA as a measure of endoplasmic reticulum (ER) stress. Cell Stress Chaperon 2011, 17, 275–279. [Google Scholar] [CrossRef] [Green Version]
- Oda, S.; Uchida, Y.; Aleo, M.D.; Koza-Taylor, P.H.; Matsui, Y.; Hizue, M.; Marroquin, L.D.; Whritenour, J.; Uchida, E.; Yokoi, T. An in vitro coculture system of human peripheral blood mononuclear cells with hepatocellular carcinoma-derived cells for predicting drug-induced liver injury. Arch. Toxicol. 2020, 95, 149–168. [Google Scholar] [CrossRef]
- Doktorova, T.Y.; Yildirimman, R.; Vinken, M.; Vilardell, M.; Vanhaecke, T.; Gmuender, H.; Brolén, G.; Holmgren, G.; Li, R.; Van Delft, J.; et al. Transcriptomic responses generated by hepatocarcinogens in a battery of liver-based in vitro models. Carcinogenesis 2013, 34, 1393–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uehara, T.; Kiyosawa, N.; Shimizu, T.; Omura, K.; Hirode, M.; Imazawa, T.; Mizukawa, Y.; Ono, A.; Miyagishima, T.; Nagao, T.; et al. Species-specific differences in coumarin-induced hepatotoxicity as an example toxicogenomics-based approach to assessing risk of toxicity to humans. Hum. Exp. Toxicol. 2008, 27, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Komulainen, T.; Lodge, T.; Hinttala, R.; Bolszak, M.; Pietilä, M.; Koivunen, P.; Hakkola, J.; Poulton, J.; Morten, K.J.; Uusimaa, J. Sodium valproate induces mitochondrial respiration dysfunction in HepG2 in vitro cell model. Toxicology 2015, 331, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Le Hellard, S.; Theisen, F.M.; Haberhausen, M.; Raeder, M.B.; Fernø, J.; Gebhardt, S.; Hinney, A.; Remschmidt, H.; Krieg, J.C.; Mehler-Wex, C.; et al. Association between the insulin-induced gene 2 (INSIG2) and weight gain in a German sample of antipsychotic-treated schizophrenic patients: Perturbation of SREBP-controlled lipogenesis in drug-related metabolic adverse effects? Mol. Psychiatry 2008, 14, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.H.; Han, D.; Shin, S.H.; Kim, E.-K.; Kim, H.-S. Proteomic identification of early urinary-biomarkers of acute kidney injury in preterm infants. Sci. Rep. 2020, 10, 4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.; Toprakhisar, B.; Van Haele, M.; Antoranz, A.; Boon, R.; Chesnais, F.; De Smedt, J.; Tricot, T.; Idoype, T.I.; Canella, M.; et al. A fully defined matrix to support a pluripotent stem cell derived multi-cell-liver steatohepatitis and fibrosis model. Biomaterials 2021, 276, 121006. [Google Scholar] [CrossRef]
- Wang, L.; Chen, J.; Ning, C.; Lei, D.; Ren, J. Endoplasmic Reticulum Stress Related Molecular Mechanisms in Nonalcoholic Fatty Liver Disease (NAFLD). Curr. Drug Targets 2018, 19, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Chen, S.; Zhang, J.; Doshi, U.; Li, A.P.; Guo, L. Endoplasmic Reticulum Stress Induction and ERK1/2 Activation Contribute to Nefazodone-Induced Toxicity in Hepatic Cells. Toxicol. Sci. 2016, 154, 368–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Green, R.M. Endoplasmic reticulum stress and liver diseases. Liver Res. 2019, 3, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Samuel, K.; Subramanian, R.; Braun, M.P.; Stearns, R.A.; Chiu, S.-H.L.; Evans, D.C.; Baillie, T.A. Extrapolation of Diclofenac Clearance from in Vitro Microsomal Metabolism Data: Role of Acyl Glucuronidation and Sequential Oxidative Metabolism of the Acyl Glucuronide. J. Pharmacol. Exp. Ther. 2002, 303, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Elaut, G.; Henkens, T.; Papeleu, P.; Snykers, S.; Vinken, M.; Vanhaecke, T.; Rogiers, V. Molecular Mechanisms Underlying the Dedifferentiation Process of Isolated Hepatocytes and Their Cultures. Curr. Drug Metab. 2006, 7, 629–660. [Google Scholar] [CrossRef]
- Holmgren, G.; Ulfenborg, B.; Asplund, A.; Toet, K.; Andersson, C.X.; Hammarstedt, A.; Hanemaaijer, R.; Küppers-Munther, B.; Synnergren, J. Characterization of Human Induced Pluripotent Stem Cell-Derived Hepatocytes with Mature Features and Potential for Modeling Metabolic Diseases. Int. J. Mol. Sci. 2020, 21, 469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.; Kim, H.; Park, J.Y.; Kim, G.; Han, J.; Chung, S.; Yang, J.H.; Jeon, J.S.; Woo, D.-H.; Han, C.; et al. Generation of uniform liver spheroids from human pluripotent stem cells for imaging-based drug toxicity analysis. Biomaterials 2021, 269, 120529. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Kawabata, K.; Nagamoto, Y.; Kishimoto, K.; Tashiro, K.; Sakurai, F.; Tachibana, M.; Kanda, K.; Hayakawa, T.; Furue, M.; et al. 3D spheroid culture of hESC/hiPSC-derived hepatocyte-like cells for drug toxicity testing. Biomaterials 2012, 34, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.L.; Takebe, T. Generation of multi-cellular human liver organoids from pluripotent stem cells. Methods Cell Biol. 2020, 159, 47–68. [Google Scholar] [CrossRef] [PubMed]
- Pettinato, G.; Lehoux, S.; Ramanathan, R.; Salem, M.M.; He, L.-X.; Muse, O.; Flaumenhaft, R.; Thompson, M.T.; Rouse, E.A.; Cummings, R.D.; et al. Generation of fully functional hepatocyte-like organoids from human induced pluripotent stem cells mixed with Endothelial Cells. Sci. Rep. 2019, 9, 8920. [Google Scholar] [CrossRef] [Green Version]
- Bin Ramli, M.N.; Lim, Y.S.; Koe, C.T.; Demircioglu, D.; Tng, W.; Gonzales, K.A.U.; Tan, C.P.; Szczerbinska, I.; Liang, H.; Soe, E.L.; et al. Human Pluripotent Stem Cell-Derived Organoids as Models of Liver Disease. Gastroenterology 2020, 159, 1471–1486.e12. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Hong, Y.; Xie, G.; Zhang, J.; Zhang, H.; Cai, Z. Comprehensive multi-omics approaches reveal the hepatotoxic mechanism of perfluorohexanoic acid (PFHxA) in mice. Sci. Total. Environ. 2021, 790, 148160. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Xu, Z.; Tian, L.; Yu, Z.; Wang, T.; Guo, Z.; Zhang, J.; Wang, T. Multi-Omics Integration to Reveal the Mechanism of Hepatotoxicity Induced by Dictamnine. Front. Cell Dev. Biol. 2021, 9, 700120. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghosh, S.; De Smedt, J.; Tricot, T.; Proença, S.; Kumar, M.; Nami, F.; Vanwelden, T.; Vidal, N.; Jennings, P.; Kramer, N.I.; et al. HiPSC-Derived Hepatocyte-like Cells Can Be Used as a Model for Transcriptomics-Based Study of Chemical Toxicity. Toxics 2022, 10, 1. https://doi.org/10.3390/toxics10010001
Ghosh S, De Smedt J, Tricot T, Proença S, Kumar M, Nami F, Vanwelden T, Vidal N, Jennings P, Kramer NI, et al. HiPSC-Derived Hepatocyte-like Cells Can Be Used as a Model for Transcriptomics-Based Study of Chemical Toxicity. Toxics. 2022; 10(1):1. https://doi.org/10.3390/toxics10010001
Chicago/Turabian StyleGhosh, Sreya, Jonathan De Smedt, Tine Tricot, Susana Proença, Manoj Kumar, Fatemeharefeh Nami, Thomas Vanwelden, Niels Vidal, Paul Jennings, Nynke I. Kramer, and et al. 2022. "HiPSC-Derived Hepatocyte-like Cells Can Be Used as a Model for Transcriptomics-Based Study of Chemical Toxicity" Toxics 10, no. 1: 1. https://doi.org/10.3390/toxics10010001
APA StyleGhosh, S., De Smedt, J., Tricot, T., Proença, S., Kumar, M., Nami, F., Vanwelden, T., Vidal, N., Jennings, P., Kramer, N. I., & Verfaillie, C. M. (2022). HiPSC-Derived Hepatocyte-like Cells Can Be Used as a Model for Transcriptomics-Based Study of Chemical Toxicity. Toxics, 10(1), 1. https://doi.org/10.3390/toxics10010001