The Unexplored Virome of Two Atlantic Coast Fish: Contribution of Next-Generation Sequencing to Fish Virology

 ,
, 
 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Fish Sample Collection and Nucleic Acids Extraction
2.2. Library Preparation and Sequencing
2.3. Bioinformatic Pipeline
2.4. Dataset Compilation and Phylogenetic Analysis
3. Results
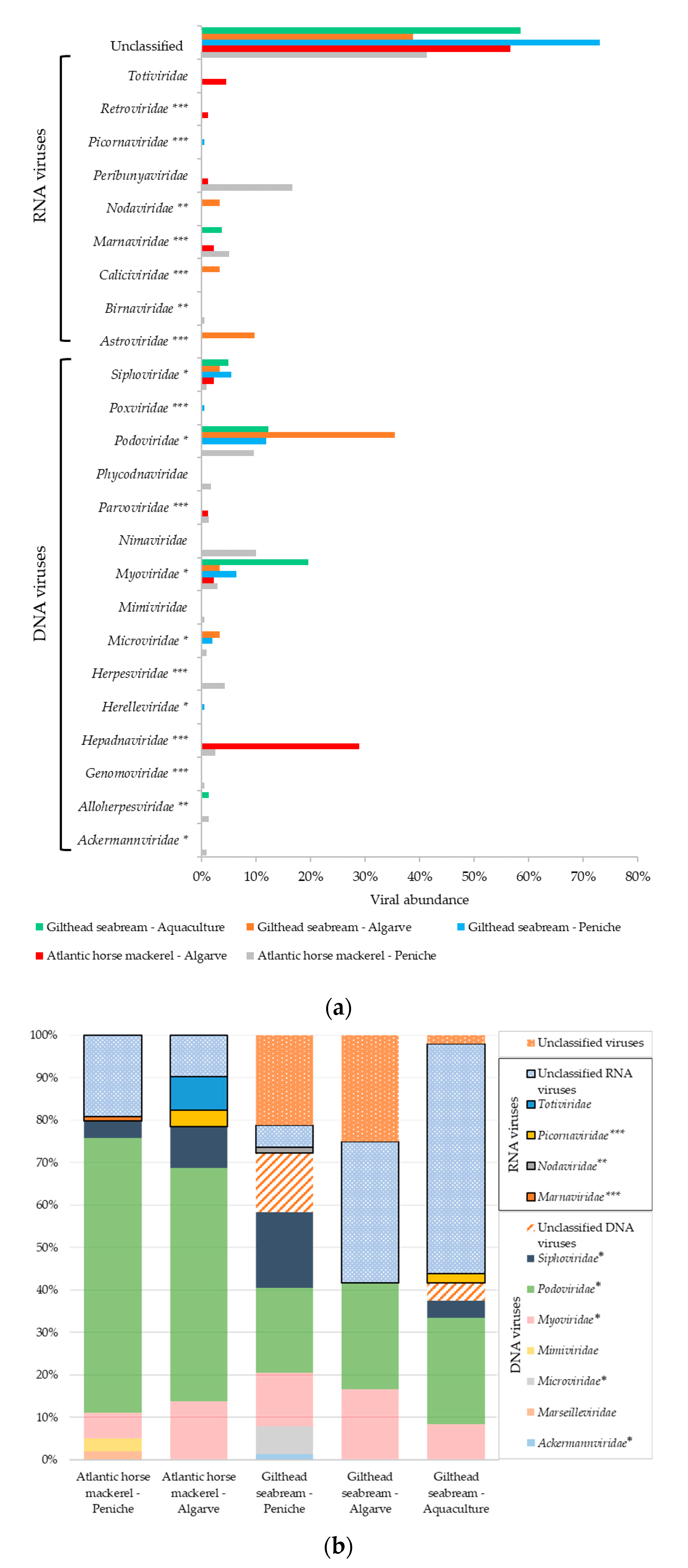
3.1. Viral Sequences Diversity
3.1.1. Viral Sequences Diversity—Global Results
3.1.2. Atlantic Horse Mackerel and Gilthead Seabream Viral Sequence Diversity
3.1.3. Viral Sequence Diversity: Fisheries vs. Aquaculture
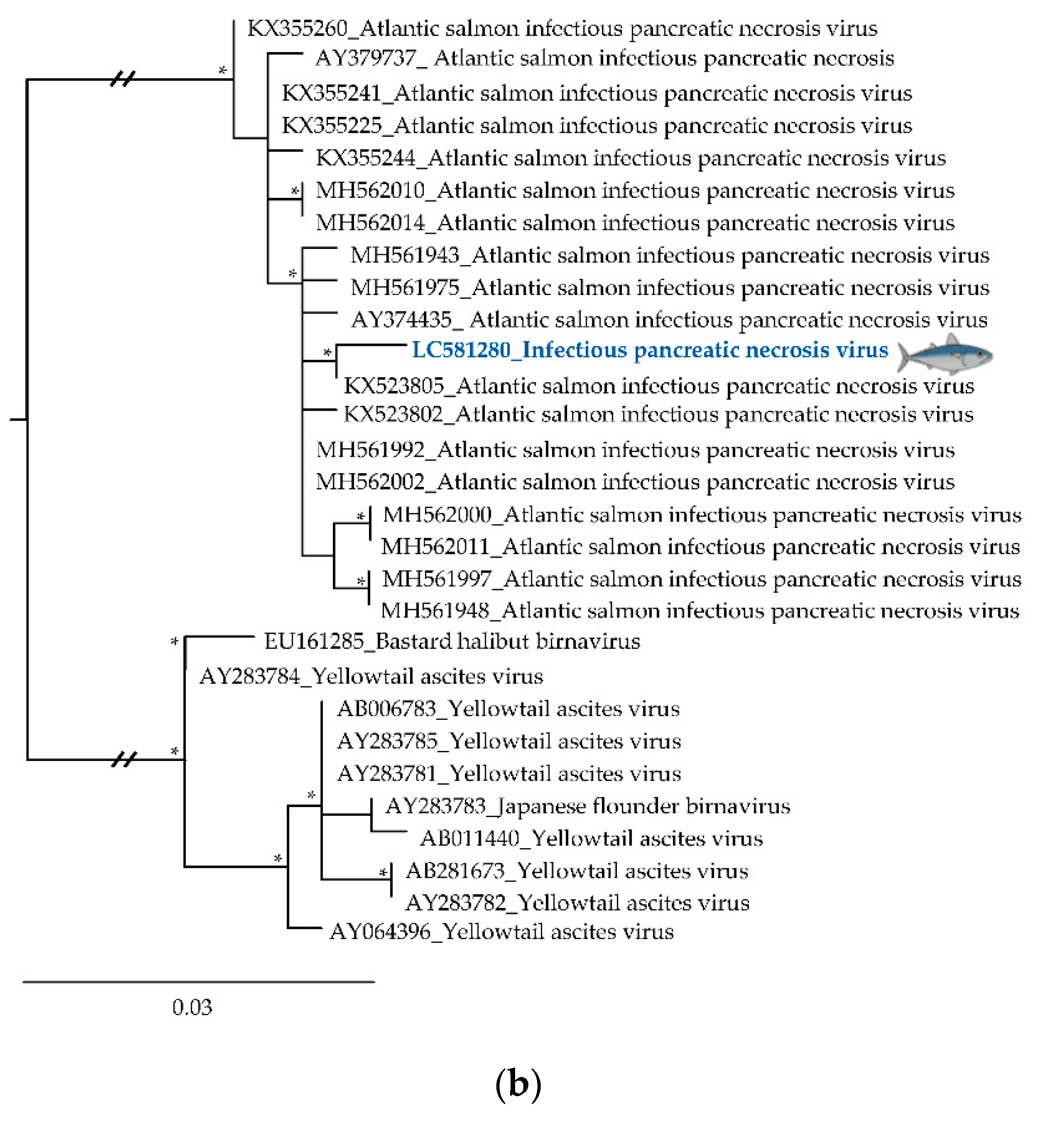
3.2. Phylogenetic Relationships of Relevant Viral Pathogenic Families
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- FAO. The State of World Fisheries and Aquaculture 2020. State World Fish. Aquac. 2020, 60–61. [Google Scholar] [CrossRef]
- Kibenge, F.S.B. Emerging viruses in aquaculture. Curr. Opin. Virol. 2019, 34, 97–103. [Google Scholar] [CrossRef]
- Geoghegan, J.L.; Di Giallonardo, F.; Cousins, K.; Shi, M.; Williamson, J.E.; Holmes, E.C. Hidden diversity and evolution of viruses in market fish. Virus Evol. 2018, 4, 1–11. [Google Scholar] [CrossRef]
- Chow, C.-E.T.; Suttle, C.A. Biogeography of Viruses in the Sea. Annu. Rev. Virol. 2015, 2, 41–66. [Google Scholar] [CrossRef]
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef]
- Weitz, J.S.; Wilhelm, S.W. Ocean viruses and their effects on microbial communities and biogeochemical cycles. F1000 Biol. Rep. 2012, 4, 2–9. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Chen, X.; Tian, J.-H.; Chen, L.-J.; Li, K.; Wang, W.; Eden, J.-S.; Shen, J.-J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef]
- Dill, J.A.; Camus, A.C.; Leary, J.H.; Di Giallonardo, F.; Holmes, E.C.; Ng, T.F.F. Distinct Viral Lineages from Fish and Amphibians Reveal the Complex Evolutionary History of Hepadnaviruses. J. Virol. 2016, 90, 7920–7933. [Google Scholar] [CrossRef]
- National Outbreak Reporting System (NORS). Available online: https://wwwn.cdc.gov/norsdashboard/ (accessed on 20 September 2020).
- Nkili-Meyong, A.A.; Bigarré, L.; Labouba, I.; Vallaeys, T.; Avarre, J.-C.; Berthet, N. Contribution of Next-Generation Sequencing to Aquatic and Fish Virology. Intervirology 2017, 59, 285–300. [Google Scholar] [CrossRef]
- Geoghegan, J.L.; Pirotta, V.; Harvey, E.; Smith, A.; Buchmann, J.P.; Ostrowski, M.; Eden, J.S.; Harcourt, R.; Holmes, E.C. Virological sampling of inaccessible wildlife with drones. Viruses 2018, 10, 300. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhang, Y.Z.; Holmes, E.C. Meta-transcriptomics and the evolutionary biology of RNA viruses. Virus Res. 2018, 243, 83–90. [Google Scholar] [CrossRef]
- Crane, M.; Hyatt, A. Viruses of fish: An overview of significant pathogens. Viruses 2011, 3, 2025–2046. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Park, S.Y.; Park, M.; Lee, S.; Lee, T.K. Seasonal dynamics and metagenomic characterization of marine viruses in Goseong Bay, Korea. PLoS ONE 2017, 12, e0169841. [Google Scholar] [CrossRef] [PubMed]
- Kurath, G.; Winton, J. Complex dynamics at the interface between wild and domestic viruses of finfish. Curr. Opin. Virol. 2011, 1, 73–80. [Google Scholar] [CrossRef]
- República Portuguesa. Available online: https://www.portugal.gov.pt/pt/gc21/governo/programa/programa-nacional-para-a-coesao-territorial-/ficheiros-coesao-territorial/programa-nacional-para-a-coesao-territorial-o-interior-em-numeros-territorio-pdf.aspx (accessed on 23 September 2020).
- Deutscher Wetterdienst (DWD), Sea Surface Temperature. Available online: https://www.dwd.de/EN/ourservices/rcccm/int/rcccm_int_sst.html (accessed on 23 September 2020).
- Fernandez-Cassi, X.; Rusiñol, M.; Martínez-Puchol, S. Viral Concentration and Amplification from Human Serum Samples Prior to Application of Next-Generation Sequencing Analysis. In The Human Virome: Methods and Protocols; Moya, A., Pérez Brocal, V., Eds.; Springer: New York, NY, USA, 2018; pp. 173–188. ISBN 978-1-4939-8682-8. [Google Scholar]
- Wang, D.; Urisman, A.; Liu, Y.T.; Springer, M.; Ksiazek, T.G.; Erdman, D.D.; Mardis, E.R.; Hickenbotham, M.; Magrini, V.; Eldred, J.; et al. Viral discovery and sequence recovery using DNA microarrays. PLoS Biol. 2003, 1, e2. [Google Scholar] [CrossRef]
- Briese, T.; Kapoor, A.; Mishra, N.; Jain, K.; Kumar, A.; Jabado, O.J.; Ian Lipkina, W. Virome capture sequencing enables sensitive viral diagnosis and comprehensive virome analysis. MBio 2015, 6, e01491-15. [Google Scholar] [CrossRef]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Alcantara, L.C.; Vanden Eynden, E.; Vandamme, A.M.; et al. Genome Detective: An automated system for virus identification from high-throughput sequencing data. Bioinformatics 2019, 35, 871–873. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. MetaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Quang Minh, B. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2009, 37, 26–31. [Google Scholar] [CrossRef]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; Dicuccio, M.; Edgar, R.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2009, 37, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Uhr, J.W.; Weissman, G. Intracellular distribution and degradation of bacteriophage in mammalian tissues. J. Immunol. 1965, 94, 544–550. [Google Scholar]
- Trigo, G.; Martins, T.G.; Fraga, A.G.; Longatto-Filho, A.; Castro, A.G.; Azeredo, J.; Pedrosa, J. Phage Therapy Is Effective against Infection by Mycobacterium ulcerans in a Murine Footpad Model. PLoS Negl. Trop. Dis. 2013, 7, e2183. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Fischer, D. Identification and investigation of ORFans in the viral world. BMC Genom. 2008, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.K.W.; Leung, C.Y.H.; Perera, H.K.K.; Ng, E.M.; Gilbert, M.; Joyner, P.H.; Grioni, A.; Ades, G.; Guan, Y.; Peiris, J.S.M.; et al. A Novel Group of Avian Astroviruses in Wild Aquatic Birds. J. Virol. 2012, 86, 13772–13778. [Google Scholar] [CrossRef]
- Rivera, R.; Nollens, H.H.; Venn-Watson, S.; Gulland, F.M.D.; Wellehan, J.F.X. Characterization of phylogenetically diverse astroviruses of marine mammals. J. Gen. Virol. 2010, 91, 166–173. [Google Scholar] [CrossRef]
- Sahul Hameed, A.S.; Ninawe, A.S.; Nakai, T.; Chi, S.C.; Johnson, K.L. ICTV virus taxonomy profile: Nodaviridae. J. Gen. Virol. 2019, 100, 3–4. [Google Scholar] [CrossRef]
- Toffan, A.; Pascoli, F.; Pretto, T.; Panzarin, V.; Abbadi, M.; Buratin, A.; Quartesan, R.; Gijon, D.; Padros, F. Viral nervous necrosis in gilthead sea bream (Sparus aurata) caused by reassortant betanodavirus RGNNV/SJNNV: An emerging threat for Mediterranean aquaculture. Sci. Rep. 2017, 7, 46755. [Google Scholar] [CrossRef]
- Berzak, R.; Scheinin, A.; Davidovich, N.; Regev, Y.; Diga, R.; Tchernov, D.; Morick, D. Prevalence of nervous necrosis virus (NNV) and Streptococcus species in wild marine fish and crustaceans from the Levantine Basin, Mediterranean Sea. Dis. Aquat. Organ. 2019, 133, 7–17. [Google Scholar] [CrossRef]
- Giacopello, C.; Foti, M.; Bottari, T.; Fisichella, V.; Barbera, G. Detection of viral encephalopathy and retinopathy virus (VERV) in wild marine fish species of the South Tyrrhenian Sea (Central Mediterranean). J. Fish Dis. 2013, 36, 819–821. [Google Scholar] [CrossRef]
- Luna, G.M.; Corinaldesi, C.; Dell’Anno, A.; Pusceddu, A.; Danovaro, R. Impact of aquaculture on benthic virus-prokaryote interactions in the Mediterranean Sea. Water Res. 2013, 47, 1156–1168. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.L.; Gabrielsen, M.; Beh, P.L.; Kueh, C.L.; Thong, Q.X.; Streetley, J.; Tan, W.S.; Bhella, D. Structure of the Macrobrachium rosenbergii nodavirus: A new genus within the Nodaviridae? PLoS Biol. 2018, 16, e3000038. [Google Scholar] [CrossRef] [PubMed]
- Isshiki, T.; Nagano, T.; Suzuki, S. Infectivity of aquabirnavirus strains to various marine fish species. Dis. Aquat. Organ. 2001, 46, 109–114. [Google Scholar] [CrossRef]
- Hirayama, T.; Nagano, I.; Shinmoto, H.; Yagyu, K.I.; Oshima, S.I. Isolation and characterization of virulent yellowtail ascites virus. Microbiol. Immunol. 2007, 51, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Manríquez, R.A.; Vera, T.; Villalba, M.V.; Mancilla, A.; Vakharia, V.N.; Yañez, A.J.; Cárcamo, J.G. Molecular characterization of infectious pancreatic necrosis virus strains isolated from the three types of salmonids farmed in Chile. Virol. J. 2017, 14, 1–16. [Google Scholar] [CrossRef]
- Mutoloki, S.; Evensen, Ø. Sequence similarities of the capsid gene of Chilean and European isolates of infectious pancreatic necrosis virus point towards a common origin. J. Gen. Virol. 2011, 92, 1721–1726. [Google Scholar] [CrossRef]
- Lun, J.H.; Hewitt, J.; Yan, G.J.H.; Tuipulotu, D.E.; Rawlinson, W.D.; White, P.A. Recombinant GII.P16/GII.4 sydney 2012 was the dominant norovirus identified in Australia and New Zealand in 2017. Viruses 2018, 10, 548. [Google Scholar] [CrossRef]
- Ruis, C.; Roy, S.; Brown, J.R.; Allen, D.J.; Goldstein, R.A.; Breuer, J. The emerging GII.P16-GII.4 Sydney 2012 norovirus lineage is circulating worldwide, arose by late-2014 and contains polymerase changes that may increase virus transmission. PLoS ONE 2017, 12, e0179572. [Google Scholar] [CrossRef]
- Ramesh, A.; Nakielny, S.; Hsu, J.; Kyohere, M.; Byaruhanga, O.; de Bourcy, C.; Egger, R.; Dimitrov, B.; Juan, Y.-F.; Sheu, J.; et al. Etiology of fever in Ugandan children: Identification of microbial pathogens using metagenomic next-generation sequencing and IDseq, a platform for unbiased metagenomic analysis. bioRxiv 2018, 385005. [Google Scholar] [CrossRef]
- McIntyre, C.L.; Knowles, N.J.; Simmonds, P. Proposals for the classification of human rhinovirus species A, B and C into genotypically assigned types. J. Gen. Virol. 2013, 94, 1791–1806. [Google Scholar] [CrossRef]
- Abeles, S.R.; Pride, D.T. Molecular bases and role of viruses in the human microbiome. J. Mol. Biol. 2014, 426, 3892–3906. [Google Scholar] [CrossRef]
- Łusiak-Szelachowska, M.; Weber-Dąbrowska, B.; Jończyk-Matysiak, E.; Wojciechowska, R.; Górski, A. Bacteriophages in the gastrointestinal tract and their implications. Gut Pathog. 2017, 9, 1–5. [Google Scholar] [CrossRef]
- Dąbrowska, K. Phage therapy: What factors shape phage pharmacokinetics and bioavailability? Systematic and critical review. Med. Res. Rev. 2019, 39, 2000–2025. [Google Scholar] [CrossRef]
- Geier, M.R.; Trigg, M.E.; Merril, C.R. Fate of bacteriophage lambda in Non-immune germ-free mice. Nature 1973, 246, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Ezenwa, V.O.; Price, S.A.; Altizer, S.; Vitone, N.D.; Cook, K.C. Host traits and parasite species richness in even and odd-toed hoofed mammals, Artiodactyla and Perissodactyla. Oikos 2006, 115, 526–536. [Google Scholar] [CrossRef]
- Lindenfors, P.; Nunn, C.L.; Jones, K.E.; Cunningham, A.A.; Sechrest, W.; Gittleman, J.L. Parasite species richness in carnivores: Effects of host body mass, latitude, geographical range and population density. Glob. Ecol. Biogeogr. 2007, 16, 496–509. [Google Scholar] [CrossRef]
- Webber, Q.M.R.; Fletcher, Q.E.; Willis, C.K.R. Viral Richness is Positively Related to Group Size, but Not Mating System, in Bats. EcoHealth 2017, 14, 652–661. [Google Scholar] [CrossRef]
- May, R.M.; Anderson, R.M. Population biology of infectious diseases: Part II. Nature 1979, 280, 455–461. [Google Scholar] [CrossRef]
- Munang’andu, H.M. Environmental viral metagenomics analyses in aquaculture: Applications in epidemiology and disease control. Front. Microbiol. 2016, 7, 1986. [Google Scholar] [CrossRef]
- Uren Webster, T.M.; Consuegra, S.; Hitchings, M.; Garcia de Leaniz, C. Interpopulation Variation in the Atlantic Salmon Microbiome Reflects Environmental and Genetic Diversity. Appl. Environ. Microbiol. 2018, 84, e00691-18. [Google Scholar] [CrossRef]
- Alavandi, S.V.; Poornima, M. Viral metagenomics: A tool for virus discovery and diversity in aquaculture. Indian J. Virol. 2012, 23, 88–98. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Source | Fishery Type | Nº of Specimens |
|---|---|---|---|
| Sparus aurata (gilthead seabream) | Peniche fish market | Wild fisheries | 5 |
| Sparus aurata (gilthead seabream) | Algarve fish market | Wild fisheries | 5 |
| Sparus aurata (gilthead seabream) | Algarve fish market | Aquaculture | 5 |
| Trachurus trachurus (Atlantic horse mackerel) | Peniche fish market | Wild fisheries | 10 |
| Trachurus trachurus (Atlantic horse mackerel) | Algarve fish market | Wild fisheries | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filipa-Silva, A.; Parreira, R.; Martínez-Puchol, S.; Bofill-Mas, S.; Barreto Crespo, M.T.; Nunes, M. The Unexplored Virome of Two Atlantic Coast Fish: Contribution of Next-Generation Sequencing to Fish Virology. Foods 2020, 9, 1634. https://doi.org/10.3390/foods9111634
Filipa-Silva A, Parreira R, Martínez-Puchol S, Bofill-Mas S, Barreto Crespo MT, Nunes M. The Unexplored Virome of Two Atlantic Coast Fish: Contribution of Next-Generation Sequencing to Fish Virology. Foods. 2020; 9(11):1634. https://doi.org/10.3390/foods9111634
Chicago/Turabian StyleFilipa-Silva, Andreia, Ricardo Parreira, Sandra Martínez-Puchol, Sílvia Bofill-Mas, Maria Teresa Barreto Crespo, and Mónica Nunes. 2020. "The Unexplored Virome of Two Atlantic Coast Fish: Contribution of Next-Generation Sequencing to Fish Virology" Foods 9, no. 11: 1634. https://doi.org/10.3390/foods9111634
APA StyleFilipa-Silva, A., Parreira, R., Martínez-Puchol, S., Bofill-Mas, S., Barreto Crespo, M. T., & Nunes, M. (2020). The Unexplored Virome of Two Atlantic Coast Fish: Contribution of Next-Generation Sequencing to Fish Virology. Foods, 9(11), 1634. https://doi.org/10.3390/foods9111634