A Real-Time PCR Screening Assay for Rapid Detection of Listeria Monocytogenes Outbreak Strains

 , , , , , , , ,
, , , , , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Isolates DNA Extraction
2.2. Serogroup
2.3. PFGE
2.4. Next Generation Sequencing
2.5. Real-Time PCR
3. Results
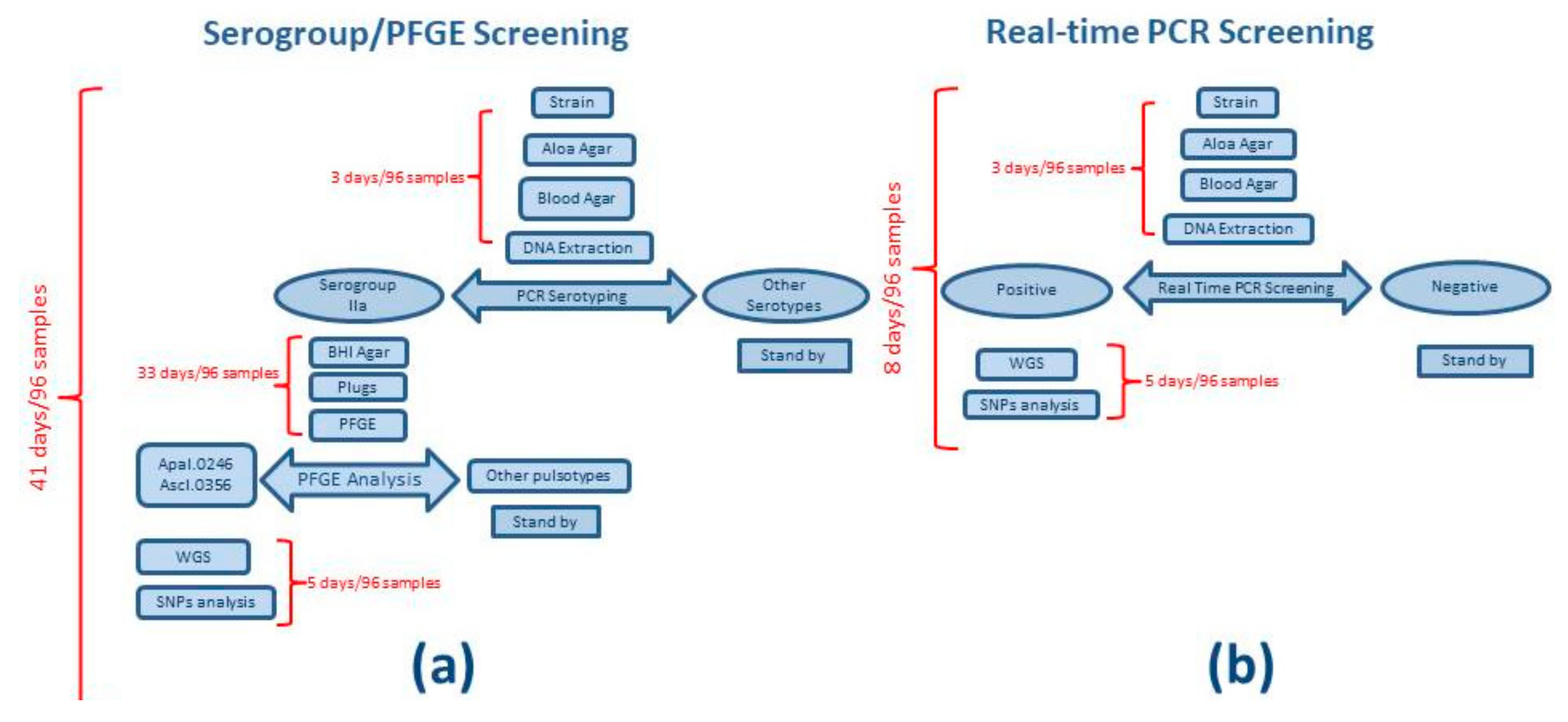
3.1. Workflow
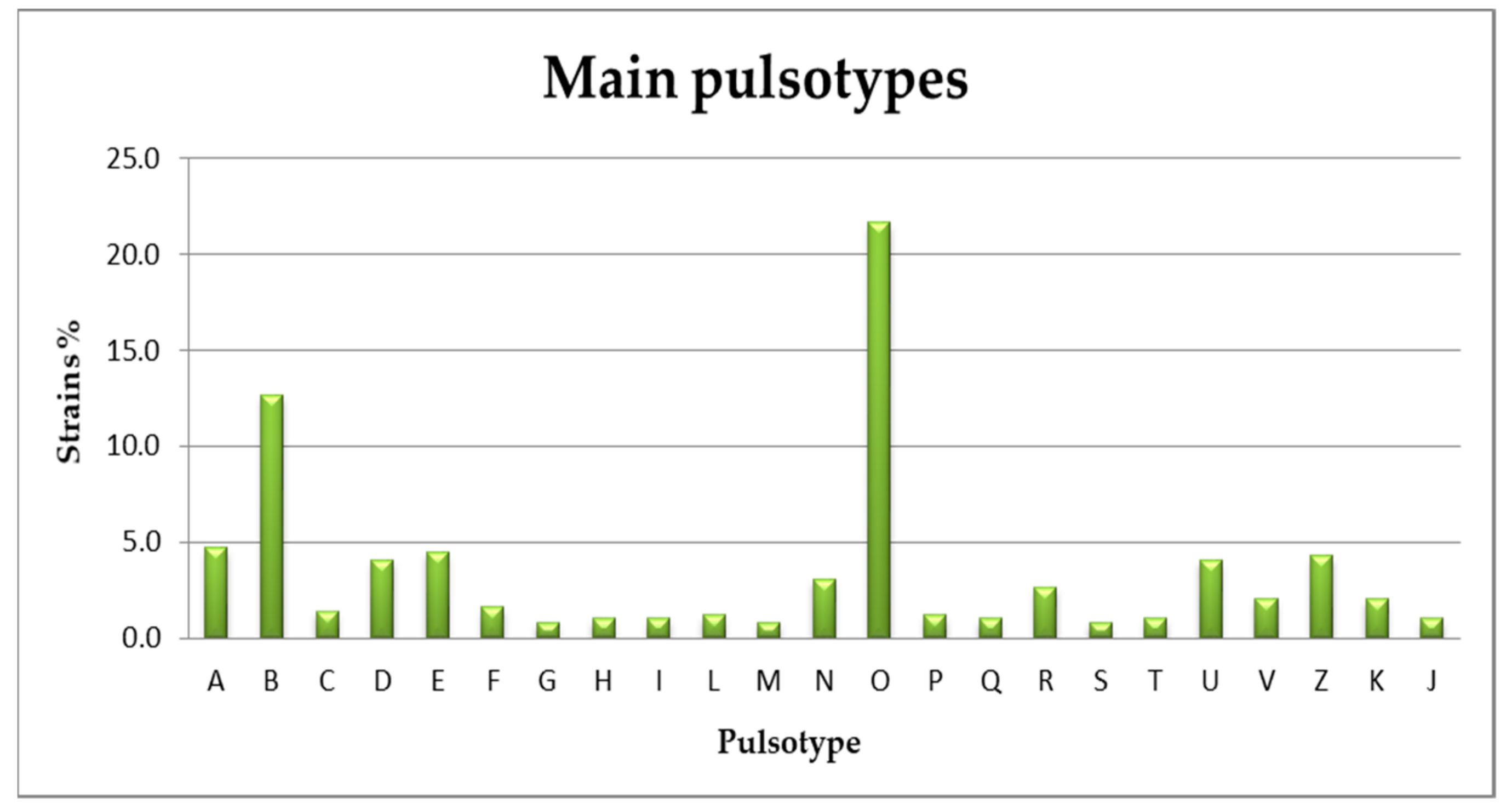
3.2. Serogrouping and PFGE
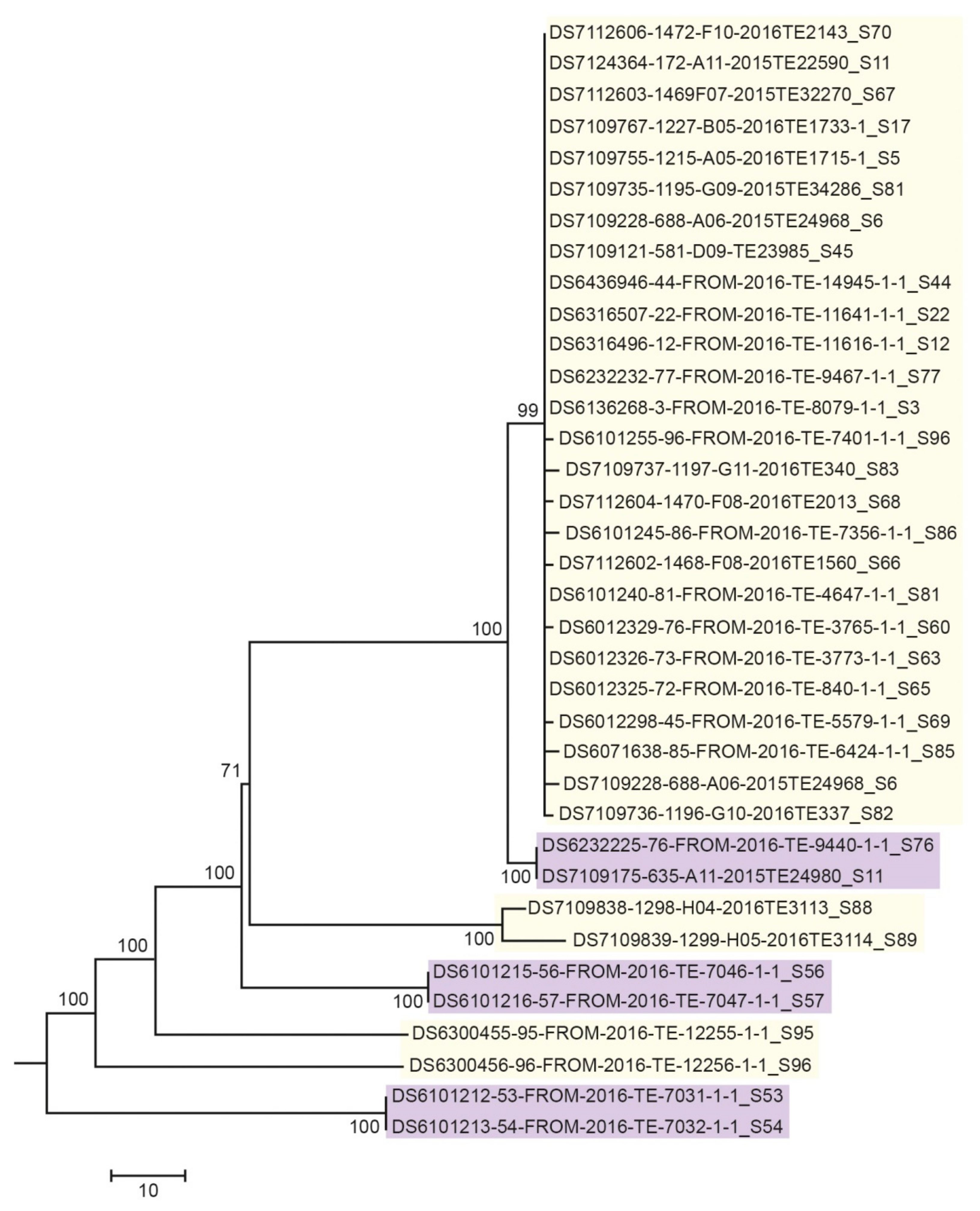
3.3. Next Generation Sequencing
3.4. Real-Time PCR
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Radoshevich, L.; Cossart, P. Listeria monocytogenes: Towards a complete picture of its physiology and pathogenesis. Nat. Rev. Microbiol. 2018, 16, 32–46. [Google Scholar] [CrossRef]
- Chlebicz, A.; Śliżewska, K. Campylobacteriosis, Salmonellosis, Yersiniosis, and Listeriosis as Zoonotic Foodborne Diseases: A Review. Int. J. Environ. Res. Public Health 2018, 15, 863. [Google Scholar] [CrossRef]
- European Food Safety Authority (EFSA) and European Centre for Disease Prevention and Control. (ECDC). The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2018. EFSA J. 2019, 17, 5926. Available online: https://efsa.onlinelibrary.wiley.com/doi/epdf/10.2903/j.efsa.2019.5926 (accessed on 27 December 2019).
- Duranti, A.; Sabbatucci, M.; Blasi, G.; Acciari, V.A.; Ancora, M.; Bella, A.; Busani, L.; Centorame, P.; Cammà, C.; Conti, F.; et al. A severe outbreak of listeriosis in central Italy with a rare pulsotype associated with processed pork products. J. Med. Microbiol. 2018, 67, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Pietzka, A.; Allerberger, F.; Murer, A.; Lennkh, A.; Stöger, A.; Cabal Rosel, A.; Huhulescu, S.; Maritschnik, S.; Springer, B.; Lepuschitz, S.; et al. Whole Genome Sequencing Based Surveillance of L. monocytogenes for Early Detection and Investigations of Listeriosis Outbreaks. Front. Public Health 2019, 7, 139. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.R.; Burall, L.S. Serotype to genotype: The changing landscape of listeriosis outbreak investigations. Food Microb. 2018, 75, 18–27. [Google Scholar] [CrossRef]
- Struelens, M.J.; Brisse, S. From molecular to genomic epidemiology: Transforming surveillance and control of infectious diseases. Eurosurveillance 2013, 18, 20386. [Google Scholar] [CrossRef]
- Quainoo, S.; Coolen, J.P.M.; van Hijum, S.A.F.T.; Huynen, M.A.; Melchers, W.J.G.; van Schaik, W.; Wertheim, H.F.L. Whole-Genome Sequencing of Bacterial Pathogens: The Future of Nosocomial Outbreak Analysis. Clin. Microbiol. Rev. 2017, 30, 1015–1063. [Google Scholar] [CrossRef]
- Doumith, M.; Buchrieser, C.; Glaser, P.; Jacquet, C.; Martin, P. Differentiation of the Major Listeria monocytogenes Serovars by Multiplex PCR. J. Clin. Microbiol. 2004, 42, 3819–3822. [Google Scholar] [CrossRef]
- Kérouanton, A.; Marault, M.; Petit, L.; Grout, J.; Dao, T.T.; Brisabois, A. Evaluation of a multiplex PCR assay as an alternative method for Listeria monocytogenes serotyping. J. Microbiol. Methods 2010, 80, 134–137. [Google Scholar] [CrossRef]
- PulseNet-International. Standard Operating Procedure for Pulsenet PFGE of Listeria monocytogenes. 2013. Available online: http://www.cdc.gov/pulsenet/PDF/listeria-pfge-protocol-508c.pdf (accessed on 21 November 2019).
- Acciari, V.A.; Torresi, M.; Iannetti, L.; Scattolini, S.; Pomilio, F.; Decastelli, L.; Colmegna, S.; Muliari, R.; Bossù, T.; Proroga, Y.; et al. Listeria monocytogenes in smoked salmon and other smoked fish at retail in Italy: Frequency of contamination and strain characterization in products from different manufacturers. J. Food Prot. 2017, 80, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.L.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Gardner, S.N.; Slezak, T.; Hall, B.G. kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genomes. Bioinformatics 2015, 31, 2877–2878. [Google Scholar] [CrossRef]
- Morganti, M.; Scaltriti, E.; Cozzolino, P.; Bolzoni, L.; Casadei, G.; Pierantoni, M.; Foni, E.; Pongolini, S. Processing-Dependent and Clonal Contamination Patterns of Listeria monocytogenes in the Cured Ham Food Chain Revealed by Genetic Analysis. Appl. Environ. Microbiol. 2015, 82, 822–831. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 2725–2729. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Vaerman, J.L.; Saussoy, P.; Ingargiola, I. Evaluation of Real Time PCR data. J. Biol. Regul. Homeost. Agents 2004, 18, 212–214. [Google Scholar]
- Lee, S.; Chen, Y.; Gorski, L.; Ward, T.J.; Osborne, J.; Kathariou, S. Listeria monocytogenes Source Distribution Analysis Indicates Regional Heterogeneity and Ecological Niche Preference among Serotype 4b Clones. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Orsi, R.H.; den Bakker, H.C.; Wiedmann, M. Listeria monocytogenes lineages: Genomics, evolution, ecology, and phenotypic characteristics. Int. J. Med. Microbiol. 2011, 301, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, F.; Palma, F.; Guillier, L.; Lucchi, A.; De Cesare, A.; Manfreda, G. Listeria monocytogenes Sequence Types 121 and 14 Repeatedly Isolated within One Year of Sampling in a Rabbit Meat Processing Plant: Persistence and Ecophysiology. Front. Microbiol. 2018, 9, 596. [Google Scholar] [CrossRef]
- Michelon, D.; Félix, B.; Vingadassalon, N.; Mariet, J.-F.; Larsson, J.T.; Møller-Nielsen, E.; Roussel, S. PFGE Standard Operating Procedures for Listeria monocytogenes: Harmonizing the Typing of Food and Clinical Strains in Europe. Foodborne Pathog. Dis. 2015, 12, 244–252. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Oligonucleotide | Sequence 5′-3′ | Size |
|---|---|---|
| Rec-fwd | AAATAATGCGGAGTTAAAAGGTGAA | 74 bp |
| Rec-rev | TGGACTGCATTTGGTATGTGAGT | |
| Rec-probe | FAM-TACGGATTGCCGTCCCCGAAAGT-BHQ1 | |
| Trans-fwd | CTCATTACGTTGATTGGCATACG | 79 bp |
| Trans-rev | GGTTCGTGGTCTCCTTTTACAATAA | |
| Trans-probe | JOE-AACGAAGAAAAGGGAAAAACTCCCACCC-BHQ1 |
| Assay | Slope | R2 | Efficiency |
|---|---|---|---|
| Rec | −3.34 | 0.998 | 99% |
| Trans | −3.47 | 0.999 | 94% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torresi, M.; Ruolo, A.; Acciari, V.A.; Ancora, M.; Blasi, G.; Cammà, C.; Centorame, P.; Centorotola, G.; Curini, V.; Guidi, F.; et al. A Real-Time PCR Screening Assay for Rapid Detection of Listeria Monocytogenes Outbreak Strains. Foods 2020, 9, 67. https://doi.org/10.3390/foods9010067
Torresi M, Ruolo A, Acciari VA, Ancora M, Blasi G, Cammà C, Centorame P, Centorotola G, Curini V, Guidi F, et al. A Real-Time PCR Screening Assay for Rapid Detection of Listeria Monocytogenes Outbreak Strains. Foods. 2020; 9(1):67. https://doi.org/10.3390/foods9010067
Chicago/Turabian StyleTorresi, Marina, Anna Ruolo, Vicdalia Aniela Acciari, Massimo Ancora, Giuliana Blasi, Cesare Cammà, Patrizia Centorame, Gabriella Centorotola, Valentina Curini, Fabrizia Guidi, and et al. 2020. "A Real-Time PCR Screening Assay for Rapid Detection of Listeria Monocytogenes Outbreak Strains" Foods 9, no. 1: 67. https://doi.org/10.3390/foods9010067
APA StyleTorresi, M., Ruolo, A., Acciari, V. A., Ancora, M., Blasi, G., Cammà, C., Centorame, P., Centorotola, G., Curini, V., Guidi, F., Marcacci, M., Orsini, M., Pomilio, F., & Di Domenico, M. (2020). A Real-Time PCR Screening Assay for Rapid Detection of Listeria Monocytogenes Outbreak Strains. Foods, 9(1), 67. https://doi.org/10.3390/foods9010067