Validation Including Uncertainty Estimation of a GC–MS/MS Method for Determination of Selected Halogenated Priority Substances in Fish Using Rapid and Efficient Lipid Removing Sample Preparation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Standards and Reagents
2.2. Fish Samples
2.3. Lipid and Moisture Determination
2.4. Sample Preparation
2.5. Instrumental Analysis
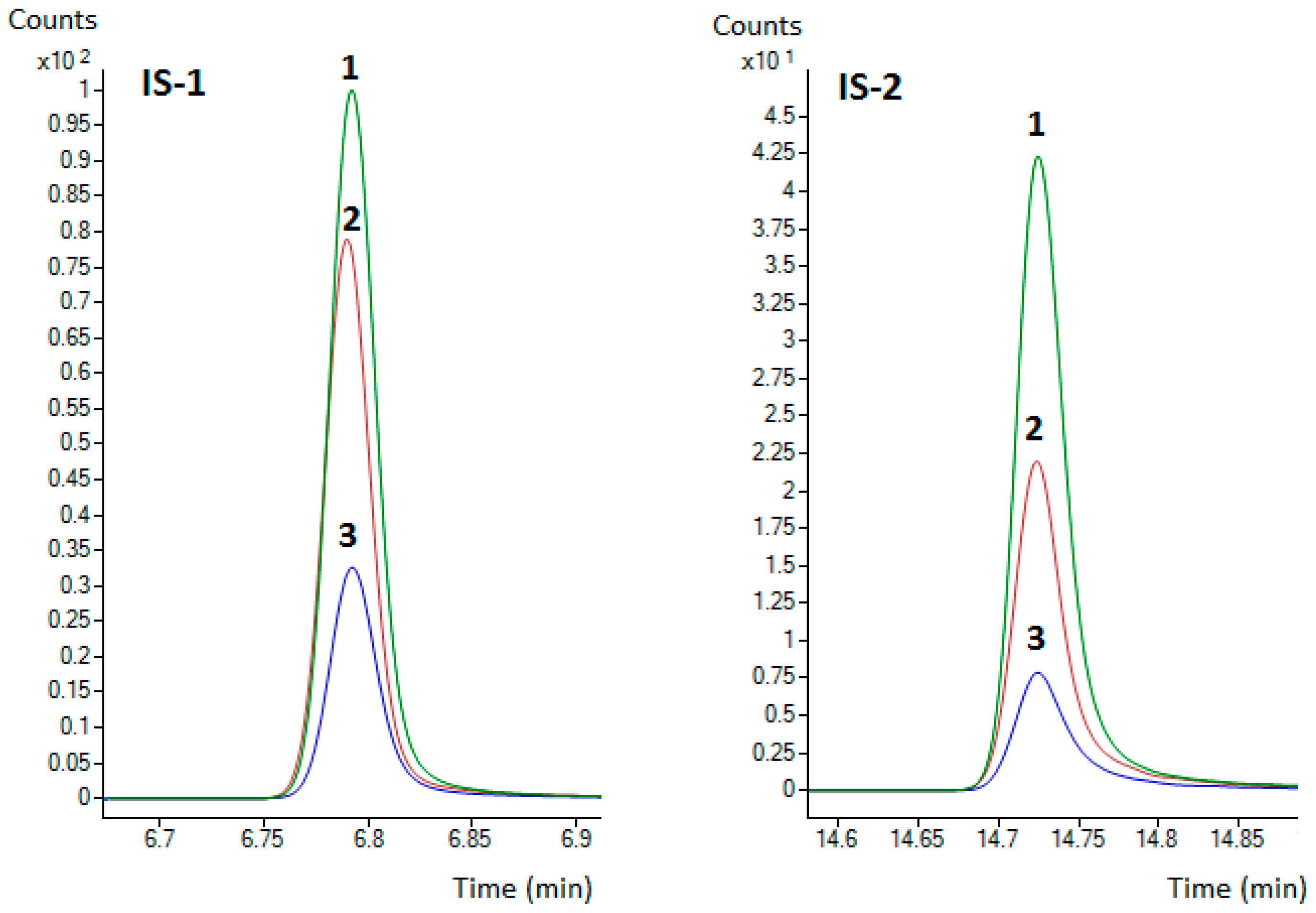
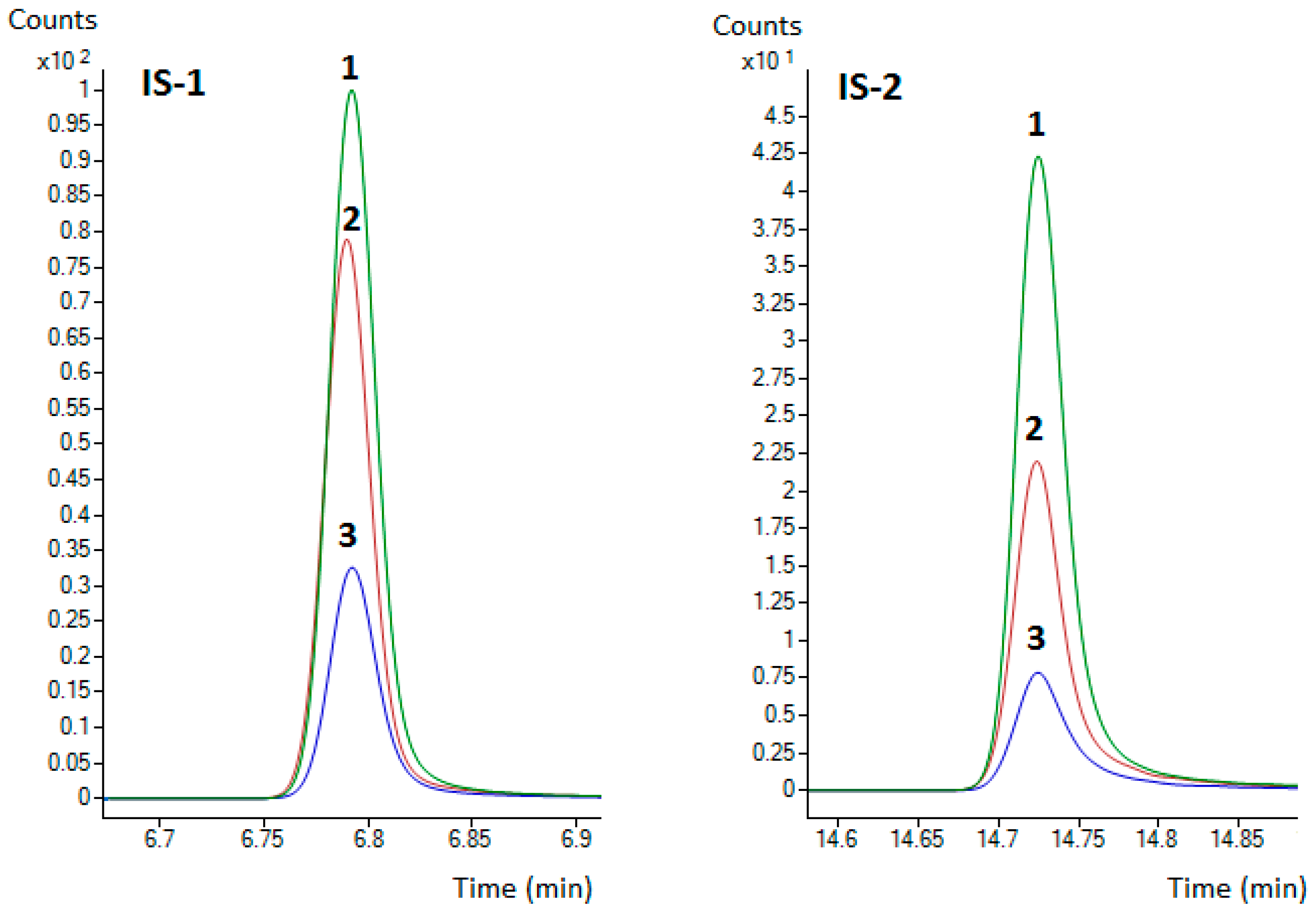
2.6. Matrix Effect Evaluation
2.7. Measurement Uncertainty Calculation
3. Results and Discussion
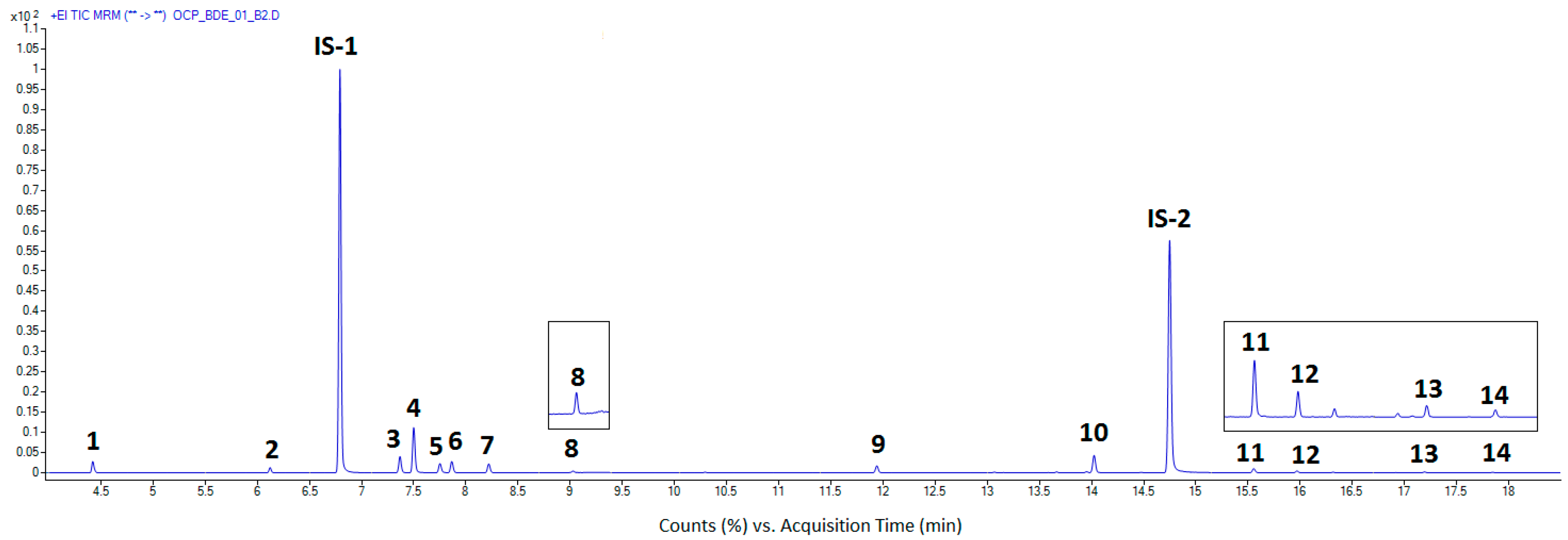
3.1. Instrumental Analysis
3.2. ME
3.3. Method Validation
3.3.1. Linearity
3.3.2. Limits of the Method
3.3.3. Recovery
3.3.4. Accuracy
3.3.5. Uncertainty of Measurement
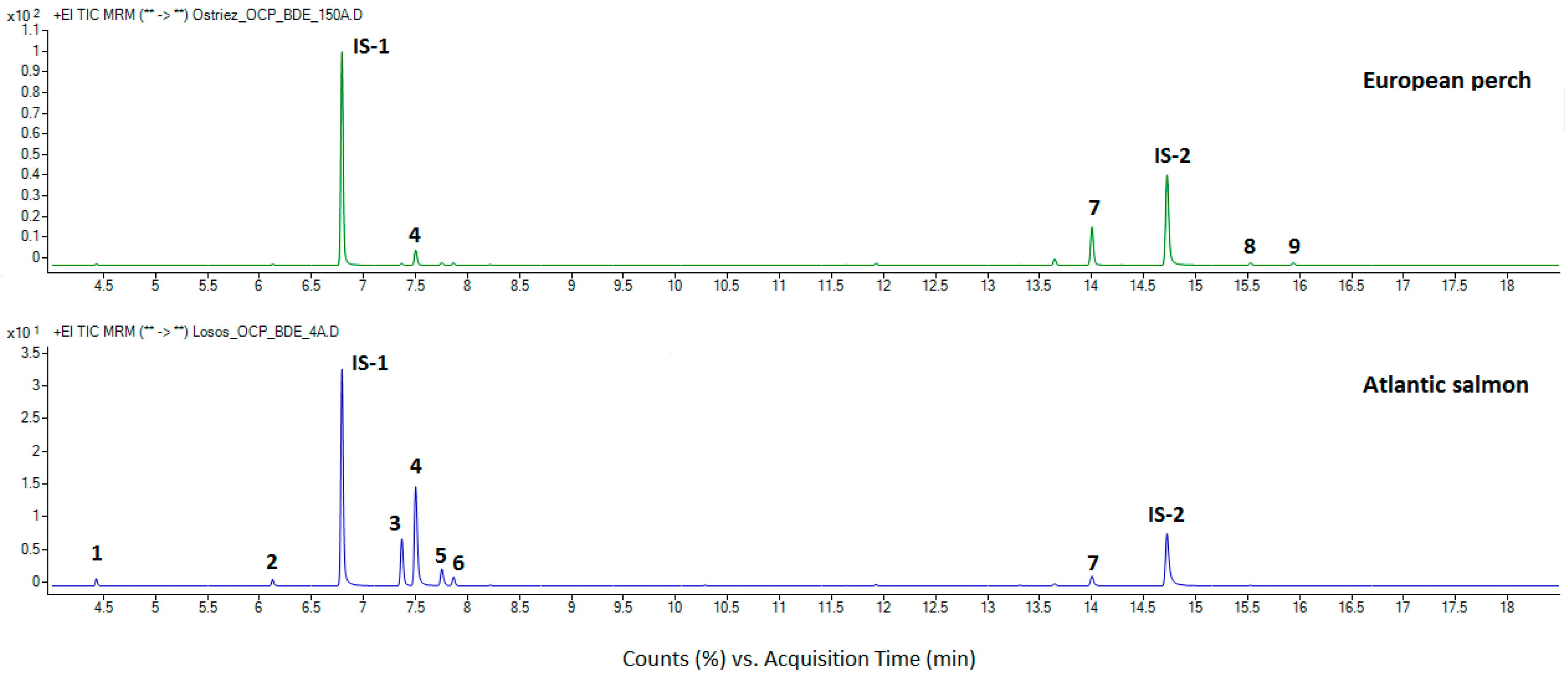
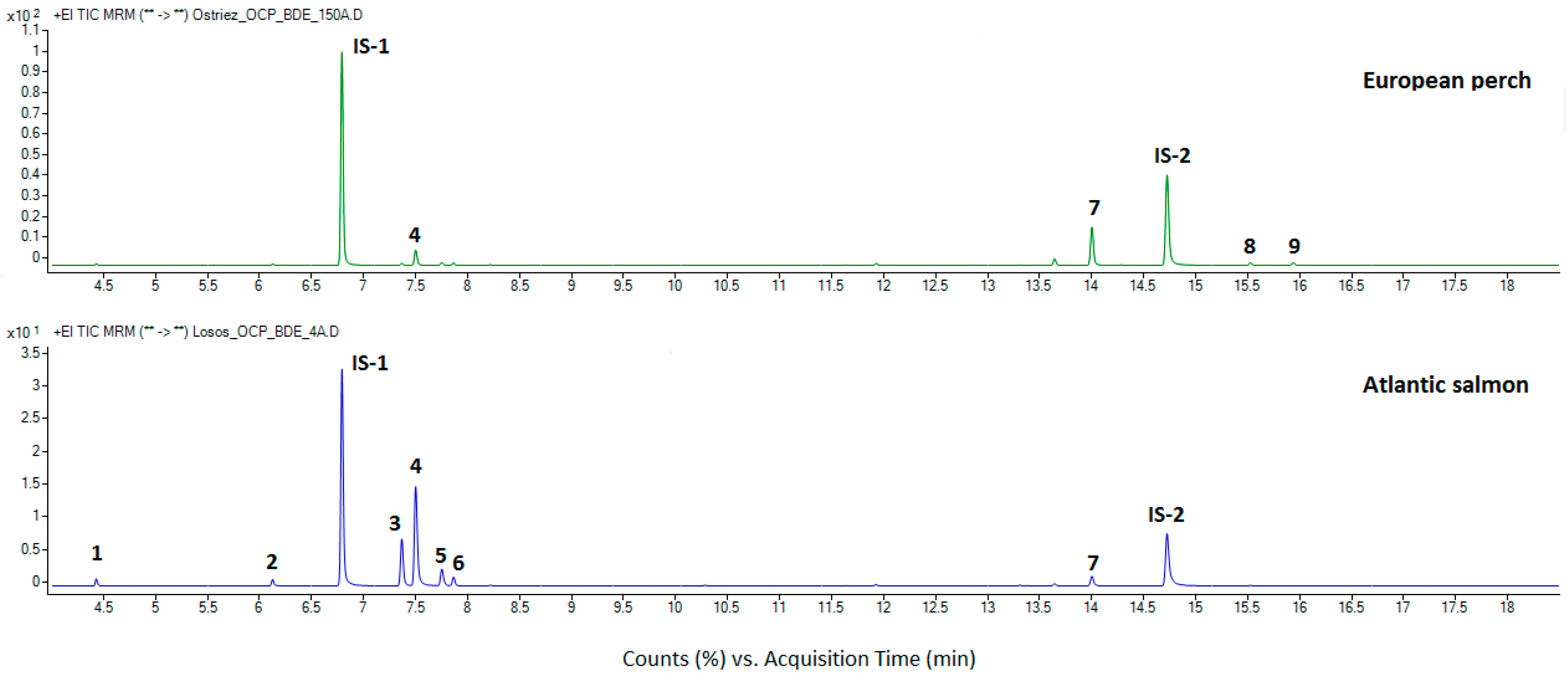
3.4. Application of the Method to Real Samples
3.5. Method’s Analytical Eco-Scale Evaluation
3.6. Comparison of the Proposed Method with Other Reported QuEChERS Based Methods
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Haggblom, M.M.; Bossert, I.D. Halogenated organic compounds: A global perspective. In Dehalogenation: Microbial Processes and Environmental Applications; Haggblom, M.M., Bossert, I.D., Eds.; Kluwer Academic Publishers: Norwell, MA, USA, 2003; pp. 3–32. [Google Scholar]
- United Nations Environment Program (UNEP); Stockholm Convention on Persistent Organic Pollutants. Adoption of Amendments of Annexes A, B and C; United Nations Environment Program: Geneva, Switzerland, 2009. [Google Scholar]
- European Commission. Regulation (EC) No 396/2005 of the European Parliament and of the Council of 23 February 2005 on maximum residue levels of pesticides in or on food and feed of plant and animal origin and amending Council Directive 91/414/EEC. Offic. J. Eur. Commun. 2005, L 70, 1–16. [Google Scholar]
- European Commission. 2014/118/EU: Commission Recommendation of 3 March 2014 on the monitoring of traces of brominated flame retardants in food. Offic. J. 2014, L 65, 39–40. [Google Scholar]
- European Commission. Decision No 2455/2001/EC of the European Parliament and of the Council of 20 November 2001 establishing the list of priority substances in the field of water policy and amending Directive 2000/60/EC. Offic. J. Eur. Commun. 2001, L 331, 1–5. [Google Scholar]
- US EPA. List of Priority Pollutants. Available online: http://water.epa.gov/scitech/methods/cwa/ pollutants.cfm (accessed on 17 July 2018).
- Chung, S.W.C.; Chen, B.L.S. Determination of organochlorine pesticide residues in fatty foods: A critical review on the analytical methods and their testing capabilities. J. Chromatogr. A 2011, 1218, 5555–5567. [Google Scholar] [CrossRef] [PubMed]
- Berton, P.; Lana, N.B.; Ríos, J.M.; García-Reyes, J.F.; Altamirano, J.C. State of the art of environmentally friendly sample preparation approaches for determination of PBDEs and metabolites in environmental and biological samples: A critical review. Anal. Chim. Acta 2016, 905, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Tölgyessy, P.; Miháliková, Z.; Matulová, M. Determination of selected chlorinated priority substances in fish using QuEChERS method with dual dSPE clean-up and gas chromatography. Chromatographia 2016, 79, 1561–1568. [Google Scholar] [CrossRef]
- Pietroń, W.J.; Małagocki, P. Quantification of polybrominated diphenyl ethers (PBDEs) in food. A review. Talanta 2017, 167, 411–427. [Google Scholar] [CrossRef] [PubMed]
- Rejczak, T.; Tuzimski, T. A review of recent developments and trends in the QuEChERS sample preparation approach. Open Chem. 2015, 13, 980–1010. [Google Scholar] [CrossRef]
- Norli, H.R.; Christiansen, A.; Deribe, E. Application of QuEChERS method for extraction of selected persistent organic pollutants in fish tissue and analysis by gas chromatography mass spectrometry. J. Chromatogr. A 2011, 1218, 7234–7241. [Google Scholar] [CrossRef]
- Molina-Ruiz, J.M.; Cieslik, E.; Cieslik, I.; Walkowska, I. Determination of pesticide residues in fish tissues by modified QuEChERS method and dual-d-SPE clean-up coupled to gas chromatography-mass spectrometry. Environ. Sci. Pollut. Res. Int. 2015, 22, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.S.; Utture, S.; Banerjee, K.; Ahammed Shabeer, T.P.; Kamble, N.; Mathew, S.; Ashok Kumar, K. Multiresidue analysis of multiclass pesticides and polyaromatic hydrocarbons in fatty fish by gas chromatography tandem mass spectrometry and evaluation of matrix effect. Food Chem. 2016, 196, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.A.; Sieve, K.K.; Ratajczak, R.E.; Bringolf, R.B.; Belden, J.B. Simultaneous extraction and cleanup of high-lipid organs from white sturgeon (Acipenser transmontanus) for multiple legacy and emerging organic contaminants using QuEChERS sample preparation. Talanta 2016, 146, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, P.L.; Fortin, F.; Groleau, P.E.; Brousseau, P.; Fournier, M.; Desrosiers, M. QuEChERS extraction for multi-residue analysis of PCBs, PAHs, PBDEs and PCDD/Fs in biological samples. Talanta 2017, 165, 332–338. [Google Scholar] [CrossRef]
- Han, L.; Matarrita, J.; Sapozhnikova, Y.; Lehotay, S.J. Evaluation of a recent product to remove lipids and other matrix co-extractives in the analysis of pesticide residues and environmental contaminants in foods. J. Chromatogr. A 2016, 1449, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Tölgyessy, P.; Nagyová, S. Rapid sample preparation method with high lipid removal efficiency for determination of sulphuric acid stable organic compounds in fish samples. Food Anal. Methods 2018, 11, 2485–2496. [Google Scholar] [CrossRef]
- Tölgyessy, P.; Miháliková, Z. Rapid determination of total lipids in fish samples employing extraction/partitioning with acetone/ethyl acetate solvent mixture and gravimetric quantification. Food Control 2016, 60, 44–49. [Google Scholar] [CrossRef]
- Frenich, A.G.; Martínez Vidal, J.L.; Fernández Moreno, J.L.; Romero-González, R. Compensation for matrix effects in gas chromatography–tandem mass spectrometry using a single point standard addition. J. Chromatogr. A 2009, 1216, 4798–4808. [Google Scholar] [CrossRef] [PubMed]
- Suchánek, M.; Friedecký, B.; Kratochvíla, J.; Budina, M.; Bartoš, V. Recommendations for the determination of uncertainties in the results of measurements/clinical tests in clinical laboratories. Klin. Biochem. Metab. 2006, 14, 43–53. (In Czech) [Google Scholar]
- L’Homme, B.; Scholl, G.; Eppe, G.; Focant, J.F. Validation of a gas chromatography–triple quadrupole mass spectrometry method for confirmatory analysis of dioxins and dioxin-like polychlorobiphenyls in feed following new EU Regulation 709/2014. J. Chromatogr. A 2015, 1376, 149–158. [Google Scholar] [CrossRef]
- Munaretto, J.S.; Ferronato, G.; Ribeiro, L.C.; Martins, M.L.; Adaime, M.B.; Zanella, R. Development of a multiresidue method for the determination of endocrine disrupters in fish fillet using gas chromatography–triple quadrupole tandem mass spectrometry. Talanta 2013, 116, 827–834. [Google Scholar] [CrossRef]
- Sapozhnikova, Y.; Lehotay, S.J. Multi-class, multi-residue analysis of pesticides, polychlorinated biphenyls, polycyclic aromatic hydrocarbons, polybrominated diphenyl ethers and novel flame retardants in fish using fast, low-pressure gas chromatography–tandem mass spectrometry. Anal. Chim. Acta 2013, 758, 80–92. [Google Scholar] [CrossRef]
- European Commission. Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticides Residues Analysis in Food and Feed, SANTE/11813/2017. Supersedes SANTE/11945/2015, Implemented by 01/01/2018. Available online: https://ec.europa.eu/food/sites/food/files/plant/docs/pesticides_mrl_guidelines_wrkdoc_2017-11813.pdf (accessed on 16 November 2018).
- ISO 5725-1. Accuracy (Trueness and Precision) of Measurement Methods and Results—Part 1: General Principles and Definitions; International Organization for Standardization: Geneva, Switzerland, 1994. [Google Scholar]
- ConsultGLP. Measurement Uncertainty—Comparing GUM and Top down Approaches. Available online: https://consultglp.com/2017/04/27/measurement-uncertainty-comparing-gum-and-top-down-approaches/ (accessed on 2 March 2019).
- Olivares, I.R.B.; Costa, S.P.; Camargo, R.S.; Pacces, V.H.P. Development of a rapid and sensitive routine method of analyses for organochlorine compounds in fish: A metrological approach. Pharm. Anal. Acta 2016, 7, 502. [Google Scholar]
- Dimitrova, R.T.; Stoykova, I.I.; Yankovska-Stefanova, T.T.; Yaneva, S.A.; Stoyanchev, T.T. Development of analytical method for determination of organochlorine pesticides residues in meat by GC-ECD. Revue Méd. Vét. 2018, 169, 77–86. [Google Scholar]
- Neugebauer, F.; Dreyer, A.; Lohmann, N.; Koschorreck, J. Determination of halogenated flame retardants by GC-API-MS/MS and GC-EI-MS: A multi-compound multi-matrix method. Anal. Bioanal. Chem. 2018, 410, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Gałuszka, A.; Migaszewski, Z.M.; Konieczka, P.; Namieśnik, J. Analytical Eco-Scale for assessing the greenness of analytical procedures. Trends Anal. Chem. 2012, 37, 61–72. [Google Scholar] [CrossRef]
- Baduel, C.; Mueller, J.F.; Tsai, H.; Gomez Ramos, M.J. Development of sample extraction and clean-up strategies for target and non-target analysis of environmental contaminants in biological matrices. J. Chromatogr. A 2015, 1426, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Cruz, R.; Marques, A.; Casal, S.; Cunha, S.C. Fast and environmental-friendly methods for the determination of polybrominated diphenyl ethers and their metabolites in fish tissues and feed. Sci. Total Environ. 2019, 646, 1503–1515. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Analyte | tR (min) | MRM Transitions (m/z) | |||
|---|---|---|---|---|---|
| Quantifier | CE (V) | Qualifier | CE (V) | ||
| Hexachloro-1,3-butadiene | 4.42 | 225→190 | 15 | 260→225 | 15 |
| Pentachlorobenzene | 6.12 | 248→213 | 25 | 250→180 | 20 |
| Tetrachloro-m-xylene (IS-1) | 6.79 | 244→209 | 15 | 171→136 | 15 |
| alpha-HCH | 7.36 | 219→183 | 5 | 217→181 | 15 |
| Hexachlorobenzene | 7.50 | 284→214 | 35 | 284→249 | 20 |
| beta-HCH | 7.75 | 219→183 | 5 | 217→181 | 15 |
| Lindane | 7.86 | 219→183 | 5 | 217→181 | 15 |
| delta-HCH | 8.22 | 219→183 | 5 | 217→181 | 15 |
| Heptachlor | 9.02 | 272→237 | 25 | 272→117 | 35 |
| BDE-28 | 11.94 | 246→139 | 30 | 406→246 | 20 |
| BDE-47 | 14.02 | 326→217 | 30 | 486→326 | 20 |
| BDE-77 (IS-2) | 14.74 | 326→217 | 30 | 486→326 | 20 |
| BDE-100 | 15.55 | 564→404 | 20 | 404→297 | 30 |
| BDE-99 | 15.97 | 564→404 | 20 | 404→297 | 30 |
| BDE-154 | 17.19 | 644→484 | 20 | 484→324 | 40 |
| BDE-153 | 17.84 | 644→484 | 20 | 484→324 | 40 |
| Analyte | ME (%) | RSD (%) |
|---|---|---|
| Hexachloro-1,3-butadiene | −5.1 | 11 |
| Pentachlorobenzene | −1.9 | 11 |
| Tetrachloro-m-xylene (IS-1) | −1.3 | 12 |
| alpha-HCH | −1.2 | 11 |
| Hexachlorobenzene | 3.8 | 13 |
| beta-HCH | 9.3 | 10 |
| Lindane | 2.3 | 10 |
| delta-HCH | 3.6 | 11 |
| Heptachlor | 1.0 | 12 |
| BDE-28 | 1.6 | 14 |
| BDE-47 | 5.7 | 16 |
| BDE-77 (IS-2) | 1.3 | 15 |
| BDE-100 | 3.9 | 16 |
| BDE-99 | 10 | 14 |
| BDE-154 | 7.0 | 14 |
| BDE-153 | 11 | 14 |
| Analyte | Linear Range (µg/kg) | R2 | RRF | RRF_RSD (%) | LOD (µg/kg) | LOQ (µg/kg) | Accuracy | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Precision | Trueness | |||||||||
| Preintra | Preinter | R | Br | |||||||
| RSD (%) | RSD (%) | (%) | (%) | |||||||
| Hexachlorobutatadiene | 0.1–60 | 0.99997 | 1.7 | 6.7 | 0.028 | 0.092 | 3.0 | 5.7 | 95 | −4.7 |
| Pentachlorobenzene | 0.1–60 | 0.99975 | 1.0 | 5.8 | 0.036 | 0.12 | 2.3 | 4.9 | 95 | −5.2 |
| alpha-HCH | 0.1–60 | 0.99989 | 3.3 | 7.1 | 0.029 | 0.096 | 0.5 | 6.7 | 89 | −11 |
| Hexachlorobenzene | 0.1–60 | 0.99986 | 1.4 | 5.9 | 0.052 | 0.17 | 0.8 | 3.2 | 107 | 7.4 |
| beta-HCH | 0.1–60 | 0.99973 | 2.1 | 11 | 0.036 | 0.12 | 3.4 | 7.2 | 88 | −12 |
| Lindane | 0.1–60 | 0.99987 | 2.4 | 12 | 0.040 | 0.13 | 1.6 | 7.0 | 87 | −13 |
| delta-HCH | 0.1–60 | 0.99982 | 2.0 | 12 | 0.037 | 0.12 | 4.7 | 8.3 | 91 | −9.4 |
| Heptachlor | 0.1–60 | 0.99952 | 0.46 | 6.9 | 0.039 | 0.13 | 9.2 | 16 | 94 | −6.2 |
| BDE-28 | 0.1–60 | 0.99984 | 5.3 | 5.6 | 0.037 | 0.12 | 8.9 | 9.0 | 99 | −1.3 |
| BDE-47 | 0.1–60 | 0.99994 | 3.2 | 13 | 0.028 | 0.092 | 4.8 | 6.6 | 102 | 1.9 |
| BDE-100 | 0.1–60 | 0.99984 | 1.6 | 10 | 0.028 | 0.092 | 2.9 | 9.0 | 100 | 0.22 |
| BDE-99 | 0.1–60 | 0.99986 | 1.2 | 5.9 | 0.021 | 0.072 | 6.6 | 9.2 | 99 | −0.89 |
| BDE-154 | 0.1–60 | 0.99939 | 0.49 | 12 | 0.049 | 0.16 | 9.0 | 13 | 101 | 1.2 |
| BDE-153 | 0.1–60 | 0.99912 | 0.26 | 8.1 | 0.042 | 0.14 | 11 | 13 | 105 | 5.1 |
| Analyte | Recovery (RSD) a (%) | ||||
|---|---|---|---|---|---|
| 1 µg/kg | 5 µg/kg | 15 µg/kg | 30 µg/kg | 60 µg/kg | |
| Hexachloro-1,3-butadiene | 101 (6) | 96 (4) | 124 (5) | 98 (6) | 97 (6) |
| Pentachlorobenzene | 98 (4) | 88 (2) | 103 (10) | 103 (15) | 90 (13) |
| alpha-HCH | 99 (7) | 86 (5) | 101 (17) | 91 (17) | 62 (15) |
| Hexachlorobenzene | 103 (5) | 100 (4) | 101 (16) | 97 (6) | 94 (6) |
| beta-HCH | 95 (10) | 82 (7) | 86 (16) | 92 (18) | 58 (14) |
| Lindane | 94 (10) | 88 (5) | 96 (16) | 90 (18) | 59 (15) |
| delta-HCH | 91 (10) | 85 (16) | 89 (16) | 88 (18) | 57 (14) |
| Heptachlor | 91 (7) | 94 (8) | 89 (12) | 86 (9) | 83 (12) |
| BDE-28 | 101 (6) | 93 (10) | 94 (18) | 94 (3) | 93 (14) |
| BDE-47 | 98 (6) | 110 (2) | 95 (6) | 94 (3) | 92 (15) |
| BDE-100 | 100 (5) | 100 (5) | 100 (14) | 96 (5) | 96 (10) |
| BDE-99 | 101 (9) | 100 (8) | 101 (15) | 95 (12) | 96 (8) |
| BDE-154 | 105 (4) | 104 (12) | 99 (11) | 94 (6) | 94 (7) |
| BDE-153 | 98 (11) | 98 (12) | 102 (9) | 94 (12) | 93 (5) |
| Analyte | Certified Value a (μg/kg) | Determined Value a (μg/kg) | Trueness (RSD) (%) |
|---|---|---|---|
| Hexachlorobenzene | 7.25 ± 0.83 | 6.47 ± 1.5 | 89 (2) |
| alpha-HCH | 5.72 ± 0.65 | 5.44 ± 1.4 | 95 (7) |
| Lindane | 1.14 ± 0.18 | 0.89 ± 0.26 | 78 (5) |
| BDE-28 | 0.742 ± 0.027 | 0.467 ± 0.067 | 63 (5) |
| BDE-47 | 29.9 ± 2.3 | 30.2 ± 5.1 | 101 (5) |
| BDE-99 | 18.5 ± 2.1 | 22.0 ± 3.7 | 119 (16) |
| BDE-100 | 8.57 ± 0.52 | 9.04 ± 1.8 | 105 (9) |
| BDE-153 | 2.81 ± 0.41 | 3.16 ± 0.69 | 112 (9) |
| BDE-154 | 5.77 ± 0.80 | 6.57 ± 1.2 | 114 (12) |
| Analyte | ur,repro (%) | Br (%) | ur,cm (%) | ur,ref (%) | ur,tot (%) | Ur,tot (%) |
|---|---|---|---|---|---|---|
| Hexachloro-1,3-butadiene | 8.01 | −4.66 | 0.909 | 1.15 | 9.38 | 18.8 |
| Pentachlorobenzene | 9.90 | −5.15 | 0.676 | 0.250 | 11.2 | 22.4 |
| alpha-HCH | 7.83 | −10.6 | 0.154 | 0.475 | 13.2 | 26.3 |
| Hexachlorobenzene | 8.42 | 7.38 | 0.274 | 1.00 | 11.2 | 22.5 |
| beta-HCH | 8.13 | −11.6 | 0.954 | 0.150 | 14.2 | 28.4 |
| Lindane | 6.04 | −13.0 | 0.451 | 0.866 | 14.4 | 28.7 |
| delta-HCH | 7.15 | −9.40 | 1.34 | 0.330 | 11.9 | 23.8 |
| Heptachlor | 7.60 | −6.23 | 2.74 | 0.250 | 10.2 | 20.4 |
| BDE-28 | 6.48 | −1.28 | 2.78 | 0.295 | 7.18 | 14.4 |
| BDE-47 | 7.98 | 1.90 | 1.56 | 0.300 | 8.36 | 16.7 |
| BDE-100 | 9.75 | 0.192 | 0.913 | 0.300 | 9.80 | 19.6 |
| BDE-99 | 8.01 | −0.879 | 2.06 | 0.300 | 8.32 | 16.6 |
| BDE-154 | 8.50 | 1.17 | 2.89 | 0.300 | 9.06 | 18.1 |
| BDE-153 | 9.00 | 5.08 | 3.50 | 0.300 | 10.9 | 21.8 |
| Analyte | European chub Concentr. a/RR b (µg/kg/%) | Crucian carp Concentr. a/RR b (µg/kg/%) | European perch Concentr. a/RR b (µg/kg/%) | Northern pike Concentr. a/RR b (µg/kg/%) | Zander Concentr. a/RR b (µg/kg/%) | Brown trout Concentr. a/RR b (µg/kg/%) | Atlantic salmon Concentr. a/RR b (µg/kg/%) | Alaska pollock Concentr. a/RR b (µg/kg/%) |
|---|---|---|---|---|---|---|---|---|
| Hexachloro-1,3-butadiene | <0.09/96 (2) | <0.09/86 (6) | <0.09/87 (1) | <0.09/95 (1) | <0.09/93 (1) | <0.09/116 (1) | 0.90 ± 0.01/98 (4) | 0.22 ± 0.01/104 (3) |
| Pentachlorobenzene | <0.12/92 (3) | <0.12/96 (6) | <0.12/90 (2) | <0.12/105 (4) | <0.12/87 (3) | <0.12/108 (3) | 0.22 ± 0.01/95 (1) | <0.12/104 (3) |
| alpha-HCH | <0.10/85 (2) | <0.10/98 (8) | <0.10/84 (2) | <0.10/92 (8) | <0.10/83 (6) | <0.10/105 (4) | 0.23 ± 0.01/86 (3) | <0.10/95 (2) |
| Hexachlorobenzene | 1.00 ± 0.01/95 (2) | 0.35 ± 0.01/92 (3) | 0.48 ± 0.01/93 (1) | 1.84 ± 0.02/96 (3) | 0.70 ± 0.01/95 (2) | 1.12 ± 0.01/118 (2) | 2.68 ± 0.05/96 (4) | 0.18 ± 0.01/99 (1) |
| beta-HCH | <0.12/82 (3) | <0.12/93 (8) | <0.12/82 (2) | <0.12/97 (11) | <0.12/76 (7) | 0.80 ± 0.02/113 (7) | 0.12 ± 0.01/81 (4) | <0.12/93 (3) |
| Lindane | <0.13/83 (3) | <0.13/93 (8) | <0.13/82 (2) | <0.13/91 (9) | <0.13/79 (6) | <0.13/101 (6) | <0.13/84 (4) | <0.13/91 (2) |
| delta-HCH | <0.12/82 (2) | <0.12/92 (8) | <0.12/82 (3) | <0.12/102 (10) | <0.12/78 (7) | 0.13 ± 0.003/100 (7) | <0.12/82 (4) | <0.12/90 (3) |
| Heptachlor | <0.13/78 (6) | <0.13/64 (12) | <0.13/98 (3) | <0.13/71 (6) | <0.13/114 (8) | <0.13/106 (8) | <0.13/83 (4) | <0.13/65 (10) |
| BDE-28 | <0.12/89 (7) | <0.12/128 (9) | <0.12/88 (2) | <0.12/110 (5) | <0.12/99 (8) | <0.12/87 (4) | <0.12/86 (4) | <0.12/114 (9) |
| BDE-47 | 0.69 ± 0.01/95 (2) | <0.09/100 (2) | 1.33 ± 0.04/103 (5) | 0.21 ± 0.01/104 (6) | 1.45 ± 0.01/103 (3) | 0.36 ± 0.01/94 (4) | 0.43 ± 0.02/97 (5) | <0.09/94 (0.4) |
| BDE-100 | 0.20 ± 0.002/100 (9) | <0.09/94 (8) | 0.26 ± 0.02/111 (5) | <0.09/105 (9) | 0.18 ± 0.01/96 (6) | 0.10 ± 0.01/97 (6) | 0.09 ± 0.01/105 (10) | <0.09/71 (12) |
| BDE-99 | <0.07/96 (9) | <0.07/91 (8) | 0.41 ± 0.02/113 (6) | <0.07/111 (14) | <0.07/96 (6) | 0.25 ± 0.01/94 (5) | <0.07/104 (9) | <0.07/67 (4) |
| BDE-154 | <0.16/103 (10) | <0.16/100 (21) | <0.16/120 (3) | <0.16/128 (15) | <0.16/99 (9) | <0.16/97 (7) | <0.16/112 (13) | <0.16/53 (15) |
| BDE-153 | <0.14/99 (14) | <0.14/103 (23) | <0.14/119 (3) | <0.14/114 (19) | <0.14/95 (7) | <0.14/95 (6) | <0.14/100 (2) | <0.14/53 (16) |
| Lipid content (%) | 3.5 | 0.96 | 3.0 | 2.4 | 1.5 | 8.2 | 16 | 0.63 |
| Moisture content (%) | 78 | 74 | 74 | 78 | 77 | 71 | 58 | 81 |
| Penalty Points | |
|---|---|
| Reagents | |
| MeCN (5 mL) | 4 |
| CHCl3 (50 µL) | 2 |
| Hexane (80 µL) | 8 |
| Analytes standard solution | 4 |
| H2O (4 mL) | 0 |
| H2SO4 (1 mL) | 2 |
| MgSO4 (2 g) | 0 |
| NaCl (0.5 g) | 0 |
| CH3COONa | 0 |
| Instruments | |
| Vortex | 1 |
| Centrifuge | 1 |
| GC–MS/MS | 3 |
| Occupational hazard | 3 |
| Waste | 4 |
| Total penalty points | Σ 32 |
| Analytical Eco-Scale total score | 68 |
| Analytes | Extractant | Clean-Up | Analysis | Recoveries (%) | LOQs (µg/kg) | Reference |
|---|---|---|---|---|---|---|
| Pesticides | MeCN | Dual dSPE (1. PSA + C18 + MgSO4; 2. PSA + C18 + MgSO4) | GC–ECD | 57–98 | 1.5–3.5 | [9] |
| Pesticides | MeCN or MeCN/THF (3:1) | Freezing (2 h), dual dSPE (1. CaCl2; 2. PSA + MgSO4) | GC–MS | 43–113 | 1–10 | [12] |
| Pesticides | MeCN + CHCl3 (10:1) | Dual dSPE (1. PSA + SAX +NH2 + MgSO4; 2. C18), freezing (overnight) | GC–MS | 61–102 | 4–6 | [13] |
| Pesticides | MeCN + hexane (15:2) | Freezing (20 min), dual dSPE (1. CaCl2 + MgSO4; 2. PSA + florisil + C18 +MgSO4) | GC–MS/MS | 60–127 | 2–13 | [14] |
| PBDEs | MeCN (sonication) | Dual dSPE (1. PSA + C18 + MgSO4; 2. PSA + C18 + MgSO4) | GC–MS | 60–107 | <15 | [15] |
| PBDEs | Ethyl acetate | GPC, SPE (silica + Na2SO4) | GC–MS | 88–140 | 0.09–2.2 | [16] |
| Pesticides | MeCN | Freezing (min. 4 h), dSPE (Z-Sep + MgSO4), filtration (0.2 µm PTFE filter) | GC–MS/MS | 86–101 | 0.08–0.15 | [32] |
| PBDEs | MeCN + toluene (4:1) | Dual dSPE (1. EMR-Lipid; 2. Z-Sep + MgSO4) | GC–MS/MS | 79–116 (muscle) 89–107 (liver) | 0.015–0.065 0.85–1.1 | [33] |
| Pesticides, PBDEs | MeCN | pH-tuned DLLME (0.5 M CH3COONa, CHCl3), H2SO4 clean-up | GC–MS/MS | 57–124 (pesticides) 93–110 (PBDEs) | 0.09–0.17 0.07–0.16 | This work |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagyová, S.; Tölgyessy, P. Validation Including Uncertainty Estimation of a GC–MS/MS Method for Determination of Selected Halogenated Priority Substances in Fish Using Rapid and Efficient Lipid Removing Sample Preparation. Foods 2019, 8, 101. https://doi.org/10.3390/foods8030101
Nagyová S, Tölgyessy P. Validation Including Uncertainty Estimation of a GC–MS/MS Method for Determination of Selected Halogenated Priority Substances in Fish Using Rapid and Efficient Lipid Removing Sample Preparation. Foods. 2019; 8(3):101. https://doi.org/10.3390/foods8030101
Chicago/Turabian StyleNagyová, Slávka, and Peter Tölgyessy. 2019. "Validation Including Uncertainty Estimation of a GC–MS/MS Method for Determination of Selected Halogenated Priority Substances in Fish Using Rapid and Efficient Lipid Removing Sample Preparation" Foods 8, no. 3: 101. https://doi.org/10.3390/foods8030101
APA StyleNagyová, S., & Tölgyessy, P. (2019). Validation Including Uncertainty Estimation of a GC–MS/MS Method for Determination of Selected Halogenated Priority Substances in Fish Using Rapid and Efficient Lipid Removing Sample Preparation. Foods, 8(3), 101. https://doi.org/10.3390/foods8030101