
Nitric Oxide and Lutein: Function, Performance, and Protection of Neural Tissue

{kind=link}
{kind=link}
{kind=link}
Abstract
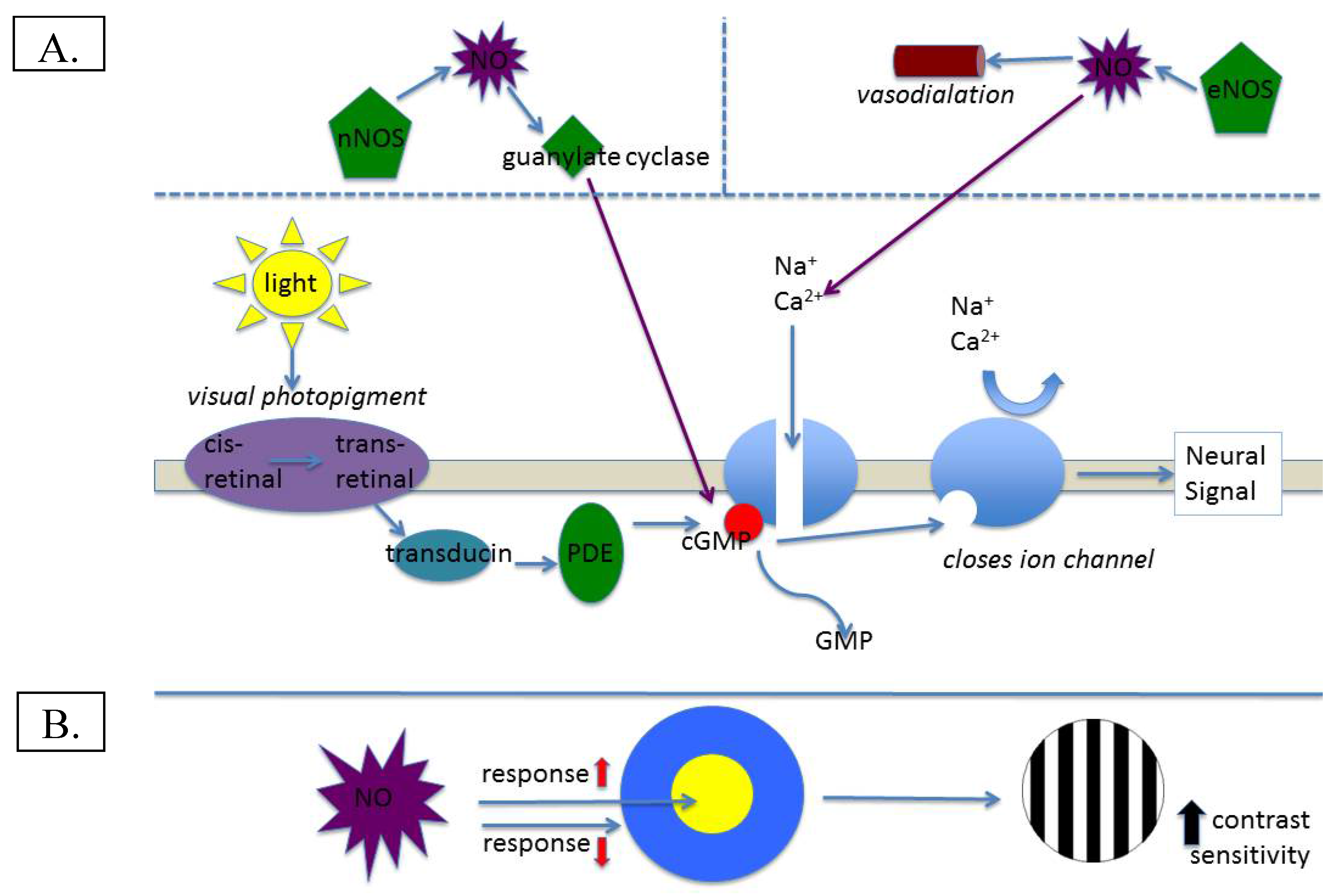
:1. Nitric Oxide in Normal Retinal Function

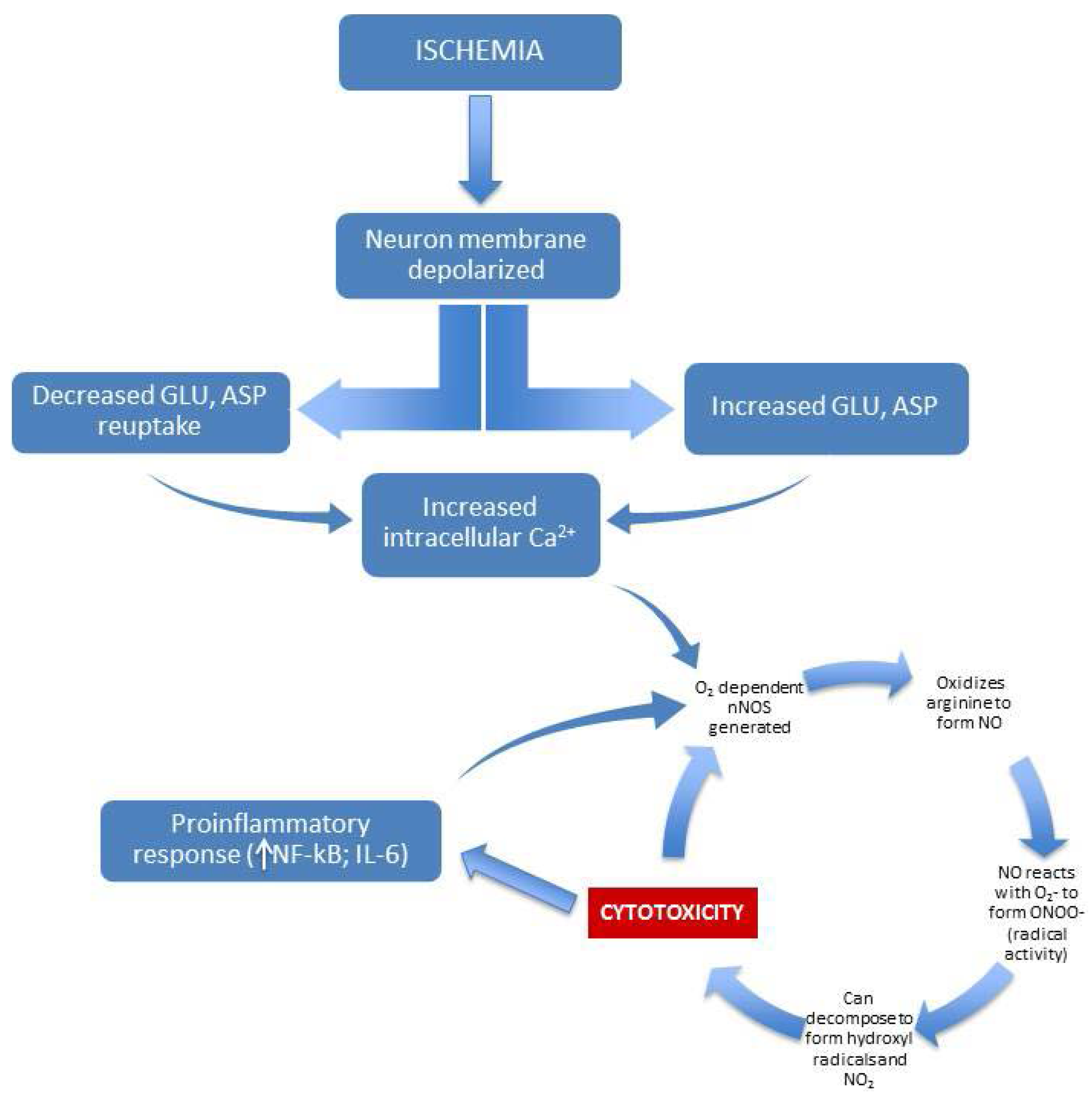
2. Nitric Oxide in Retinal Disease

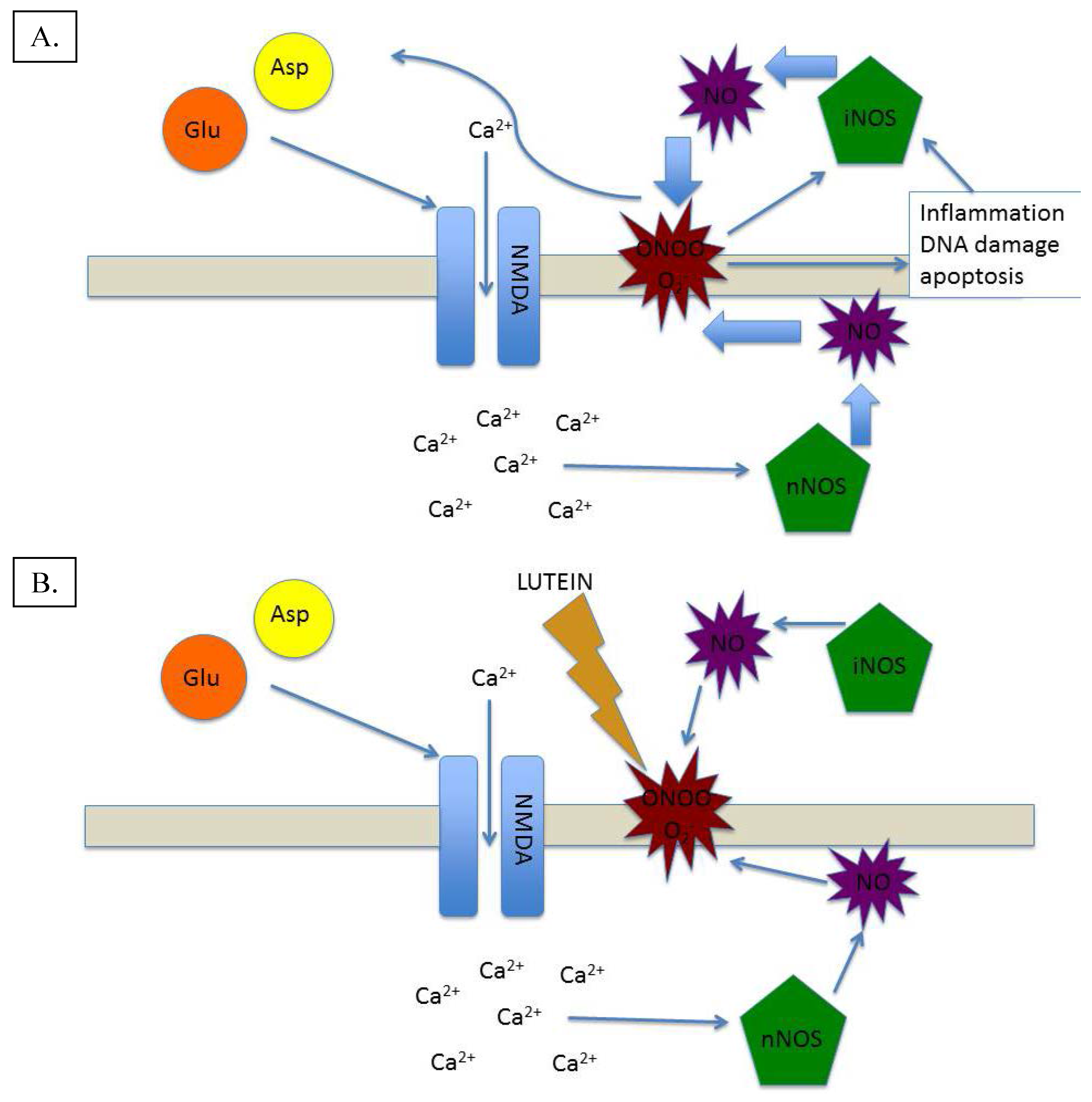
3. Lutein and Nitric Oxide: A Homeostatic Symbiosis
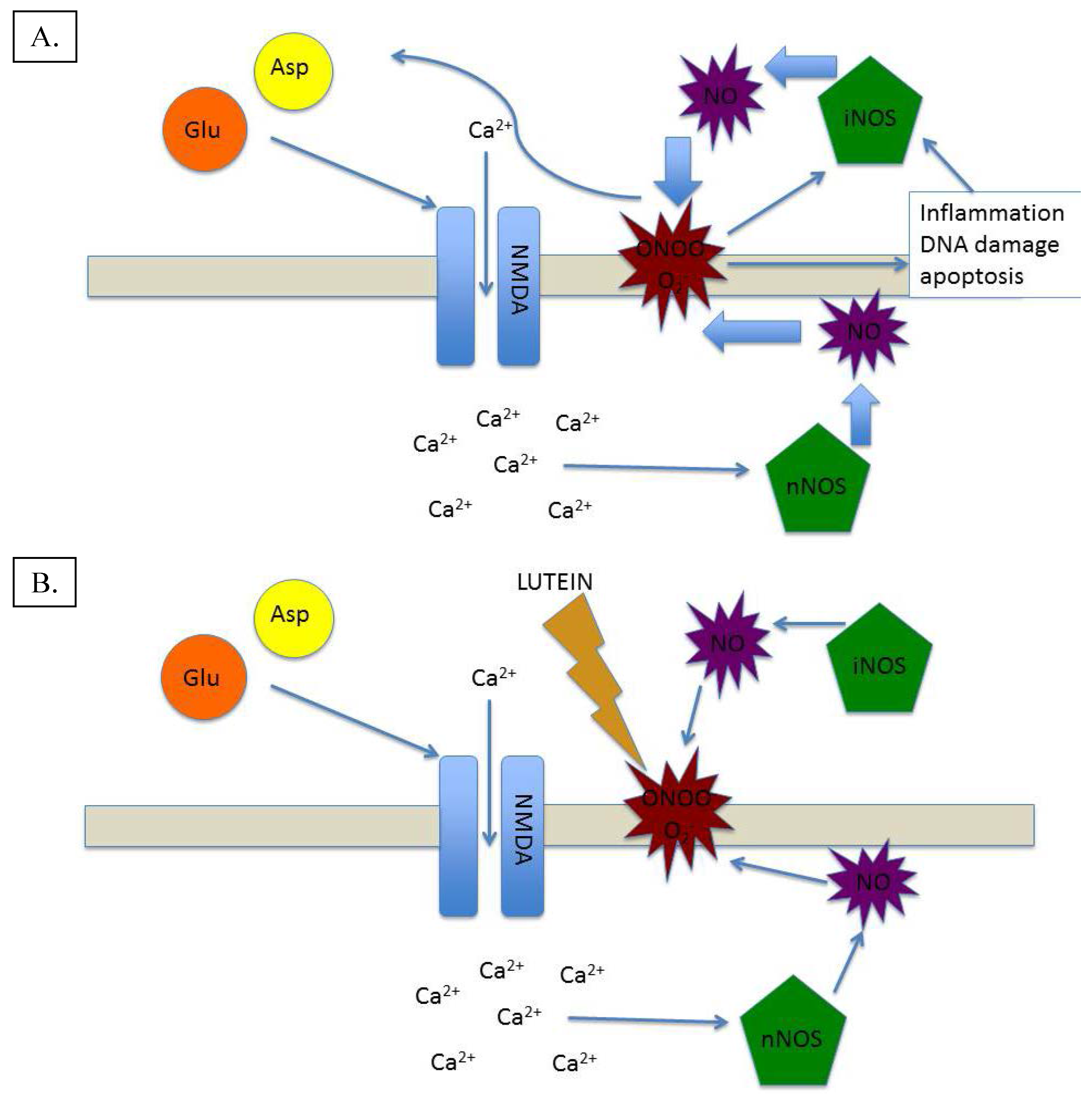
4. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Futterman, S. Metabolism and Photochemistry in the Retina. In Adler’s Physiology of the Eye, 6th ed.; Moses, R.A., Ed.; C.V. Mosby Company: St. Louis, MO, USA, 1975; pp. 406–419. [Google Scholar]
- Whikehart, D.R. Biochemistry of the Eye; Butterworth-Heinemann: Boston, MA, USA, 1994; p. 73. [Google Scholar]
- Bredt, D.S.; Hwang, P.M.; Snyder, S.H. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature 1990, 347, 768–770. [Google Scholar] [CrossRef] [PubMed]
- Erez, A.; Nagamani, S.C.; Shchelochkov, O.A.; Premkumar, M.H.; Campeau, P.M.; Chen, Y.; Garg, H.K.; Li, L.; Mian, A.; Bertin, T.K.; et al. Requirement of argininosuccinate lyase for systemic nitric oxide production. Nat. Med. 2011, 17, 1619–1626. [Google Scholar] [CrossRef] [PubMed]
- Knowles, R.G.; Moncada, S. Nitric oxide synthases in mammals. Biochem. J. 1994, 298(Pt 2), 249–258. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.M.; Ostwald, P.; Roth, S. Nitric oxide: A review of its role in retinal function and disease. Vis. Res. 1996, 36, 2979–2994. [Google Scholar] [CrossRef]
- Schmetterer, L.; Polak, K. Role of nitric oxide in control of ocular blood flow. Prog. Retin. Eye Res. 2001, 20, 823–847. [Google Scholar] [CrossRef]
- Blute, T.A.; Lee, H.K.; Huffmaster, T.; Haverkamp, S.; Eldred, W.D. Localization of natriuretic peptides and their activation of particulate guanylate cyclase and nitric oxide synthase in the retina. J. Comp. Neurol. 2000, 424, 689–700. [Google Scholar] [CrossRef]
- Hirooka, K.; Kourennyi, D.E.; Barnes, S. Calcium channel activation facilitated by nitric oxide in retinal ganglion cells. J. Neurophysiol. 2000, 83, 198–206. [Google Scholar] [PubMed]
- Vielma, A.H.; Retamal, M.A.; Schmachtenberg, O. Nitric oxide signaling in the retina: What have we learned in two decades? Brain Res. 2011, 1430, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Ostwald, P.; Goldstein, I.M.; Pachnanda, A.; Roth, S. Effect of nitric oxide synthase inhibition on blood flow after retinal ischemia in cats. Investig. Ophthalmol. Vis. Sci. 1995, 36, 2396–2403. [Google Scholar]
- Vielma, A.; Delgado, L.; Elgueta, C.; Osorio, R.; Palacios, A.G.; Schmachtenberg, O. Nitric oxide amplifies the rat electroretinogram. Exp. Eye Res. 2010, 91, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Tilton, R.G.; Chang, K.; Hasan, K.S.; Smith, S.R.; Petrash, J.M.; Misko, T.P.; Moore, W.M.; Currie, M.G.; Corbett, J.A.; McDaniel, M.L.; et al. Prevention of diabetic vascular dysfunction by guanidines. Inhibition of nitric oxide synthase versus advanced glycation end-product formation. Diabetes 1993, 42, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Bhutto, I.A.; Baba, T.; Merges, C.; McLeod, D.S.; Lutty, G.A. Low nitric oxide synthases (NOSs) in eyes with age-related macular degeneration (AMD). Exp. Eye Res. 2010, 90, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Ikeda, Y.; Notomi, S.; Ishikawa, K.; Murakami, Y.; Hisatomi, T.; Enaida, H.; Ishibashi, T. Clinical evidence of sustained chronic inflammatory reaction in retinitis pigmentosa. Ophthalmology 2013, 120, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Fu, Z.J.; Ma, H.; Jang, W.C.; So, K.F.; Wong, D.; Lo, A.C. Effect of lutein on retinal neurons and oxidative stress in a model of acute retinal ischemia/reperfusion. Investig. Ophthalmol. Vis. Sci. 2009, 50, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhu, D.Y. Neuronal nitric oxide synthase: Structure, subcellular localization, regulation, and clinical implications. Nitric Oxide 2009, 179, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A. Transport and metabolism of glutamate and GABA in neurons and glial cells. Int. Rev. Neurobiol. 1981, 22, 1–45. [Google Scholar] [PubMed]
- Lipton, S.A.; Rosenberg, P.A. Excitatory amino acids as a final common pathway for neurologic disorders. N. Engl. J. Med. 1994, 330, 613–622. [Google Scholar] [PubMed]
- Choi, D.W. Methods for antagonizing glutamate neurotoxicity. Cerebrovasc. Brain Metab. Rev. 1990, 2, 105–147. [Google Scholar] [PubMed]
- Bredt, D.S.; Snyder, S.H. Nitric oxide mediates glutamate-linked enhancement of cGMP levels in the cerebellum. Proc. Natl. Acad. Sci. USA 1989, 86, 9030–9033. [Google Scholar] [CrossRef] [PubMed]
- Garthwaite, J.; Charles, S.L.; Chess-Williams, R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggest role as intracellular messenger in the brain. Nature 1988, 336, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Garthwaite, J.; Garthwaite, G.; Palmer, R.M.; Moncada, S. NMDA receptor activation induces nitric oxide synthesis from arginine in rat brain slices. Eur. J. Pharmacol. 1989, 172, 413–416. [Google Scholar] [CrossRef]
- Betteridge, D.J. What is oxidative stress? Metabolism 2000, 49 (Suppl. S1), 3–8. [Google Scholar] [CrossRef]
- Kuo, P.C.; Schroeder, R.A. The emerging multifaceted roles of nitric oxide. Ann. Surg. 1995, 221, 220–235. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.M.; Billiar, T.R. New insights into the regulation of inducible nitric oxide synthesis. Am. J. Physiol. 1994, 266 (6 Pt 1), E829–E839. [Google Scholar] [PubMed]
- Pellegrini-Giampietro, D.E.; Cherici, G.; Alesiani, M.; Carla, V.; Moroni, F. Excitatory amino acid release and free radical formation may cooperate in the genesis of ischemia-induced neuronal damage. J. Neurosci. 1990, 10, 1035–1041. [Google Scholar] [PubMed]
- Osborne, N.N.; Casson, R.J.; Wood, J.P.; Chidlow, G.; Graham, M.; Melena, J. Retinal ischemia: Mechanisms of damage and potential therapeutic strategies. Prog. Retin. Eye Res. 2004, 23, 91–147. [Google Scholar] [CrossRef] [PubMed]
- Dawson, V.L.; Dawson, T.M.; London, E.D.; Bredt, D.S.; Snyder, S.H. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc. Natl. Acad. Sci. USA 1991, 88, 6368–6371. [Google Scholar] [CrossRef] [PubMed]
- Lefer, A.M.; Lefer, D.J. Pharmacology of the endothelium in ischemia-reperfusion and circulatory shock. Annu. Rev. Pharmacol. Toxicol. 1993, 33, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Santocono, M.; Zurria, M.; Berrettini, M.; Fedeli, D.; Falcioni, G. Lutein, zeaxanthin and astaxanthin protect against DNA damage in SK-N-SH human neuroblastoma cells induced by reactive nitrogen species. J. Photochem. Photobiol. B 2007, 88, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S. Nitric oxide and cell respiration: Physiology and pathology. Verh. K. Acad. Geneeskd. Belg. 2000, 62, 171–179. [Google Scholar] [PubMed]
- Rudolph, V.; Rudolph, T.K.; Schopfer, F.J.; Bonacci, G.; Woodcock, S.R.; Cole, M.P.; Baker, P.R.; Ramani, R.; Freeman, B.A. Endogenous generation and protective effects of nitro-fatty acids in a murine model of focal cardiac ischaemia and reperfusion. Cardiovasc. Res. 2010, 85, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.C.; Finkelstein, M.A. Visual sensitivity. In Handbook of Perception and Human Performance (Volume 1); Boff, K., Kaufman, L., Thomas, J., Eds.; Wiley: New York, NY, USA, 1986; pp. 5–32. [Google Scholar]
- Bone, R.A.; Landrum, J.T. Dose-dependent response of serum lutein and macular pigment optical density to supplementation with lutein esters. Arch. Biochem. Biophys. 2010, 504, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Neuringer, M.; Sandstrom, M.M.; Johnson, E.J.; Snodderly, D.M. Nutritional manipulation of primate retinas, I: Effects of lutein or zeaxanthin supplements on serum and macular pigment in xanthophyll-free rhesus monkeys. Investig. Ophthalmol. Vis. Sci. 2004, 45, 3234–3243. [Google Scholar] [CrossRef] [PubMed]
- Krinsky, N.I.; Landrum, J.T.; Bone, R.A. Biologic mechanisms of the protective role of lutein and zeaxanthin in the eye. Annu. Rev. Nutr. 2003, 23, 171–201. [Google Scholar] [CrossRef] [PubMed]
- Snodderly, D.M.; Brown, P.K.; Delori, F.C.; Auran, J.D. The macular pigment. I. Absorbance spectra, localization, and discrimination from other yellow pigments in primate retinas. Investig. Ophthalmol. Vis. Sci. 1984, 25, 660–673. [Google Scholar]
- Ham, W.T., Jr.; Mueller, H.A.; Sliney, D.H. Retinal sensitivity to damage from short wavelength light. Nature 1976, 260, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Beatty, S.; Koh, H.H.; Henson, D.; Boulton, M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000, 45, 115–134. [Google Scholar] [CrossRef]
- Hammond, B.R., Jr.; Wooten, B.R.; Snodderly, D.M. Preservation of visual sensitivity of older subjects: Association with macular pigment density. Investig. Ophthalmol. Vis. Sci. 1998, 39, 397–406. [Google Scholar]
- Humphries, J.M.; Khachik, F. Distribution of lutein, zeaxanthin, and related geometrical isomers in fruit, vegetables, wheat, and pasta products. J. Agric. Food Chem. 2003, 51, 1322–1327. [Google Scholar] [CrossRef] [PubMed]
- Ruban, A.V.; Pascal, A.; Lee, P.J.; Robert, B.; Horton, P. Molecular configuration of xanthophyll cycle carotenoids in photosystem II antenna complexes. J. Biol. Chem. 2002, 277, 42937–42942. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.L. A theory of sensitized luminescence. J. Chem. Phys. 1953, 21, 836. [Google Scholar] [CrossRef]
- Sindhu, E.R.; Preethi, K.C.; Kuttan, R. Antioxidant activity of carotenoid lutein in vitro and in vivo. Indian J. Exp. Biol. 2010, 48, 843–848. [Google Scholar] [PubMed]
- Perrone, S.; Longini, M.; Marzocchi, B.; Picardi, A.; Bellieni, C.V.; Proietti, F.; Rodriguez, A.; Turrisi, G.; Buonocore, G. Effects of lutein on oxidative stress in the term newborn: A pilot study. Neonatology 2010, 97, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Perrone, S.; Tei, M.; Longini, M.; Santacroce, A.; Turrisi, G.; Proietti, F.; Felici, C.; Picardi, A.; Bazzini, F.; Vasarri, P.; et al. Lipid and protein oxidation in newborn infants after lutein administration. Oxid. Med. Cell. Longev. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Kijlstra, A.; van der Veen, R.L.; Makridaki, M.; Murray, I.J.; Berendschot, T.T. The effect of lutein supplementation on blood plasma levels of complement factor D, C5a, and C3d. PLoS ONE 2013, 8, e73387. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Kim, D.; Hong, Y.M.; Mizuno, S.; Joo, C.K. Inhibition of nNOS and COX-2 expression by lutein in acute retinal ischemia. Nutrition 2006, 22, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.B.; Graham-Engeland, J.E.; Engeland, C.G.; Smyth, J.M.; Almeida, D.M.; Katz, M.J.; Lipton, R.B.; Mogle, J.A.; Munoz, E.; Ram, N.; et al. The effects of stress on cognitive aging, physiology and emotion (ESCAPE) project. BMC Psychiatry 2015, 15, 146. [Google Scholar] [CrossRef] [PubMed]
- Craft, N.E.; Haitema, T.B.; Garnett, K.M.; Fitch, K.A.; Dorey, C.K. Carotenoid, tocopherol, and retinol concentrations in elderly human brain. J. Nutr. Health Aging 2004, 8, 156–162. [Google Scholar] [PubMed]
- Vishwanathan, R.; Kuchan, M.J.; Sen, S.; Johnson, E.J. Lutein and preterm infants with decreased concentrations of brain carotenoids. J. Pediatr. Gastroenterol. Nutr. 2014, 59, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Vishwanathan, R.; Neuringer, M.; Snodderly, D.M.; Schalch, W.; Johnson, E.J. Macular lutein and zeaxanthin are related to brain lutein and zeaxanthin in primates. Nutr. Neurosci. 2013, 16, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.J.; McDonald, K.; Caldarella, S.M.; Chung, H.Y.; Troen, A.M.; Snodderly, D.M. Cognitive findings of an exploratory trial of docosahexaenoic acid and lutein supplementation in older women. Nutr. Neurosci. 2008, 11, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Feeney, J.; Finucane, C.; Savva, G.M.; Cronin, H.; Beatty, S.; Nolan, J.M.; Kenny, R.A. Low macular pigment optical density is associated with lower cognitive performance in a large population-based sample of older adults. Neurobiol. Aging 2013, 34, 2449–2456. [Google Scholar] [CrossRef] [PubMed]
- Vishwanathan, R.; Iannaccone, A.; Scott, T.M.; Kritchevsky, S.B.; Jennings, B.J.; Carboni, G.; Forma, G.; Satterfield, S.; Harris, T.; Johnson, K.C.; et al. Macular pigment optical density is related to cognitive function in older people. Age Ageing 2014, 43, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Rafi, M.M.; Shafaie, Y. Dietary lutein modulates inducible nitric oxide synthase (iNOS) gene and protein expression in mouse macrophage cells (RAW 264.7). Mol. Nutr. Food Res. 2007, 51, 333–430. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Li, Y.; Wu, Y.; Zhang, Y.; Wang, Z.; Liu, X. Lutein suppresses inflammatory responses through Nrf2 activation and NF-κB inactivation in lipopolysaccharide-stimulated BV-2 microglia. Mol. Nutr. Food Res. 2015, 59, 163–1673. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Hong, J.S. Chronic microglial activation and progressive dopaminergic neurotoxicity. Biochem. Soc. Trans. 2007, 35 Pt 5, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Stahl, W.; Ale-Agha, N.; Polidori, M.C. Non-antioxidant properties of carotenoids. Biol. Chem. 2002, 383, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Stringham, J.M.; Garcia, P.V.; Smith, P.A.; Hiers, P.L.; McLin, L.N.; Kuyk, T.K.; Foutch, B.K. Macular pigment and visual performance in low-light conditions. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2459–2468. [Google Scholar] [CrossRef] [PubMed]
- Hammond, B.R., Jr.; Wooten, B.R. CFF thresholds: Relation to macular pigment optical density. Ophthalmic Physiol. Opt. 2005, 25, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Stringham, N.T.; Stringham, J.M. Temporal visual mechanisms may mediate compensation for macular pigment. Perception 2015, in press. [Google Scholar] [CrossRef]
- Johnson, E.J.; Maras, J.E.; Rasmussen, H.M.; Tucker, K.L. Intake of lutein and zeaxanthin differ with age, sex, and ethnicity. J. Am. Diet. Assoc. 2010, 110, 1357–1362. [Google Scholar] [CrossRef] [PubMed]
- Nolan, J.M.; Loskutova, E.; Howard, A.N.; Moran, R.; Mulcahy, R.; Stack, J.; Bolger, M.; Dennison, J.; Akuffo, K.O.; Owens, N.; et al. Macular pigment, visual function, and macular disease among subjects with Alzheimer’s disease: An exploratory study. J. Alzheimer’s Dis. 2014, 42, 1191–1202. [Google Scholar]
- Neuringer, M.; Anderson, G.J.; Connor, W.E. The essentiality of N-3 fatty acids for the development and function of the retina and brain. Ann. Rev. Nutr. 1988, 8, 517–541. [Google Scholar] [CrossRef] [PubMed]
- Steinert, J.R.; Chernova, T.; Forsythe, I.D. Nitric oxide signaling in brain function, dysfunction, and dementia. Neuroscientist 2010, 16, 435–452. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S. The double-edged role of nitric oxide in brain function and superoxide-mediated injury. J. Dev. Physiol. 1991, 15, 53–59. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stringham, J.M.; Stringham, N.T. Nitric Oxide and Lutein: Function, Performance, and Protection of Neural Tissue. Foods 2015, 4, 678-689. https://doi.org/10.3390/foods4040678
Stringham JM, Stringham NT. Nitric Oxide and Lutein: Function, Performance, and Protection of Neural Tissue. Foods. 2015; 4(4):678-689. https://doi.org/10.3390/foods4040678
Chicago/Turabian StyleStringham, James M., and Nicole T. Stringham. 2015. "Nitric Oxide and Lutein: Function, Performance, and Protection of Neural Tissue" Foods 4, no. 4: 678-689. https://doi.org/10.3390/foods4040678
APA StyleStringham, J. M., & Stringham, N. T. (2015). Nitric Oxide and Lutein: Function, Performance, and Protection of Neural Tissue. Foods, 4(4), 678-689. https://doi.org/10.3390/foods4040678