
Genomic and Transcriptomic Analysis of Mutant Bacillus subtilis with Enhanced Nattokinase Production via ARTP Mutagenesis
 ,
,  , ,
, , 

Abstract
1. Introduction
2. Materials and Methods
2.1. Strain and Culture Conditions
2.2. ARTP Mutagenesis
2.3. Determination of the Enzymatic Activity of Nattokinase (NK)
2.3.1. Fibrin Plate Analysis
2.3.2. Fibrinolytic Activity Determination
2.4. Strain Screening Methods
2.5. Determination of the Genetic Stability of Mutant Strain
2.6. Determination of the Indicators of the Fermentation Progress
2.7. Whole Genome Sequencing and Functional Annotation
2.8. Genome Resequencing
2.9. Transcriptome Sequencing, Annotation and Analysis
2.10. Statistical Analysis
3. Results and Discussion
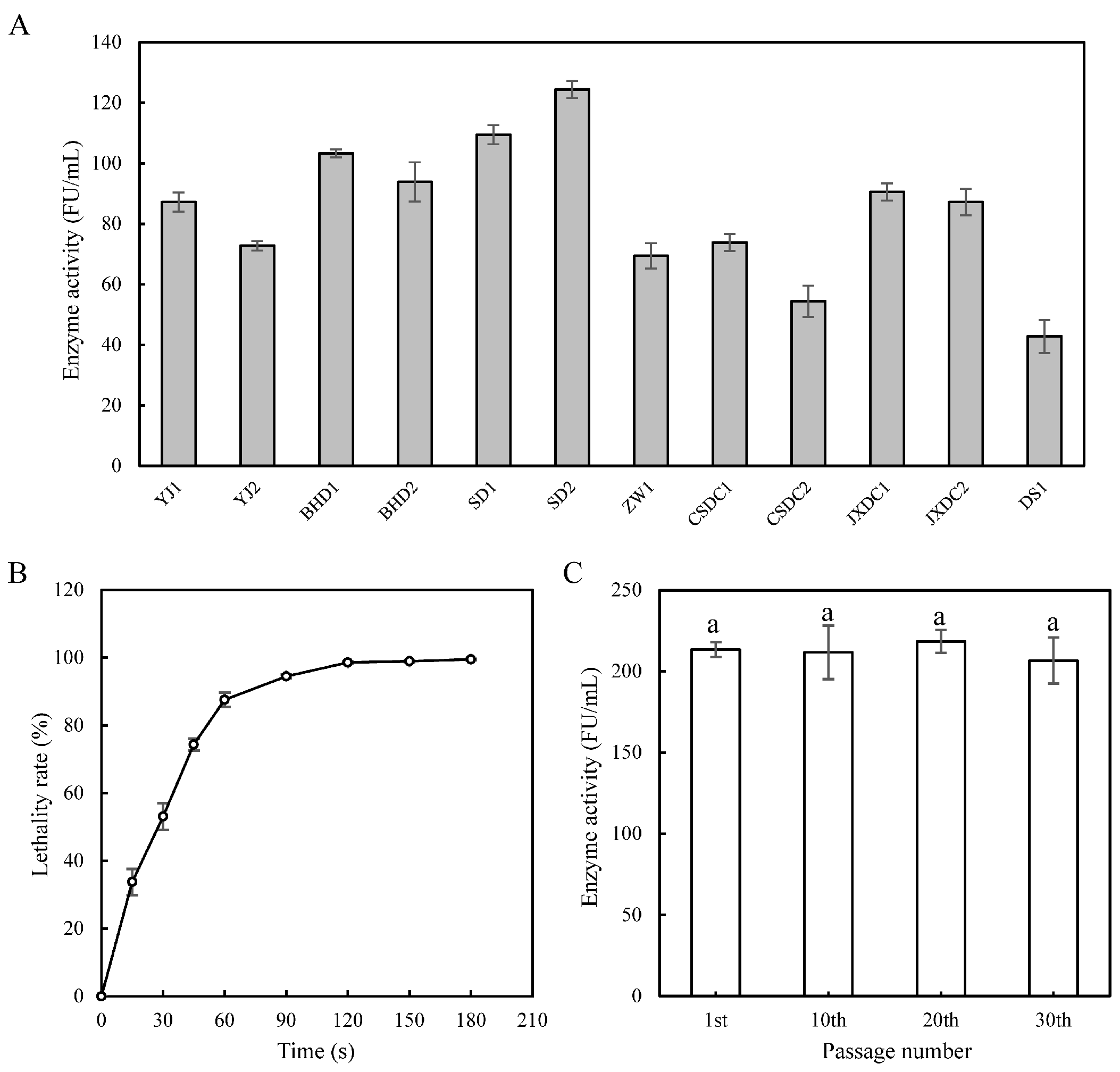
3.1. Strains Screening and ARTP Mutagenesis
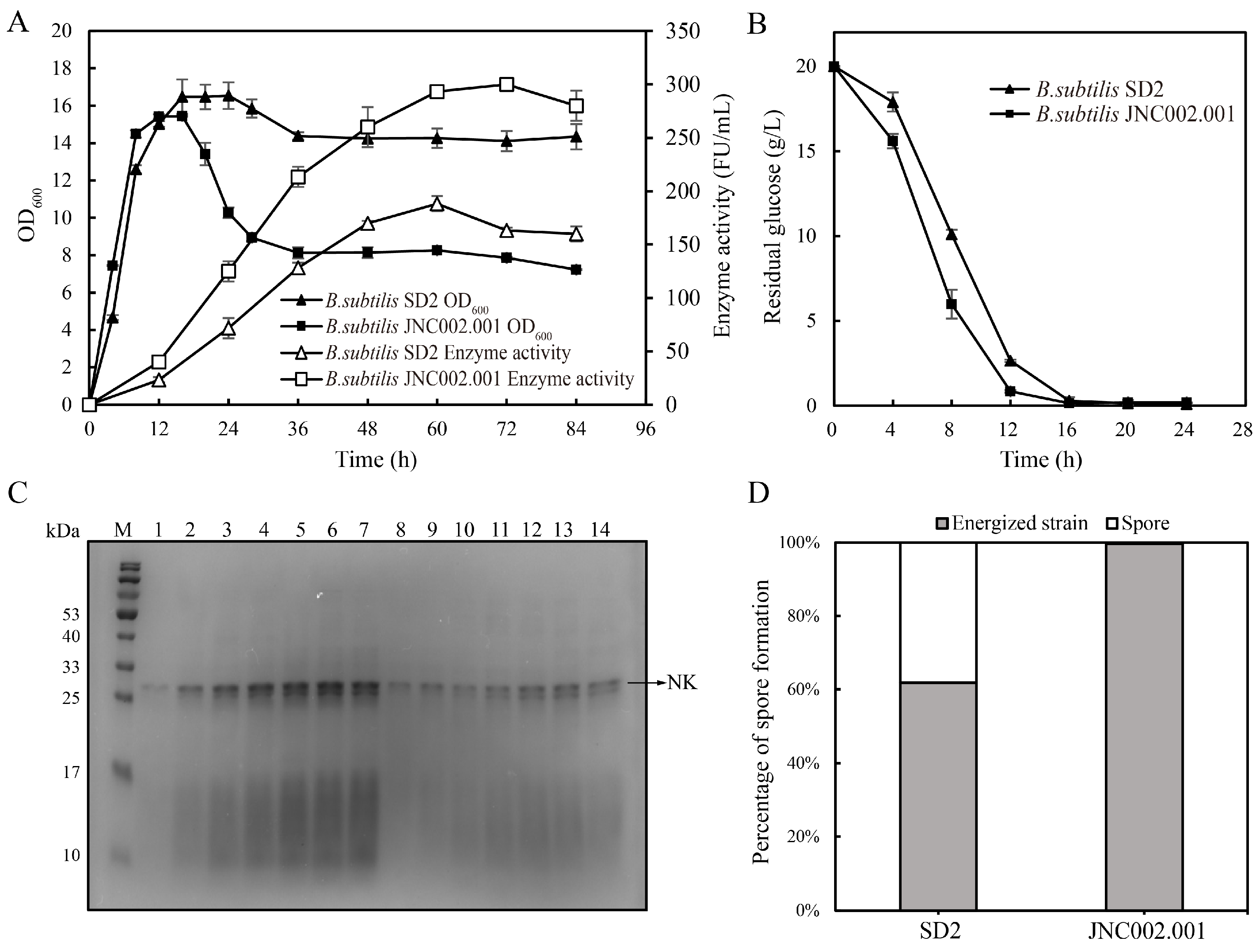
3.2. Comparison of the Fermentations of the Initial and Mutant Strains
3.3. Genomic Analysis
3.3.1. B. subtilis SD2 Genome Characterization and Functional Classification
3.3.2. Resequencing Results of B. subtilis JNC002.001
3.4. Transcriptomic Analysis
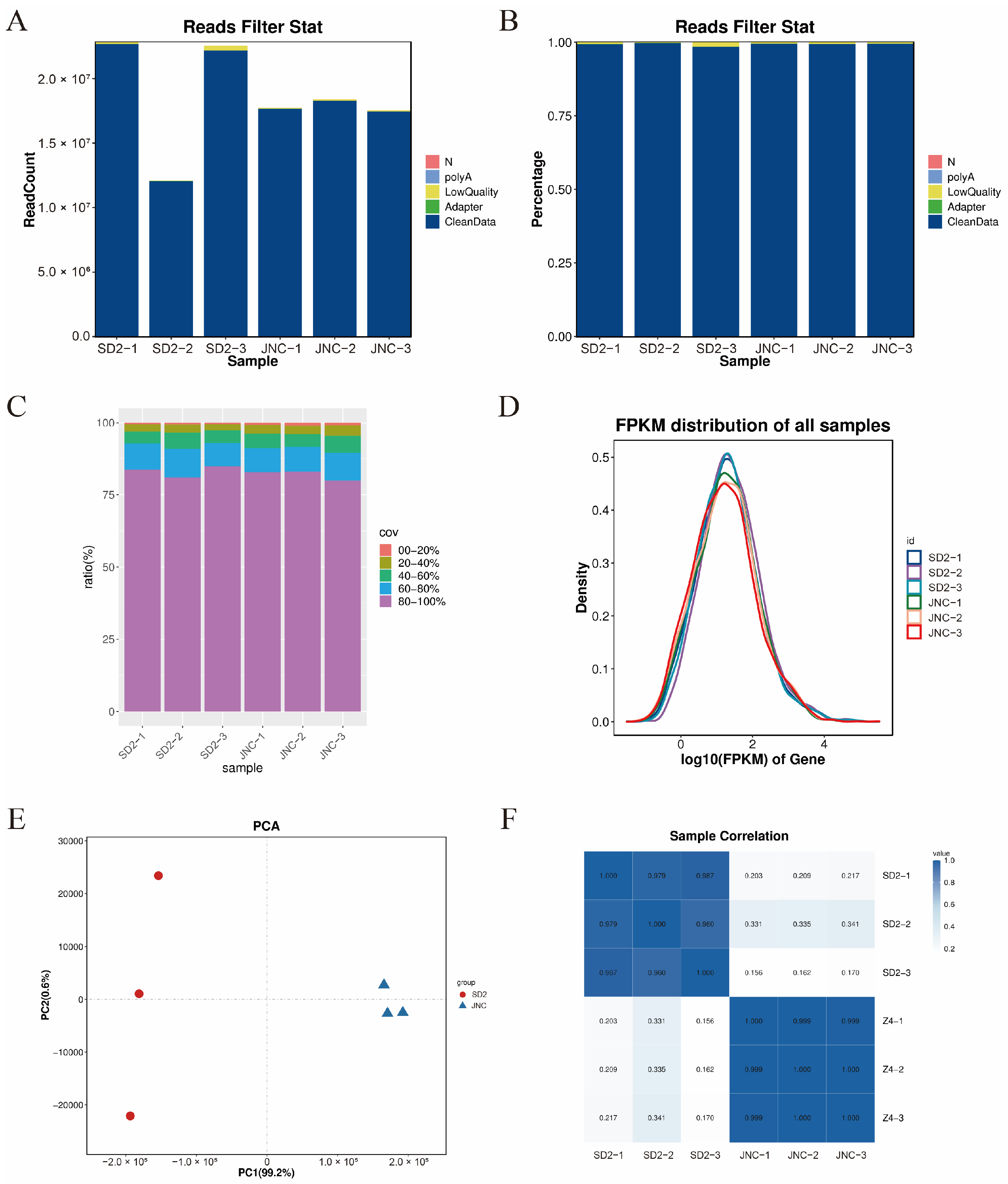
3.4.1. Quality Evaluation of the RNA Sequencing and Assembling
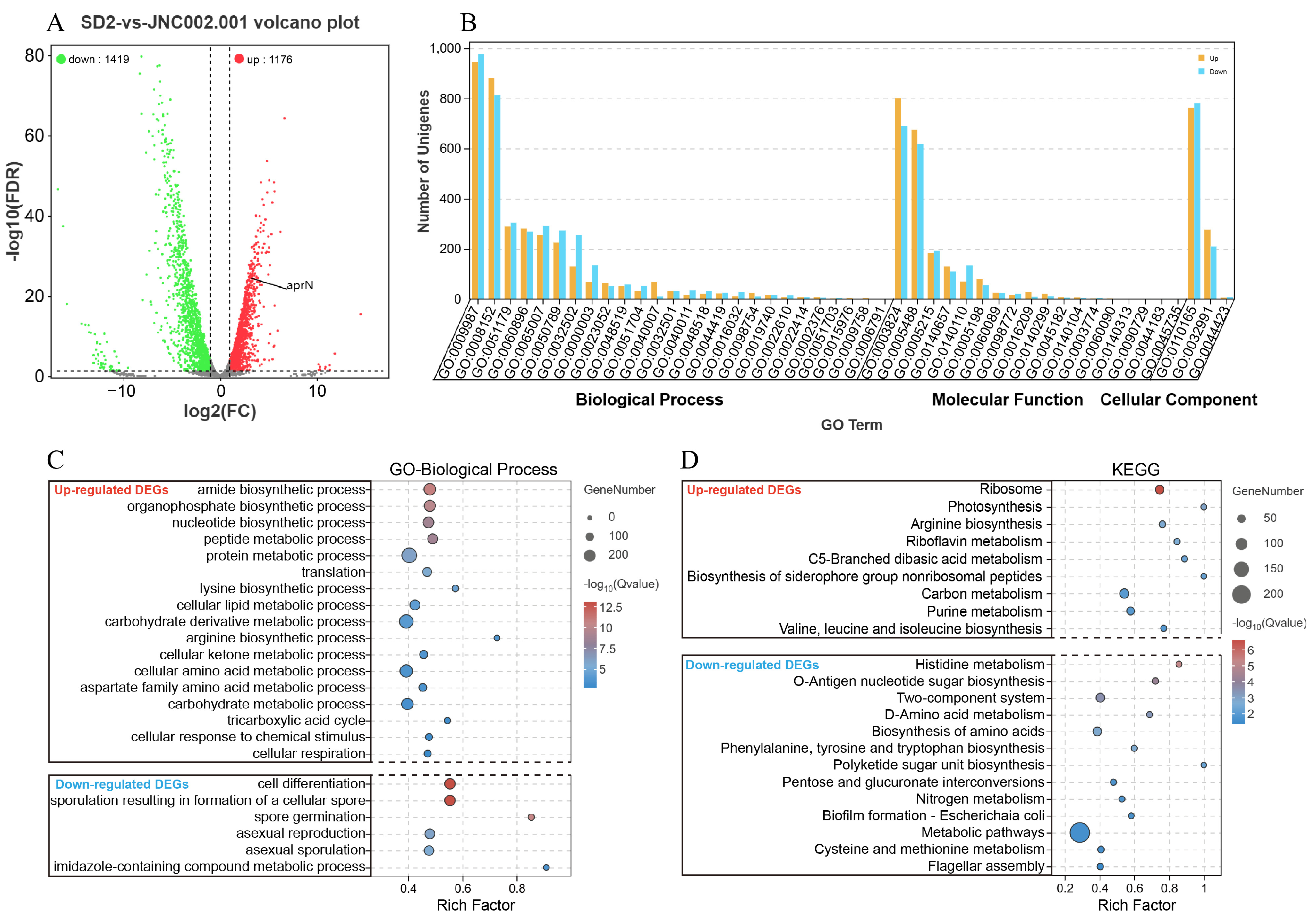
3.4.2. Statistics Analysis of the DEGs
3.4.3. GO and KEGG Analysis of the DEGs
3.5. Pathway Analysis of the Key DEGs
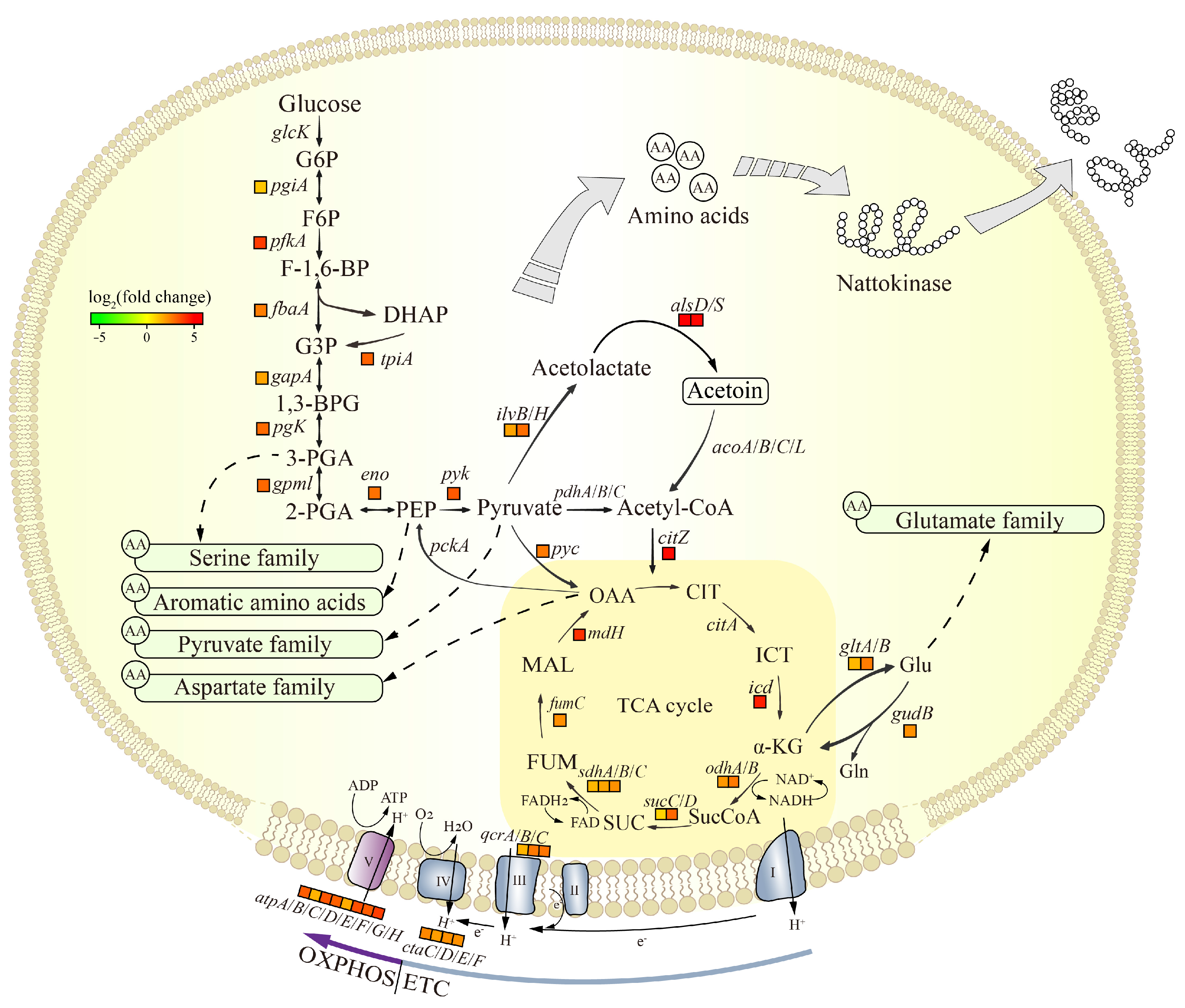
3.5.1. Analysis of the DEGs in Central Carbon Metabolism
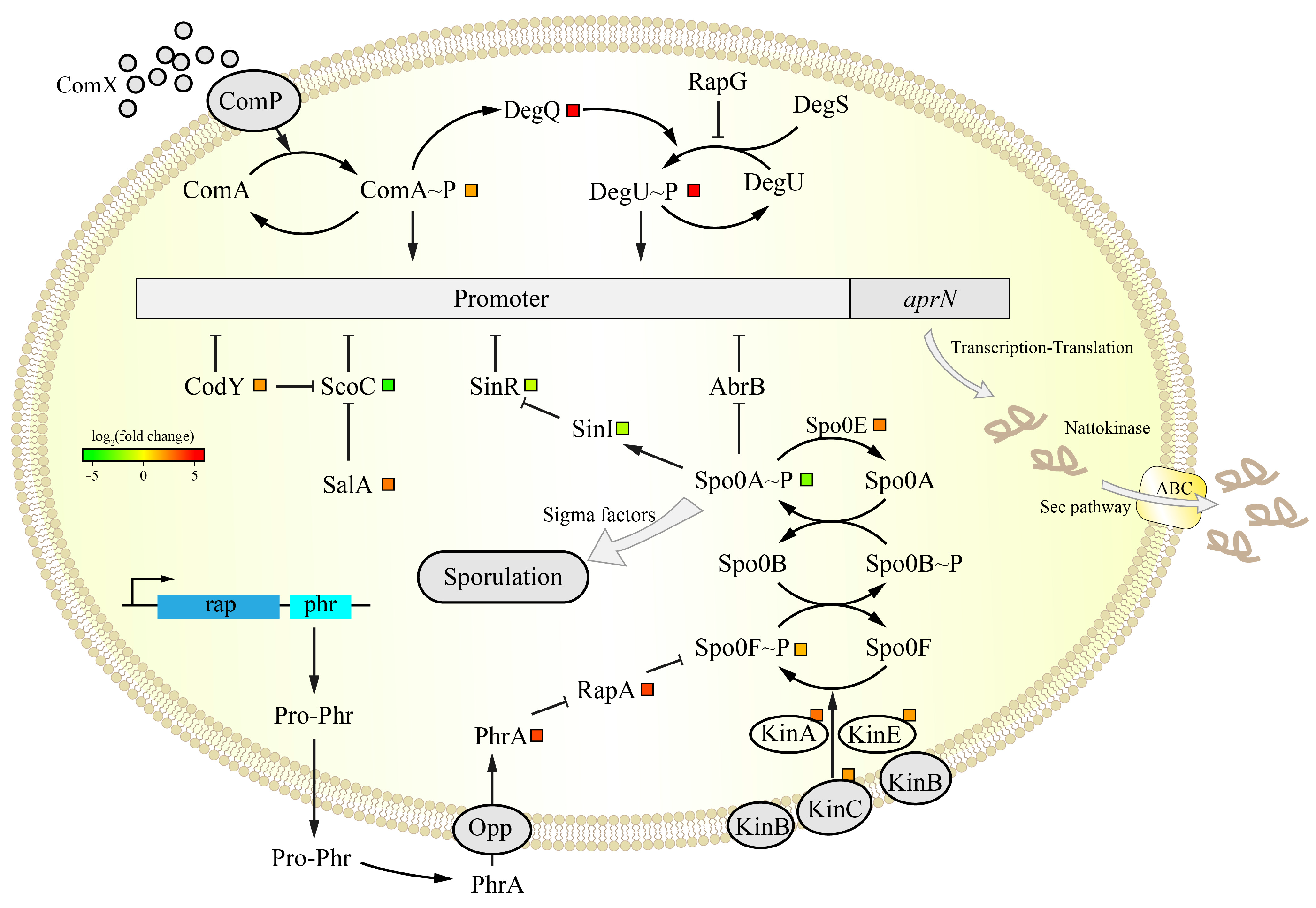
3.5.2. Analysis of the DEGs in the aprN Gene Transcription Process
3.5.3. Analysis of the DEGs in Sporulation
3.5.4. Analysis of the DEGs in NK Transport and Folding
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ARTP | atmospheric and room temperature plasma |
| DEG | differentially expressed gene |
| ETC | electron transport chain |
| GO | Gene Ontology |
| Ins | insertion |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LB | Luria–Bertani |
| NK | nattokinase |
| OXPHOS | oxidative phosphorylation |
| SNP | single nucleotide polymorphism |
References
- Sumi, H.; Hamada, H.; Tsushima, H.; Mihara, H.; Muraki, H. A novel fibrinolytic enzyme (nattokinase) in the vegetable cheese Natto; a typical and popular soybean food in the Japanese diet. Experientia 1987, 43, 1110–1111. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, L.; Tang, X.; Li, J. Biotechnology, Bioengineering and Applications of Bacillus Nattokinase. Biomolecules 2022, 12, 980. [Google Scholar] [CrossRef]
- Nakamura, T.; Yamagata, Y.; Ichishima, E. Nucleotide Sequence of the Subtilisin NAT Gene, aprN, of Bacillus subtilis (natto). Biosci. Biotechnol. Biochem. 1992, 56, 1869–1871. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ban, Y.-H.; Cha, Y.; An, E.S.; Choi, J.; Seo, D.W.; Park, D.; Choi, E.-K.; Kim, Y.-B. Comparative anti-thrombotic activity and haemorrhagic adverse effect of nattokinase and tissue-type plasminogen activator. Food Sci. Biotechnol. 2019, 28, 1535–1542. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, Y.; Zhang, Y.; Xu, F.; Chen, J.; Duan, L.; Zhang, T.; Wang, J.; Zhang, F. Breaking the vicious loop between inflammation, oxidative stress and coagulation, a novel anti-thrombus insight of nattokinase by inhibiting LPS-induced inflammation and oxidative stress. Redox Biol. 2020, 32, 101500. [Google Scholar] [CrossRef]
- Gallelli, G.; Di Mizio, G.; Palleria, C.; Siniscalchi, A.; Rubino, P.; Muraca, L.; Cione, E.; Salerno, M.; De Sarro, G.; Gallelli, L. Data Recorded in Real Life Support the Safety of Nattokinase in Patients with Vascular Diseases. Nutrients 2021, 13, 2031. [Google Scholar] [CrossRef]
- Jang, J.-Y.; Kim, T.-S.; Cai, J.; Kim, J.; Kim, Y.; Shin, K.; Kim, K.S.; Park, S.K.; Lee, S.-P.; Choi, E.-K.; et al. Nattokinase improves blood flow by inhibiting platelet aggregation and thrombus formation. Lab. Anim. Res. 2013, 29, 221. [Google Scholar] [CrossRef]
- Chen, H.; Chen, J.; Zhang, F.; Li, Y.; Wang, R.; Zheng, Q.; Zhang, X.; Zeng, J.; Xu, F.; Lin, Y. Effective management of atherosclerosis progress and hyperlipidemia with nattokinase: A clinical study with 1062 participants. Front. Cardiovasc. Med. 2022, 9, 964977. [Google Scholar] [CrossRef]
- Li, X.; Long, J.; Gao, Q.; Pan, M.; Wang, J.; Yang, F.; Zhang, Y. Nattokinase Supplementation and Cardiovascular Risk Factors: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Rev. Cardiovasc. Med. 2023, 24, 234. [Google Scholar] [CrossRef]
- Sheng, Y.; Yang, J.; Wang, C.; Sun, X.; Yan, L. Microbial nattokinase: From synthesis to potential application. Food Funct. 2023, 14, 2568–2585. [Google Scholar] [CrossRef]
- Wu, H.; Wang, H.; Xu, F.; Chen, J.; Duan, L.; Zhang, F. Acute toxicity and genotoxicity evaluations of Nattokinase, a promising agent for cardiovascular diseases prevention. Regul. Toxicol. Pharmacol. 2019, 103, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Suo, F.; Cheng, J.; Han, L.; Hao, W.; Guo, J.; Zhou, Z. Stepwise modifications of genetic parts reinforce the secretory production of nattokinase in Bacillus subtilis. Microb. Biotechnol. 2018, 11, 930–942. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zheng, W.; Ge, C.; Cui, W.; Zhou, L.; Zhou, Z. High-level extracellular production of recombinant nattokinase in Bacillus subtilis WB800 by multiple tandem promoters. BMC Microbiol. 2019, 19, 89. [Google Scholar] [CrossRef]
- Galano, M.; van den Dungen, M.W.; van Rij, T.; Abbas, H.E. Safety evaluation of food enzymes produced by a safe strain lineage of Bacillus subtilis. Regul. Toxicol. Pharmacol. 2021, 126, 105030. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Jiang, H.; Yang, G.; Ou, Y.; Lu, S.; Jiang, J.; Lei, R.; Su, L. Regulation and management of the biosecurity for synthetic biology. Synth. Syst. Biotechnol. 2022, 7, 784–790. [Google Scholar] [CrossRef]
- Goormans, A.R.; Snoeck, N.; Decadt, H.; Vermeulen, K.; Peters, G.; Coussement, P.; Van Herpe, D.; Beauprez, J.J.; De Maeseneire, S.L.; Soetaert, W.K. Comprehensive study on Escherichia coli genomic expression: Does position really matter? Metab. Eng. 2020, 62, 10–19. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Zhang, X.; Tong, Y.; Yang, R. Genomic and transcriptomic analysis of Bacillus subtilis JNFE1126 with higher nattokinase production through ultraviolet combined 60Co-γ ray mutagenesis. LWT 2021, 147, 111652. [Google Scholar] [CrossRef]
- Sheng, Y.; Zhang, S.; Li, X.; Wang, S.; Liu, T.; Wang, C.; Yan, L. Phenotypic and genomic insights into mutant with high nattokinase-producing activity induced by carbon ion beam irradiation of Bacillus subtilis. Int. J. Biol. Macromol. 2024, 271, 132398. [Google Scholar] [CrossRef]
- Gao, X.; Liu, E.; Yin, Y.; Yang, L.; Huang, Q.; Chen, S.; Ho, C.-T. Enhancing Activities of Salt-Tolerant Proteases Secreted by Aspergillus Oryzae Using Atmospheric and Room-Temperature Plasma Mutagenesis. J. Agric. Food Chem. 2020, 68, 2757–2764. [Google Scholar] [CrossRef]
- Li, C.; Xia, Y.; Li, M.; Zhang, T. ARTP mutagenesis of phospholipase D-producing strain Streptomyces hiroshimensis SK43.001, and its enzymatic properties. Heliyon 2022, 8, 12587. [Google Scholar] [CrossRef]
- Yang, Y.; Okoye, C.O.; Xiang, J.; Huang, W.; Liu, Y.; He, R.; Huang, G.; Ma, H. Genome-wide transcriptome analyses reveal changes in glutathione-overproducing yeast obtained by ARTP mutagenesis for rice wine brewing. Food Biosci. 2024, 60, 104388. [Google Scholar] [CrossRef]
- Zhang, G.; Zabed, H.M.; Zhang, Y.; Li, J.; Yun, J.; Qi, X. Random mutagenesis and transcriptomics-guided rational engineering in Zygosaccharomyces rouxii for elevating D-arabitol biosynthesis. Bioresour. Technol. 2024, 400, 130685. [Google Scholar] [CrossRef]
- Chen, L.; Liu, X.; Li, C.; Li, H.; Chen, W.; Li, D. Transcriptome analyses reveal the DHA enhancement mechanism in Schizochytrium limacinum LD11 mutant. Algal Res. 2022, 67, 102868. [Google Scholar] [CrossRef]
- Zhang, A.; Ma, Y.; Deng, Y.; Zhou, Z.; Cao, Y.; Yang, B.; Bai, J.; Sun, Q. Enhancing Protease and Amylase Activities in Bacillus licheniformis XS-4 for Traditional Soy Sauce Fermentation Using ARTP Mutagenesis. Foods 2023, 12, 2381. [Google Scholar] [CrossRef]
- Liu, D.; Han, Z.; Hu, Z.; Yu, C.; Wang, Y.; Tong, J.; Fang, X.; Yue, W.; Nie, G. Comparative analysis of the transcriptome of Bacillus subtilis natto incubated in different substrates for nattokinase production. Process Biochem. 2023, 129, 30–43. [Google Scholar] [CrossRef]
- Tian, Z.; Hou, L.; Hu, M.; Gao, Y.; Li, D.; Fan, B.; Wang, F.; Li, S. Optimization of Sporulation Conditions for Bacillus subtilis BSNK-5. Processes 2022, 10, 1133. [Google Scholar] [CrossRef]
- Chin, C.S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A.; et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, 112963. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Shen, S.; Li, W.; Wang, J. Bioherbicidal potential of Bacillus altitudinis D30202 on Avena fatua L.: A whole-genome sequencing analysis. J. Appl. Genet. 2023, 64, 809–817. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Zhu, X.; Luo, H.; Yu, X.; Lv, H.; Su, L.; Zhang, K.; Wu, J. Genome-Wide CRISPRi Screening of Key Genes for Recombinant Protein Expression in Bacillus Subtilis. Adv. Sci. 2024, 11, 2404313. [Google Scholar] [CrossRef]
- Yu, L.; Li, F.; Ni, J.; Qin, X.; Lai, J.; Su, X.; Li, Z.; Zhang, M. UV-ARTP compound mutagenesis breeding improves macrolactins production of Bacillus siamensis and reveals metabolism changes by proteomic. J. Biotechnol. 2024, 381, 36–48. [Google Scholar] [CrossRef]
- Lagage, V.; Chen, V.; Uphoff, S. Adaptation delay causes a burst of mutations in bacteria responding to oxidative stress. EMBO Rep. 2023, 24, e55640. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Zhou, H.; Zhang, H.; Lu, F. Optimization of alkaline protease production by rational deletion of sporulation related genes in Bacillus licheniformis. Microb. Cell Fact. 2019, 18, 127. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Yu, X.; Du, B.; Liu, K.; Yao, L.; Zhang, S.; Selin, C.; Fernando, W.G.D.; Wang, C.; Ding, Y. A single amino acid mutation in Spo0A results in sporulation deficiency of Paenibacillus polymyxa SC2. Res. Microbiol. 2016, 167, 472–479. [Google Scholar] [CrossRef]
- Leninger, M.; Sae Her, A.; Traaseth, N.J. Inducing conformational preference of the membrane protein transporter EmrE through conservative mutations. eLife 2019, 8, e48909. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Li, H.; Zhong, L.; Qin, Y.; Guo, G.; Liu, Z.; Hao, N. Analysis of heterologous expression of phaCBA promotes the acetoin stress response mechanism in Bacillus subtilis using transcriptomics and metabolomics approaches. Microb. Cell Fact. 2024, 23, 58. [Google Scholar] [CrossRef]
- Zhou, C.; Zhou, H.; Fang, H.; Ji, Y.; Wang, H.; Liu, F.; Zhang, H.; Lu, F. Spo0A can efficiently enhance the expression of the alkaline protease gene aprE in Bacillus licheniformis by specifically binding to its regulatory region. Int. J. Biol. Macromol. 2020, 159, 444–454. [Google Scholar] [CrossRef]
- Chen, P.T.; Chao, Y.-P. Enhanced production of recombinant nattokinase in Bacillus subtilis by the elimination of limiting factors. Biotechnol. Lett. 2006, 28, 1595–1600. [Google Scholar] [CrossRef]
- Tjalsma, H.; Bolhuis, A.; Jongbloed, J.D.H.; Bron, S.; van Dijl, J.M. Signal peptide-dependent protein transport in Bacillus subtilis: A genome-based survey of the secretome. Microbiol. Mol. Biol. Rev. 2000, 64, 515–547. [Google Scholar] [CrossRef]
- Chen, Y.; Li, M.; Yan, M.; Chen, Y.; Saeed, M.; Ni, Z.; Fang, Z.; Chen, H. Bacillus subtilis: Current and future modification strategies as a protein secreting factory. World J. Microbiol. Biotechnol. 2024, 40, 195. [Google Scholar] [CrossRef]
- Harwood, C.R.; Kikuchi, Y. The ins and outs of Bacillus proteases: Activities, functions and commercial significance. FEMS Microbiol. Rev. 2022, 46, fuab046. [Google Scholar] [CrossRef]
- Commichau, F.M.; Gunka, K.; Landmann, J.J.; Stülke, J. Glutamate metabolism in Bacillus subtilis: Gene expression and enzyme activities evolved to avoid futile cycles and to allow rapid responses to perturbations of the system. J. Bacteriol. 2008, 190, 3557–3564. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Zhang, H.; Fang, H.; Sun, Y.; Zhou, H.; Yang, G.; Lu, F. Transcriptome based functional identification and application of regulator AbrB on alkaline protease synthesis in Bacillus licheniformis 2709. Int. J. Biol. Macromol. 2021, 166, 1491–1498. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, G.; Albertini, A.M.; Ferrari, E.; Sonenshein, A.L.; Belitsky, B.R. Interplay of CodY and ScoC in the Regulation of Major Extracellular Protease Genes of Bacillus subtilis. J. Bacteriol. 2016, 198, 907–920. [Google Scholar] [CrossRef]
- Gallegos-Monterrosa, R.; Christensen, M.N.; Barchewitz, T.; Koppenhöfer, S.; Priyadarshini, B.; Bálint, B.; Maróti, G.; Kempen, P.J.; Dragoš, A.; Kovács, Á.T. Impact of Rap-Phr system abundance on adaptation of Bacillus subtilis. Commun. Biol. 2021, 4, 468. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Han, L.-L.; Chen, T.-Y.; Lu, Y.-B.; Feng, H. Characterization of a Protease Hyper-Productive Mutant of Bacillus pumilus by Comparative Genomic and Transcriptomic Analysis. Curr. Microbiol. 2020, 77, 3612–3622. [Google Scholar] [CrossRef] [PubMed]
- Derouiche, A.; Shi, L.; Bidnenko, V.; Ventroux, M.; Pigonneau, N.; Franz-Wachtel, M.; Kalantari, A.; Nessler, S.; Noirot-Gros, M.; Mijakovic, I. Bacillus subtilis SalA is a phosphorylation-dependent transcription regulator that represses scoC and activates the production of the exoprotease AprE. Mol. Microbiol. 2015, 97, 1195–1208. [Google Scholar] [CrossRef]
- Perego, M. Forty years in the making: Understanding the molecular mechanism of peptide regulation in bacterial development. PLoS Biol. 2013, 11, 1001516. [Google Scholar] [CrossRef]
- Bartels, J.; Blüher, A.; López Castellanos, S.; Richter, M.; Günther, M.; Mascher, T. The Bacillus subtilis endospore crust: Protein interaction network, architecture and glycosylation state of a potential glycoprotein layer. Mol. Microbiol. 2019, 112, 1576–1592. [Google Scholar] [CrossRef]
- Collinson, I. SecA—A New Twist in the Tale. J. Bacteriol. 2016, 199, 00736-16. [Google Scholar] [CrossRef]
- Kontinen, V.P.; Sarvas, M. The PrsA lipoprotein is essential for protein secretion in Bacillus subtilis and sets a limit for high-level secretion. Mol. Microbiol. 1993, 8, 727–737. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CDS Region Variants | |||||
|---|---|---|---|---|---|
| Gene ID | Gene Name | Annotation | Codon | Amino Acid Mutate | Type |
| AB5991_04395 | acoA | 2,6-dichlorophenolindophenol oxidoreductase subunit alpha | CGT→CAT | R297H | missense |
| AB5991_07820 | kinA | Sporulation kinase A | ATC→GTC | I578V | missense |
| AB5991_10430 | gltA | Glutamate synthase [NADPH] large chain | GAA→GAG | E102E | silent |
| AAA→GAA | K101E | missense | |||
| AB5991_14300 | comC | Prepilin leader peptidase | GAT→TAT | D172Y | missense |
| AB5991_18005 | ganP | Galacto-oligosaccharides transport system permease protein GanP | AAA→AAG | K215K | silent |
| AB5991_18470 | mdxK | Maltose phosphorylase | TTA→TTG | L271L | silent |
| AB5991_18900 | yvyF | Uncharacterized | CAT→TAT | H78Y | missense |
| Intergenic Region Variants | |||||
| Pos | Intergenic region | Variant | Comment | ||
| 339112 | ycgL–putB | G to T | No feature found | ||
| 2331408 | mmgA–glpQB | C to CATGA | SigE-binding site | ||
| 4120715 | rpmH–dnaA | T to A | No feature found | ||
| Gene Name | Annotation | SD2_fpkm | JNC002.001_fpkm | log2FC (JNC002.001/SD2) |
|---|---|---|---|---|
| aprN | Nattokinase, subtilisin NAT | 35,684.56 | 335,260.20 | 3.23 |
| nprE | Bacillolysin, neutral protease | 1049.24 | 13,915.68 | 3.73 |
| vpr | Minor extracellular protease | 108.57 | 466.81 | 2.10 |
| bpr | Bacillopeptidase F | 381.31 | 2016.95 | 2.40 |
| wprA | Cell wall-associated protease | 22.78 | 587.41 | 4.69 |
| aprX | Serine protease | 81,411.65 | 1665.46 | −5.61 |
| isp | Intracellular serine protease | 47,239.65 | 19,476.40 | −1.28 |
| cgeB | Spore maturation protein | 97,503.94 | 3462.53 | −4.82 |
| Gene Name | Annotation | log2FC (JNC002.001/SD2) |
|---|---|---|
| abrB | Transition state regulatory protein | 0.80 |
| codY | Global transcriptional regulator | 1.98 |
| comA | Two-component system response regulator | 1.73 |
| comP | Sensor histidine kinase | −0.26 |
| degQ | Pleiotropic regulator | 5.51 |
| degU | Two-component system response regulator | 5.41 |
| sinR | Master regulator of biofilm formation | −1.46 |
| sinI | Antagonist of SinR | −1.15 |
| scoC | DNA-binding transcriptional repressor | −4.31 |
| salA | phosphorylation-dependent transcriptional regulator | 2.62 |
| spo0A | Stage 0 sporulation protein A | −2.48 |
| spo0B | Sporulation initiation phosphotransferase B | −0.22 |
| spo0F | Sporulation initiation phosphotransferase F | 1.34 |
| kinA | Sporulation kinase A | 2.79 |
| kinB | Sporulation kinase B | 0.16 |
| kinC | Sporulation kinase C | 2.02 |
| kinD | Sporulation kinase D | −0.11 |
| kinE | Sporulation kinase E | 1.87 |
| spo0E | Aspartyl-phosphate phosphatase | 2.46 |
| rapA | Response regulator aspartate phosphatase A | 3.71 |
| phrA | Phosphatase RapA inhibitor | 3.69 |
| sigF | Sporulation sigma factor | 0.26 |
| sigE | Sporulation sigma factor | −1.96 |
| sigG | Sporulation sigma factor | −3.53 |
| sigK | Sporulation sigma factor | −3.33 |
| cgeB | Spore maturation protein | −4.82 |
| cotG | Spore coat protein G | −3.36 |
| cotX | Spore coat protein X | −1.13 |
| mmgA | Acetyl-CoA acetyltransferase | −4.57 |
| Gene Name | Annotation | log2FC (JNC002.001/SD2) |
|---|---|---|
| secY | Protein translocase subunit SecY | 1.51 |
| secA | Protein translocase subunit SecA | 1.94 |
| secDF | Protein translocase subunit SecDF | 1.71 |
| secG | Probable protein-export membrane protein SecG | 2.09 |
| yrbF | Sec translocon accessory complex subunit YrbF | 2.63 |
| prsA | Foldase protein PrsA | 2.79 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, L.; Chen, Y.; He, Z.; Wang, Z.; Chen, Q.; Chen, J.; Oz, F.; Xu, Z.; Zeng, M. Genomic and Transcriptomic Analysis of Mutant Bacillus subtilis with Enhanced Nattokinase Production via ARTP Mutagenesis. Foods 2025, 14, 898. https://doi.org/10.3390/foods14050898
Guo L, Chen Y, He Z, Wang Z, Chen Q, Chen J, Oz F, Xu Z, Zeng M. Genomic and Transcriptomic Analysis of Mutant Bacillus subtilis with Enhanced Nattokinase Production via ARTP Mutagenesis. Foods. 2025; 14(5):898. https://doi.org/10.3390/foods14050898
Chicago/Turabian StyleGuo, Liuyu, Yang Chen, Zhiyong He, Zhaojun Wang, Qiuming Chen, Jie Chen, Fatih Oz, Zhimin Xu, and Maomao Zeng. 2025. "Genomic and Transcriptomic Analysis of Mutant Bacillus subtilis with Enhanced Nattokinase Production via ARTP Mutagenesis" Foods 14, no. 5: 898. https://doi.org/10.3390/foods14050898
APA StyleGuo, L., Chen, Y., He, Z., Wang, Z., Chen, Q., Chen, J., Oz, F., Xu, Z., & Zeng, M. (2025). Genomic and Transcriptomic Analysis of Mutant Bacillus subtilis with Enhanced Nattokinase Production via ARTP Mutagenesis. Foods, 14(5), 898. https://doi.org/10.3390/foods14050898