Microbial Dynamics in a Musalais Wine Fermentation: A Metagenomic Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Materials
2.1.1. Detection Methods for Fundamental Physical and Chemical Parameters
2.1.2. Wine Sample Collection and Processing
2.2. DNA Extraction and Illumina-Based Sequencing
2.3. Bioinformatic Analysis
2.3.1. Assembly, Gene Prediction, and Gene Catalog
2.3.2. Taxonomic Annotation
2.3.3. Analysis of Differences in Species or Function
2.3.4. Alpha Diversity Analysis
2.3.5. LEfSe Analysis
2.3.6. Annotation Steps for KEGG (KO) and GO
2.3.7. Redundancy Analysis (RDA)
2.3.8. Microbial Co-Occurrence Network
2.4. Analysis of Volatile Compounds in Musalais Wine
3. Results
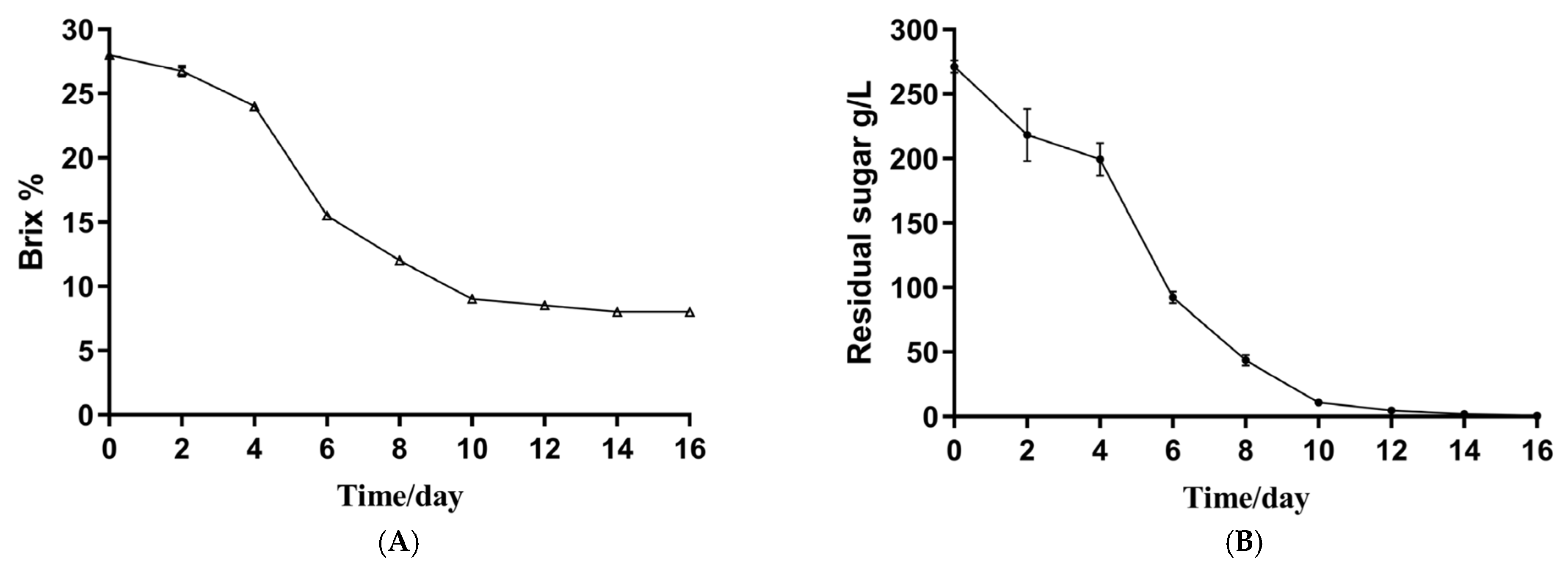
3.1. The Basic Physical and Chemical Indicators of the Musalais Wine
3.2. Analysis of Microbial Community Structure at Different Fermentation Stages of Musalais Wine
3.2.1. Alpha Diversity Analysis of Microorganisms in Musalais Wine Fermentation
3.2.2. Species Diversity Analysis of Microorganisms at Different Fermentation Stages of Musalais Wine
3.2.3. Microbial Community Structure Analysis Based on Taxonomic Levels
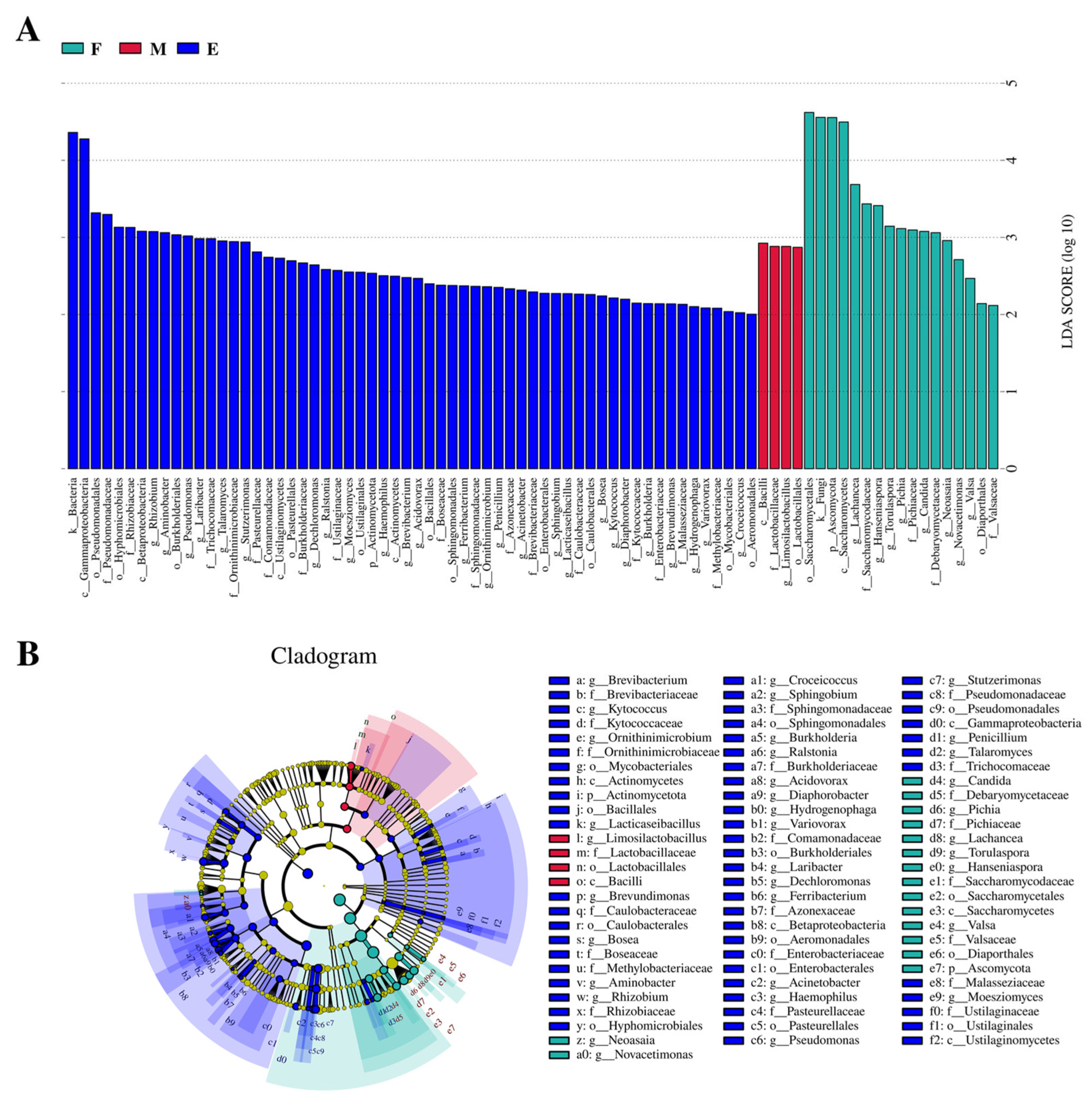
3.2.4. Analysis of Microbial Community Structure Based on Species Level
3.3. Analysis of the Correlation Between Musalais Wine Microbial Community Structure and Environmental Factors
3.4. Correlation Analysis Among Microbial Communities in Musalais Wine
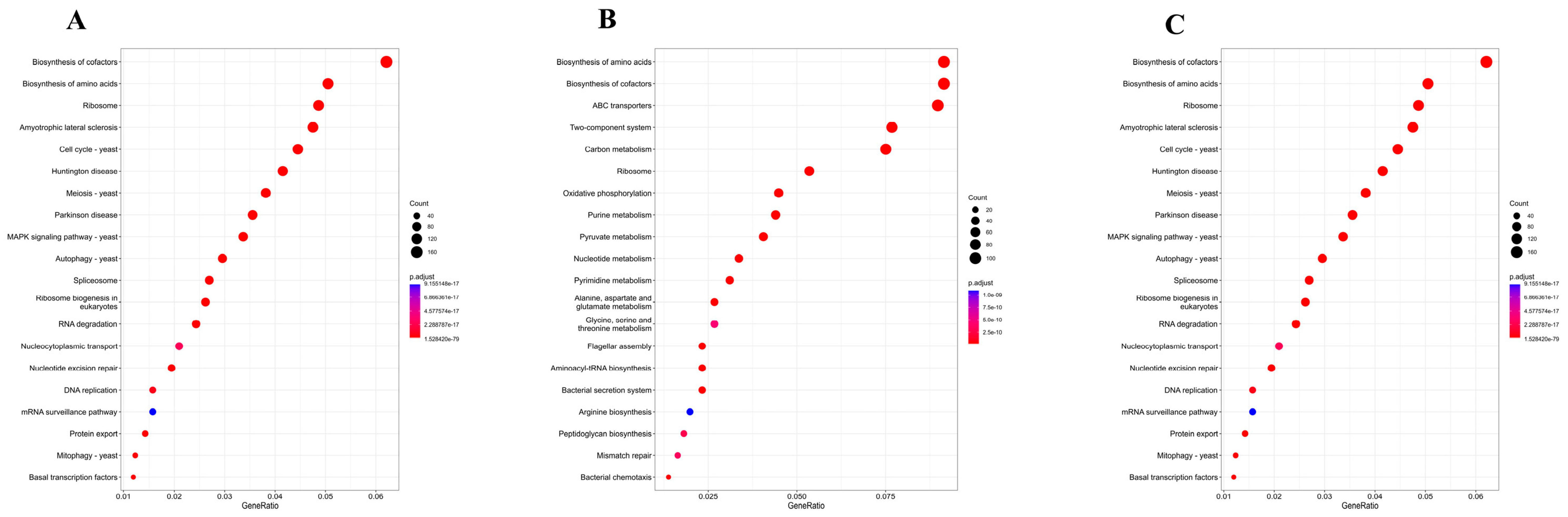
3.5. Comparative Analysis of Functional Gene Enrichment in Different Fermentation Stages
3.6. GC–MS Analysis of Volatile Compounds in Musalais Wine
4. Discussion
4.1. Microbial Origins in Musalais Wine: Resilience Beyond Heat Sterilization
4.2. Dynamic Changes in Functional Gene Enrichment During the Fermentation Process and Identification of Key Pathways
4.3. Microbial Diversity as a Catalyst for Volatile Compounds
4.4. Microbial Diversity and Associated Health Issues in Spontaneous Fermentation
4.5. Microbial Diversity from Natural Fermentation in Musalais Wine and Its Importance for Building a Microbial Resource Bank
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, L.X.; Wang, L.L.; Song, H.Z.; Guo, D.Q.; Fan, Y.G.; Hou, C.H.; Xue, J.L. Qualitative analysis of the main aroma compounds associated with traditional Musalais processing in Xinjiang, China. J. Inst. Brew. 2012, 118, 236–242. [Google Scholar] [CrossRef]
- Ma, W.R.; Yu, J.J.; Yang, F.; Zhang, X.M.; Zhang, F.J.; Jin, W.Y.; Sun, Z.W.; Zhao, Z.H.; Jia, S.R.; Zhong, C.; et al. Metagenomic analysis of the relationship between the microorganisms and the volatiles’ development in the wines during spontaneous fermentation from the eastern foothills of the Ningxia Helan mountains in China. J. Sci. Food Agric. 2023, 103, 6429–6439. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.Q.; Wang, F.F.; Tang, T.T.; Liu, Z.M.; Wang, L.L.; Wang, Q.L.; Shi, X.W.; Wang, B. A Snapshot of Microbial Succession and Volatile Component Dynamics of Marselan Wine in Xinjiang During Spontaneous Fermentation. Foods 2025, 14, 994. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.H.; Ma, Y.W.; Jiang, Z.Z.; Sam, F.E.; Peng, S.; Li, M.; Wang, J. Dynamic analysis of microbial communities and flavor properties in Merlot wines produced from inoculation and spontaneous fermentation. Food Res. Int. 2023, 164, 112379. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Xue, J. Modern technology homogenizes enological traits of indigenous Saccharomyces cerevisiae strains associated with Msalais, a traditional wine in China. World J. Microbiol. Biotechnol. 2017, 33, 63. [Google Scholar] [CrossRef] [PubMed]
- Chelliah, R.; Banan-MwineDaliri, E.; Khan, I.; Wei, S.; Elahi, F.; Yeon, S.J.; Selvakumar, V.; Ofosu, F.K.; Rubab, M.; Ju, H.H.; et al. A review on the application of bioinformatics tools in food microbiome studies. Brief. Bioinform. 2022, 23, bbac007. [Google Scholar] [CrossRef] [PubMed]
- Juszczuk-Kubiak, E.; Gregula-Kania, M.; Sokolowska, B. “Food-Omics” Applications in the Food Metagenom Profiling. Adv. Microbiol. 2021, 60, 59–75. [Google Scholar] [CrossRef]
- Bloomfield, S.J.; Hildebrand, F.; Zomer, A.L.; Palau, R.; Mather, A.E. Ecological insights into the microbiology of food using metagenomics and its potential surveillance applications. Microb. Genom. 2025, 11, 001337. [Google Scholar] [CrossRef] [PubMed]
- Salamandane, A.; Leech, J.; Almeida, R.; Silva, C.; Crispie, F.; Cotter, P.D.; Malfeito-Ferreira, M.; Brito, L. Metagenomic analysis of the bacterial microbiome, resistome and virulome distinguishes Portuguese Serra da Estrela PDO cheeses from similar non-PDO cheeses: An exploratory approach. Food Res. Int. 2024, 189, 114556. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Kamada, A.; Koyama, K.; Iwashita, K.; Goto-Yamamoto, N. Yeast diversity during the spontaneous fermentation of wine with only the microbiota on grapes cultivated in Japan. J. Biosci. Bioeng. 2023, 136, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Dhami, M.; Moukarzel, R.; Zhang, J.; On, S.; Wimalasiri, P.; Araujo, L.D.; Maxwell, D. Extending the Influence of Terroir through the Spontaneous Fermentation of Pinot Noir in the Vineyard: A Case Study of Greystone Vineyard Fermentation. J. Agric. Food Chem. 2025, 73, 8531–8542. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Manimaran, S.; Shen, Y.; Perez-Rogers, J.F.; Byrd, A.L.; Castro-Nallar, E.; Crandall, K.A.; Johnson, W.E. PathoScope 2.0: A complete computational framework for strain identification in environmental or clinical sequencing samples. Microbiome 2014, 2, 33. [Google Scholar] [CrossRef] [PubMed]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.J.; Weber, N.; Schuster, S.C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; McIver, L.J.; Rahnavard, G.; Thompson, L.R.; Schirmer, M.; Weingart, G.; Lipson, K.S.; Knight, R.; Caporaso, J.G.; Segata, N.; et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 2018, 15, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, G.; Jiang, S.; Liu, Y.X. Wekemo Bioincloud: A user-friendly platform for meta-omics data analyses. Imeta 2024, 3, e175. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Q.; Duan, J.W.; Lv, S.L.; Xu, L.; Li, H.H. Revealing the Changes in Compounds When Producing Strong-Flavor Daqu by Statistical and Instrumental Analysis. Fermentation 2022, 8, 720. [Google Scholar] [CrossRef]
- Gerard, L.M.; Corrado, M.B.; Davies, C.V.; Solda, C.A.; Dalzotto, M.G.; Esteche, S. Isolation and identification of native yeasts from the spontaneous fermentation of grape musts. Arch. Microbiol. 2023, 205, 302. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Alonso, I.; Walker, M.E.; Vallejo-Pascual, M.E.; Naharro-Carrasco, G.; Jiranek, V. Capturing yeast associated with grapes and spontaneous fermentations of the Negro Sauri minority variety from an experimental vineyard near Leon. Sci. Rep. 2021, 11, 3748. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Enriquez, L.; Vila-Crespo, J.; Rodriguez-Nogales, J.M.; Fernandez-Fernandez, E.; Ruiperez, V. Non-Saccharomyces Yeasts from Organic Vineyards as Spontaneous Fermentation Agents. Foods 2023, 12, 3644. [Google Scholar] [CrossRef] [PubMed]
- Rainieri, S.; Kodama, Y.; Kaneko, Y.; Mikata, K.; Nakao, Y.; Ashikari, T. Pure and mixed genetic lines of Saccharomyces bayanus and Saccharomyces pastorianus and their contribution to the lager brewing strain genome. Appl. Environ. Microbiol. 2006, 72, 3968–3974. [Google Scholar] [CrossRef] [PubMed]
- Rementeria, A.; Rodriguez, J.A.; Cadaval, A.; Amenabar, R.; Muguruza, J.R.; Hernando, F.L.; Sevilla, M.J. Yeast associated with spontaneous fermentations of white wines from the “Txakoli de Bizkaia” region (Basque Country, North Spain). Int. J. Food Microbiol. 2003, 86, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Steensels, J.; Verstrepen, K.J. Taming wild yeast: Potential of conventional and nonconventional yeasts in industrial fermentations. Annu. Rev. Microbiol. 2014, 68, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Vasyagin, E.A.; Urakov, V.N.; Shalamitskiy, M.Y.; Cherviak, S.N.; Ivanova, E.V.; Zagoruyko, V.I.; Beletsky, A.V.; Rakitin, A.L.; Mardanova, E.S.; Kushnirov, V.V.; et al. Development of a wine yeast strain capable of malolactic fermentation and reducing the ethyl carbamate content in wine. Foods 2024, 14, 54. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Zhou, H.; Zheng, H.; Xie, G. Investigating the role of primary fungi in Huangjiu fermentation: Insights from flavor orientation and synthetic microbiomes. Food Microbiol. 2025, 129, 104765. [Google Scholar] [CrossRef] [PubMed]
- Silva de Souza, H.L.; de Almeida, D.P.; Dos Santos, A.S.; Ramos, C.L. Physiological and fermentative performance of non-Saccharomyces yeasts isolated from kombucha for beer production. Food Sci. Technol. Int. 2025, 10820132251322288. [Google Scholar] [CrossRef] [PubMed]
- Almeida Dos Anjos, V.H.; de Brito Araujo Carvalho, A.J.; Prudencio Dutra, M.D.C.; Candido da Silva, M.C.; Santos Leite Neta, M.T.; Cardoso Viana, A.; Dos Santos Lima, M. Effect of commercial Saccharomyces cerevisiae and non-Saccharomyces yeasts on the chemical composition and bioaccessibility of pineapple wine. Food Res. Int. 2024, 194, 114888. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, Z.; Zhao, F.; Li, Y.; Liang, Y.; Zhou, R.; Shen, S.; Yu, J.; Liu, W.; Menghe, B. Dual-omics strategy for selecting optimal fermentation strains in traditional koumiss production. Food Chem. X 2025, 27, 102407. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, H.M.; Li, F.Y.; Zhao, W.H.; Yue, D.; Chi, Z.M.; Liu, G.L. Enhanced expression of glycolysis regulators KmGcr1p and KmGcr2p in Kluyveromyces marxianus: An efficient strategy for high-temperature ethanol production from inulin. Bioresour. Technol. 2025, 430, 132559. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Flores, M.; Delfino, A.V.; Rodriguez, M.E.; Lopes, C.A. Presence of Saccharomyces eubayanus in fermentative environments reveals a new adaptive scenario in Patagonia. Yeast 2023, 40, 476–492. [Google Scholar] [CrossRef] [PubMed]
- Perez, A.E.; Hernandez-Montelongo, J.; Flores, M.G.; Rodriguez, M.E.; Lopes, C.A.; Lanata, J.L. Revealing the ancient origins of blonde beers: Phylogeography and phylogenetics of cryotolerant fermentative yeast Saccharomyces eubayanus from pre-Hispanic pottery in Northwestern Patagonia, Argentina. PLoS ONE 2025, 20, e0319938. [Google Scholar] [CrossRef] [PubMed]
- Shu, M.; Yang, Y.; Pan, F.; Bian, T.; Li, Q.; Liao, F.; He, W.; Li, S.; Xu, J.; Hu, T.; et al. Effects of the multi-stress-resistant strain Zygosaccharomyces parabailii MC-5K3 bioaugmentation on microbial communities and metabolomics in tobacco waste extract. Arch. Microbiol. 2023, 205, 299. [Google Scholar] [CrossRef] [PubMed]
- Afonso, S.; Teixeira, A.L.; Escobar, E.; Ines, A.; Vilela, A. Non-Alcoholic Wines: Sensory Pleasantness and Health Benefits. Foods 2025, 14, 1356. [Google Scholar] [CrossRef] [PubMed]
- Africa, A.J.; Setati, M.E.; Hitzeroth, A.C.; Blancquaert, E.H. Exploring the evolution of microbial communities from the phyllosphere and carposphere to the grape must of Vitis vinifera L. cv’s Chardonnay and Pinot noir. Food Microbiol. 2025, 130, 104780. [Google Scholar] [CrossRef] [PubMed]
- Onetto, C.A.; Ward, C.M.; Van Den Heuvel, S.; Hale, L.; Cuijvers, K.; Borneman, A.R. Temporal and spatial dynamics within the fungal microbiome of grape fermentation. Environ. Microbiol. 2024, 26, e16660. [Google Scholar] [CrossRef] [PubMed]
- Zhadyra, S.; Tao, F.; Xu, P. Exploring the Microbiome and Functional Metabolism of Fermented Camel Milk (Shubat) Using Metagenomics. Foods 2025, 14, 1102. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Li, L.; Ma, Y.; Shen, C.H.; Ao, Z.H.; Song, C.; Mehmood, M.A.; Zhang, P.Y.; Liu, Y.; Sun, X.K.; et al. Combined transcriptomics and metabolomics analyses reveal the molecular mechanism of heat tolerance in Pichia kudriavzevii. Front. Microbiol. 2025, 16, 1572004. [Google Scholar] [CrossRef] [PubMed]
- Pei, T.W.; Gao, Z.W.; Wang, Z.H.; Wang, H.; Nwanade, C.F.; Bing, Z.Y.; Li, L.; Liang, X.J.; Zhang, Y.C.; Tang, Y.S.; et al. The genome-wide characterisation of cold shock proteins and prominent roles involved in cold response by configuring metabolic pathways in Haemaphysalis longicornis. Insect Mol. Biol. 2025. [Google Scholar] [CrossRef] [PubMed]
- Graciano, A.; Liu, A.M. Protein-derived cofactors: Chemical innovations expanding enzyme catalysis. Chem. Soc. Rev. 2025, 54, 4502–4530. [Google Scholar] [CrossRef] [PubMed]
- Reisse, S.; Haack, M.; Garbe, D.; Sommer, B.; Steffler, F.; Carsten, J.; Bohnen, F.; Sieber, V.; Brück, T. In Vitro Bioconversion of Pyruvate to n-Butanol with Minimized cofactor Utilization. Front. Bioeng. Biotechnol. 2016, 4, 74. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K. CDP-DAG synthesis by peripheral membrane-bound Tam41-type enzymes. J. Biochem. 2024, 176, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Tachezy, J.; Makki, A.; Hrdy, I. The hydrogenosome of Trichomonas vaginalis. J. Eukaryot. Microbiol. 2022, 69, e12922. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.L.; Yang, X.K.; Meng, Q.; Zhao, L.L.; Yuan, Y.X.; Chi, W.; He, L.; Shi, K.; Liu, S.W. Integrative multiomics analysis of the acid stress response of Oenococcus oeni mutants at different growth stages. Food Microbiol. 2022, 102, 103905. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.L.; Chai, X.Q.; Wang, Y.; Liu, J.; Zhou, G.H.; Wei, P.H.; Song, Y.H.; Ma, L.M. The multiple effects of REG1 deletion and SNF1 overexpression improved the production of S-adenosyl-l-methionine in Saccharomyces cerevisiae. Microb. Cell Factories 2022, 21, 174. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J. Retromer-Mediated Protein Sorting and Vesicular Trafficking. J. Genet. Genom. 2016, 43, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.; Fan, B.; Hao, J.; Yao, Y.; Ran, S.; Wang, H.; Li, H.; Wei, R. Changes in Microbial Community Diversity and the Formation Mechanism of Flavor Metabolites in Industrial-Scale Spontaneous Fermentation of Cabernet Sauvignon Wines. Foods 2025, 14, 235. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lei, X.; Sun, L.; Gao, B.; An, P.; Ye, D.; Mu, H.; Qin, Y.; Song, Y.; Liu, Y. Exploring the potentials of indigenous Saccharomyces cerevisiae and Pichia kudriavzevii for enhancing flavour and aromatic characteristics in apricot wines. Food Chem. X 2025, 25, 102178. [Google Scholar] [CrossRef] [PubMed]
- Kanter, J.P.; Benito, S.; Brezina, S.; Beisert, B.; Fritsch, S.; Patz, C.D.; Rauhut, D. The impact of hybrid yeasts on the aroma profile of cool climate Riesling wines. Food Chem. X 2020, 5, 100072. [Google Scholar] [CrossRef] [PubMed]
- Boban, A.; Vrhovsek, U.; Masuero, D.; Milanovic, V.; Budic-Leto, I. Effect of indigenous non-Saccharomyces yeasts on lipid compositions of maraština wine. Foods 2025, 14, 269. [Google Scholar] [CrossRef] [PubMed]
- Thivijan, S.; Pavalakumar, D.; Gunathunga, C.J.; Undugoda, L.J.S.; Manage, P.M.; Nugara, R.N.; Bandara, P.C.; Thambugala, K.M.; Al-Asmari, F.; Promputtha, I. Influence of indigenous non-Saccharomyces yeast strains on the physicochemical and sensory properties of wine fermentation: A promising approach to enhancing wine quality. Front. Cell. Infect. Microbiol. 2024, 14, 1495177. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Chen, Y.; Gao, B.; Zhao, X.; Sun, Q.; Qin, Y.; Song, Y.; Jiang, J.; Liu, Y. Adaptive laboratory evolution of Lachancea thermotolerans for enhanced production of 2-Phenylethanol and 2-Phenylethyl acetate in wine. Food Chem. X 2025, 27, 102483. [Google Scholar] [CrossRef] [PubMed]
- Januszek, M.; Satora, P.; Wajda, L.; Tarko, T. Saccharomyces bayanus Enhances Volatile Profile of Apple Brandies. Molecules 2020, 25, 3127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liu, D.; Liu, H.; Shen, J.; Zhang, J.; He, L.; Li, J.; Zhou, P.; Guan, X.; Liu, S.; et al. Impact of indigenous Oenococcus oeni and Lactiplantibacillus plantarum species co-culture on Cabernet Sauvignon wine malolactic fermentation: Kinetic parameters, color and aroma. Food Chem. X 2024, 22, 101369. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Feng, S.; Bi, P.; Han, J.; Li, S.; Liu, X.; Zhang, Z.; Long, F.; Guo, J. Simultaneous inoculation of non-Saccharomyces yeast and lactic acid bacteria for aromatic kiwifruit wine production. Food Microbiol. 2024, 123, 104589. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Xia, Y.; Chen, M.; Zhong, F. The availability of ethanol: A decisive factor in the biosynthesis of ethyl esters in enzyme-modified cheese flavor. Food Chem. 2025, 483, 144245. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.H.; Kim, N.H.; Shim, M.B.; Jeon, Y.W.; Ahn, J.H.; Lee, S.H.; Hwang, I.G.; Rhee, M.S. Microbiological Diversity and Prevalence of Spoilage and Pathogenic Bacteria in Commercial Fermented Alcoholic Beverages (Beer, Fruit Wine, Refined Rice Wine, and Yakju). J. Food Prot. 2015, 78, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Mehlomakulu, N.N.; Setati, M.E.; Divol, B. Characterization of novel killer toxins secreted by wine-related non-Saccharomyces yeasts and their action on Brettanomyces spp. Int. J. Food Microbiol. 2014, 188, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Belda, I.; Izquierdo-Gea, S.; Benitez-Dominguez, B.; Ruiz, J.; Vila, J.C.C. Wine Fermentation as a Model System for Microbial Ecology and Evolution. Environ. Microbiol. 2025, 27, e70092. [Google Scholar] [CrossRef] [PubMed]
- Delso, C.; Berzosa, A.; Sanz, J.; Alvarez, I.; Raso, J. Microbial Decontamination of Red Wine by Pulsed Electric Fields (PEF) after Alcoholic and Malolactic Fermentation: Effect on Saccharomyces cerevisiae, Oenococcus oeni, and Oenological Parameters during Storage. Foods 2023, 12, 278. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.B.; Liu, S.P.; Liu, T.T.; Chang, R.; Liu, G.X.; Li, M.L.; Mao, J. The Microbial Diversity and Flavor Metabolism Regulation of Xiangzao During Different Natural Fermentation Time Periods. Foods 2024, 13, 3931. [Google Scholar] [CrossRef] [PubMed]
- Lima, N. Preserving and utilizing microbial diversity for innovation and sustainability. Microbiology 2025, 171, 001544. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pei, Y.; Chen, M.; Chen, Q. Microbial Dynamics in a Musalais Wine Fermentation: A Metagenomic Study. Foods 2025, 14, 2570. https://doi.org/10.3390/foods14152570
Pei Y, Chen M, Chen Q. Microbial Dynamics in a Musalais Wine Fermentation: A Metagenomic Study. Foods. 2025; 14(15):2570. https://doi.org/10.3390/foods14152570
Chicago/Turabian StylePei, Yongzeng, Mengrong Chen, and Qiling Chen. 2025. "Microbial Dynamics in a Musalais Wine Fermentation: A Metagenomic Study" Foods 14, no. 15: 2570. https://doi.org/10.3390/foods14152570
APA StylePei, Y., Chen, M., & Chen, Q. (2025). Microbial Dynamics in a Musalais Wine Fermentation: A Metagenomic Study. Foods, 14(15), 2570. https://doi.org/10.3390/foods14152570