1. Introduction
Umami is the fifth basic taste sensation following sweet, sour, bitter, and salty [
1]. The taste-presenting properties of umami derive from the synergistic effects of free amino acids, flavor nucleotides, organic acids, and umami peptides [
2], with umami peptides being the core taste-presenting factors. These active peptides are widely found in systems of microbial interaction with food matrices, in fermented foods such as soy sauce and miso [
3], or enzymatic products of cereal proteins such as soybean and wheat [
4]. Fermented sausage, as a meat fermentation product, undergoes a synergistic effect of microbial metabolism and protein degradation during processing, releasing free amino acids and nucleotides, thus producing umami peptides with unique structures. Although direct research on umami peptides in fermented sausages is currently relatively limited, significant progress has been made in the study of fermented hams. For instance, Dang et al. [
5] identified two umami-active peptides, CCNKSV and AHSVRFY, from Jinhua ham and Parma ham. Furthermore, Cui et al. [
6] isolated and identified four umami peptides from Xuanwei ham, with sequences of MDAIKKMQ, RKYEEVAR, YVGDEAQSKRG, and VNVDEVGGEALGR. Considering that both fermented hams and fermented sausages are fermented meat products, and their flavor formation processes involve similar microbial metabolism and protein degradation, the findings and identification results of umami peptides in fermented hams can serve as an important reference for predicting and exploring potential umami peptides in fermented sausages. Consequently, fermented sausage can be regarded as a source of umami peptides.
The conventional approach for identifying umami peptides involves protein hydrolysis, followed by separation and purification using chromatography and mass spectrometry techniques. Ultimately, the confirmation of their freshness activity is achieved through artificial sensory evaluation. Liu et al. [
7] screened seven umami peptides by protein hydrolysis. In a separate study, Chen et al. [
8] identified five umami peptides by combining continuous enzymatic hydrolysis with ultrafiltration technology. However, this approach involves complicated separation and purification steps and relies on manual subjective determination, which is inefficient and expensive, and it is difficult to meet the demands of high-throughput screening.
In recent years, the rise of artificial intelligence techniques such as machine learning and deep learning has significantly boosted the field of umami peptide prediction. Jiang et al. [
9] proposed the iUmami-DRLF model, which combines mLSTM-based UniRep feature extraction with logistic regression. Charoenkwan et al. [
10] predicted umami peptides by developing the iUmami-SCM model using a scorecard approach combined with dipeptide propensity scores. Jiang et al. [
11] developed the iUP-BERT model, which used the bidirectional encoder converter BERT to extract peptide sequence features. Cui et al. [
12] predicted umami peptides using the gradient boosting decision tree model Umami_YYDS, which was validated by sensory experiments. Yue et al. [
13] proposed the Umami-MRNN model combining a recurrent neural network (RNN) and multilayer perceptron (MLP). Hansan et al. [
14] developed the UMPred-FRL model by integrating seven feature encodings using a feature learning approach. The UmamiPreDL model [
15] integrated ProtBERT’s protein sequence comprehension with CNN-driven local feature extraction, thereby demonstrating the feasibility of machine learning and deep learning in umami peptide discovery.
A key advantage of artificial intelligence lies in its ability to automatically extract complex hidden patterns from peptide sequences, thereby overcoming the reliance of traditional methods on manually designed molecular descriptors. The model integrates multi-source feature encoding and dynamic feature extraction, enabling the effective analysis of long-range dependence and spatial distribution between amino acid residues. Additionally, it addresses the issue of small-sample imbalance through data enhancement technology, enhancing the model’s generalization capability despite limited datasets. Compared with traditional hydrolysis purification methods, artificial intelligence methods help to predict umami peptides more accurately and effectively, making the high-throughput screening of novel umami peptides possible.
Protein structure prediction methods, such as AlphaFold, can aid in determining the three-dimensional shape of a target protein from its amino acid sequence. Molecular docking analyses involve simulating interactions between a ligand and a receptor to predict binding properties. The human perception of umami flavors occurs via the T1R1/T1R3 taste receptors, located on the anterior aspect of the tongue [
16]. In recent umami peptide studies, two complementary computational approaches were systematically integrated by Dang et al. [
17] for prediction. Amin et al. [
18] simulated binding patterns through LC-MS/MS technology combined with molecular docking. Wang et al. [
19], through amino acid distribution and active site analysis, proposed a new decision rule based on a scorecard to predict umami peptides. Chang et al. [
20] predicted the binding mode of the umami peptide EGTAG using this method. Ruan et al. [
21] predicted five umami peptides by analyzing their binding properties to the receptor using hydrogen bonding and hydrophobic interactions. These studies not only validate the reliability of the methods of homology modeling and molecular docking in the prediction of umami peptides but also lay the methodological foundation for the design of flavor molecules.
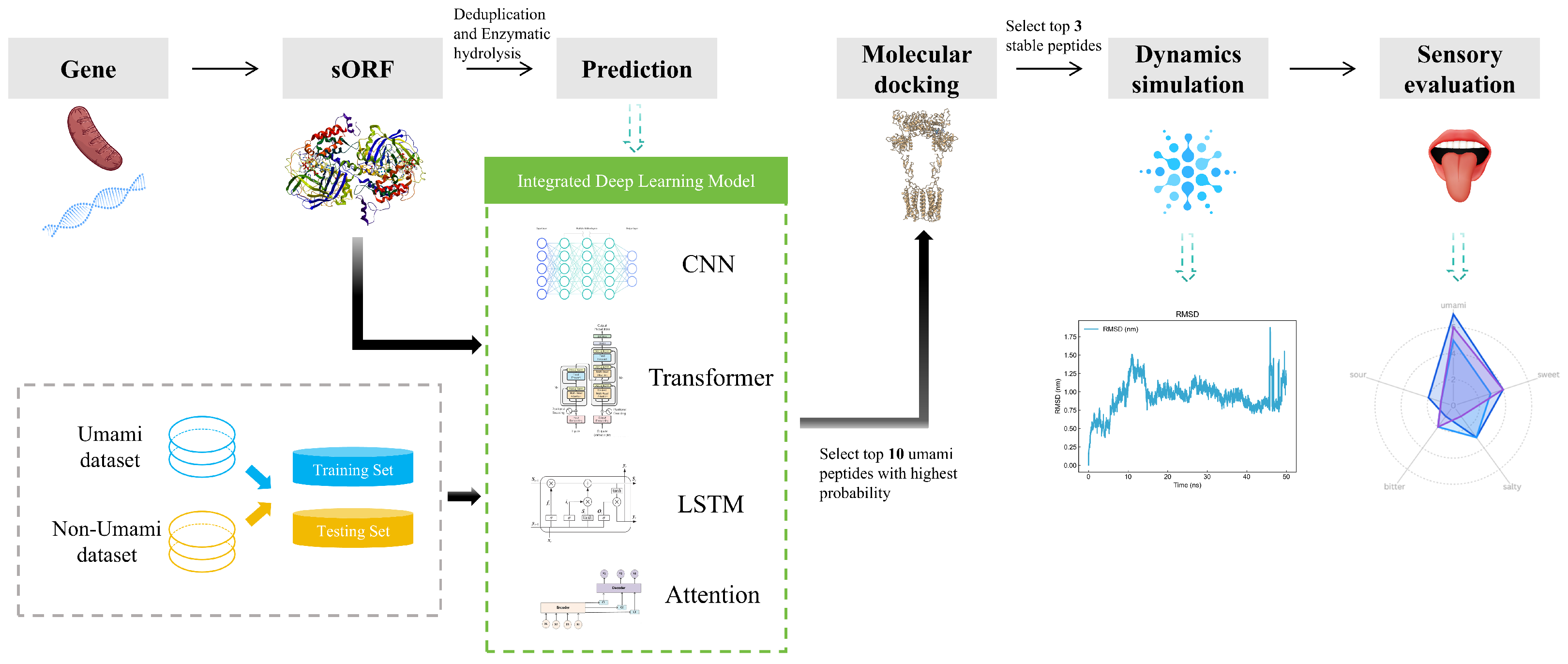
However, extant models predominantly depend on a solitary infrastructure for prediction, which is susceptible to suboptimal accuracy due to one-sided feature capture, faces challenges in balancing local sequence features and long-range dependencies, and may overlook the cross-scale association of peptide–receptor dynamic binding. In this study, we propose an integrated deep learning framework that integrates CNN, Transformer, LSTM, and Attention architectures for umami peptide prediction and ultimately integrates multidimensional information to improve the resulting model’s robustness and generalization ability. In addition, to gain a deeper understanding of the model’s prediction mechanism, we employed the SHapley Additive exPlanations (SHAP) analysis method. Through SHAP analysis, we were able to perform global interpretation to identify the amino acid positions that have the greatest impact on umami prediction; provide local explanations that clarify why specific peptide segments are predicted as umami peptides; and reveal the positive or negative contributions (i.e., feature directions) of specific amino acids at specific positions.
3. Results and Analysis
3.1. Model Construction and Evaluation Results
We performed metagenomic sequencing to acquire FASTA data for microbial communities, subsequently obtaining clean sequences by applying quality control and splice trimming. In addition, we predicted open reading frames (ORFs) with the getORF program from EMBOSS, followed by enzymatic simulations by trypsin and tryptic rennet. Peptides were de-redundantly processed using CD-HIT, and finally 150,000 non-redundant peptide data were selected for subsequent prediction analysis.
The sequences were predicted using the integrated deep learning model proposed in this paper. The precision, recall, accuracy, specificity, and F1 values for the four models and overall are shown in
Table 2.
In order to fully validate the performance and generalization ability of our model, we also performed a comparative analysis with other models (UMpred-FRL [
14], UmamiYYDS [
12], UmamiBert [
47], and mlp4umami [
48]). The performance of our model uses the average of the four sub-models.
Table 3 and
Figure 2 show that our model demonstrates significant advantages in multiple key performance indicators. Although its recall rate was slightly lower than other models, our model performed excellently in terms of precision and specificity, significantly outperforming other comparison models (especially in specificity, which was on average 17% higher than that of the other models). This indicates that our model has a stronger ability to reduce false positives and improve prediction accuracy. At the same time, in terms of overall accuracy, our model was on par with UMpred-FRL and surpassed the other models. The above data collectively indicate that the performance metrics of this model meet the requirements for umami peptide screening and have practical application value.
3.2. Model Interpretability Analysis
The SHAP analysis results provide local explanations, feature directions, and global explanation information of the model through multidimensional visualization. The force plot provides a local explanation for the prediction of a single peptide segment. Starting from the model’s average prediction value, it shows how each feature collectively pushes the prediction toward the final predicted value for that specific peptide segment through stacked red (positive contribution) and blue (negative contribution) arrows. This allows us to understand why a particular peptide segment is predicted as an umami peptide; the waterfall plot clearly indicates the direction of features. Red bars represent positive contributions of the feature to the prediction, while blue bars represent negative contributions, clearly revealing the promoting or inhibiting effects of specific amino acids at specific positions and their intensities. Furthermore, the amino acid position importance plot visually displays the global impact of amino acid positions on umami prediction. The length of its bars directly reflects the average contribution of each amino acid position to the model’s prediction.
Figure 3,
Figure 4 and
Figure 5 present the interpretability analysis of the CNN model. For local explanations, the model’s predicted value was 0.22. In terms of feature direction, the waterfall chart shows that the cumulative contribution of most features reached +0.32, indicating that the synergistic positive effects of many features have a significant impact on the prediction results. At the global explanation level, amino acids at positions Pos_1, Pos_2, and Pos_0 exhibited the highest average SHAP values, indicating that they are the most important features influencing umami prediction.
Figure 6,
Figure 7 and
Figure 8 present the interpretability analysis of the Transformer model. For local explanations, the model’s predicted value was −0.17. Although this is a negative contribution, the cumulative contribution of most features in the feature direction reached +1.46, indicating that the synergistic positive effect of these features has a significant impact on the prediction results. At the global explanation level, amino acids in the starting region exhibited the highest average SHAP values, indicating that they are the most important features influencing umami prediction.
Figure 9,
Figure 10 and
Figure 11 present the interpretability analysis of the LSTM model. For local interpretations, the model had a predicted value of 0.03. In terms of feature direction, the cumulative contribution reached +1.09, indicating that the synergistic positive effects of many features have a significant impact on the prediction results. At the global explanation level, this model was also similar to the previous two models. In summary, the model can not only identify key amino acid positions but can also provide transparent interpretability that supports its positive effectiveness in umami peptide prediction.
3.3. Umami Peptide Screening Results
Ten high-probability umami peptide candidates were obtained through integrated model screening: DDSMAATGL, DGEEDASM, ESEGESGK, DEEEVDI, ADEETGA, EEDEAK, DSDVAVAVV, DTAVSTVAQ, EEVDEAR, and EEEEKK (see
Table 4). These peptide segments ranked among the top ten in terms of the average prediction probability in the integrated model, and their prediction results were validated by four sub-models (see
Table 5 for detailed probabilities of the four sub-models). Further analysis showed that the molecular weight distribution of the candidate peptides ranged from 691.65 to 890.95 Da. DDSMAATGL, DSDVAVAVV, and DTAVSTVAQ showed a positive GRAVY value, indicating they are hydrophobic proteins, while the other seven peptides all showed negative GRAVY values, indicating they are hydrophilic proteins. Charge characterization revealed that the isoelectric points (pI) of these ten peptides ranged from 3.39 to 4.49, with all being negatively charged and less than 7.
According to the toxicity prediction assessment by ToxinPred, none of the ten candidate peptide segments showed potential toxicity risks, thereby meeting the safety requirements for food and drugs. Based on the integrated model evaluation results and physicochemical property analysis, these ten peptides were finally selected for molecular docking validation experiments.
3.4. Structural Model of the Umami Receptor T1R1/T1R3
The T1R1/T1R3 model, which was computationally generated and refined, is shown in
Figure 12. Ramachandran plot analysis indicated that the model structure is highly reliable; 100% of residues are located in allowed regions (92.7% in the most favored regions, 7.2% in additionally allowed regions, and 0.1% in generously allowed regions), with no residues in disallowed regions. It can be seen that the receptor model we constructed is structurally complete and can be used for ligand docking and subsequent functional validation studies [
49].
3.5. Molecular Docking Between Umami Peptides and Umami Receptors
Figure 13 shows the main active amino acid residues of the umami receptor, including Glu301, Ser384, Lys328, His71, Arg277, Asn69, Glu70, and Ser67. Among them, Glu301 and Arg277 are particularly prominent, as they were present in all tested umami peptides; meanwhile, Lys328 and His71 were found in seven peptides. This distribution pattern directly indicates that Glu, Arg, Lys, and His play a decisive role in umami perception through frequent binding.
Summarizing the above findings, the umami receptor T1R1/T1R3 achieves precise binding with different peptides through multiple molecular interaction modes: hydrogen bonds act as the main driving force, synergistically stabilizing the ligand–receptor complex with C-H bonds, alkyl bonds, and -alkyl bonds. The high-frequency involvement of key residues such as Glu301, Arg277, Lys328, and His71 reveals the molecular recognition mechanism in umami perception, while the dynamic distribution of binding sites reflects the adaptive regulation of binding modes by ligand structural diversity. These findings provide structural clues for elucidating the molecular basis of umami signal transduction and lay a theoretical foundation for the development of targeted receptor design strategies in food flavor engineering.
Ten umami peptides were docked with the receptor, and the results are shown in
Table 6. CDOCKER energy generally refers to the total binding energy between the ligand and the receptor, including interaction energy and ligand internal deformation energy [
50]. CDOCKER interaction energy specifically refers to the non-bonded interaction energy between the two, excluding the ligand’s own energy, more directly reflecting binding specificity and stability [
51].
Binding energy is the energy released when a ligand binds to a receptor, and the lower values indicate the more stable binding [
52]. Stable binding promotes specific interactions between the umami peptide and the umami receptor, thereby triggering the active response and functional expression of the receptor more effectively.
Based on the molecular docking results, we screened out three most promising umami peptides: DDSMAATGL, DGEEDASM, and DEEEVDI. Their molecular docking results are shown in
Figure 14. Considering two types of binding energies comprehensively, these three peptides have the lowest CDOCKER energy values, indicating the most stable overall binding conformations. At the same time, their CDOCKER interaction energies are also notably low among all tested peptides, suggesting strong ligand–receptor interaction strength. Therefore, by weighing these two indicators, we ultimately selected these three peptides as the subjects for subsequent research.
3.6. Peptide Synthesis and Purification Results
In this study, we successfully synthesized three target umami peptides—DDSMAATGL, DGEEDASM, and DEEEVDI—and obtained high-purity products after purification, with their respective purity values shown in
Table 7. These results indicate that the purity of these three peptides has been verified, making them valid for use in subsequent research.
3.7. Molecular Dynamics Simulation Results
To analyze the docking results, we also performed 50-nanosecond molecular dynamics simulations on these three peptides to further illustrate the stability of the umami peptides binding to the receptor.
A root mean square deviation plot was obtained to measure the conformational changes of ligand–receptor complexes during post-docking simulations and to assess the stability and reliability of the binding structure; smoother and less fluctuating curves usually represent a more stable system [
53]. As shown in
Figure 15, the RMSD curve of DDSMAATGL fluctuates less than 0.5 nm after 18 ns, with only a relatively high fluctuation at 15 ns; DGEEDASM fluctuates less than 0.5 nm from 10 ns to 45 ns, with a higher fluctuation only at 47 ns; DEEEVDI remains relatively stable without sudden unstable fluctuations. Therefore, the stability of the three peptides is relatively high.
The root mean square fluctuation plot reveals the flexibility changes in each residue during the simulation process, with regions of smaller fluctuations usually corresponding to more structurally stable parts [
54]. As shown in
Figure 16, the fluctuations of DDSMAATGL were basically within 0.8 nm, with higher fluctuations at 5000 and 10,000; DGEEDASM was basically within 1.0 nm, with fluctuations at 10,000; and DEEEVDI was basically within 0.6, with fluctuations at 10,000, 23,000, and 25,000. This indicates that the three peptides are relatively stable in most regions, with small fluctuations, among which DEEEVDI is the most stable.
The hydrogen bond plot exhibits the stability of the hydrogen bond network. The blue line represents the actual number of hydrogen bonds, which is the main indicator of stability; meanwhile, the red line represents the number of atomic proximities (distance < 0.35 nm), reflecting potential interactions [
55]. If the quantities of the blue and red lines remain stable over time, the system structure is relatively stable. As shown in
Figure 17, the red and blue lines of DDSMAATGL and DGEEDASM remain very stable without sudden fluctuations, with the number of hydrogen bonds staying around 10; DEEEVDI remains stable before 30,000 ps, with the number of hydrogen bonds around 7 and, although there are slight fluctuations between 30,000 and 50,000 ps, it also remains stable. Therefore, it can be concluded that the stability of the three peptide structures mainly depends on the hydrogen bonds formed with the receptor.
Based on the above molecular dynamics simulation analysis results, we successfully validated the stability of the predicted binding modes of the three umami peptides with the receptor. These data strongly support that the binding modes of these three umami peptides with the receptor are stable and reliable, providing important molecular-level insights into their mechanism of action.
3.8. Sensory Evaluation and Electronic Tongue Results of Synthetic Peptides
In order to investigate the differences in the flavor profiles of different umami peptides, the present study was conducted to jointly analyze the screened peptides using sensory evaluation and the electronic tongue technique.
A lower umami threshold value indicates that less of the substance is required to elicit a detectable sensory response, thereby signifying a stronger taste intensity. As shown in
Table 8, all the synthesized peptides possessed both umami and sweet taste. Among them, DDSMAATGL and DEEEVDI exhibited significant fresh–sweet synergistic effects and the best flavor coordination [
56]. This was primarily attributed to their low umami thresholds, indicating potent umami intensity, coupled with the absence of any detectable bitter taste. In contrast, DGEEDASM, although exhibiting the strongest sweetness intensity, was accompanied by a slight bitter taste. As shown in
Table 9, DGEEDASM had the highest bitterness among the three peptides, and its low saltiness was insufficient to mask this bitterness, thus reducing its overall flavor [
57]. The sweetness of all peptides may be due to the fact that the fresh taste receptor shares the T1R3 subunit with the sweet taste receptor, producing a composite taste sensation.
The umami threshold values of the three synthetic peptides from low to high are DDSMAATGL (0.11mg/mL), DGEEDASM (0.37 mg/mL), and DEEEVDI (0.44 mg/mL). Statistical analysis revealed that all three synthetic peptides exhibited highly significant differences in their umami thresholds compared to MSG (0.30 mg/mL) (all p < 0.001 ***). Among them, DDSMAATGL (0.11 mg/mL) had a significantly lower umami threshold than MSG, indicating its superior umami intensity. DGEEDASM (0.37 mg/mL) and DEEEVDI (0.44 mg/mL), while having higher thresholds than MSG, also showed a statistically significant difference in their umami intensities compared to MSG. In particular, DDSMAATGL had the lowest umami threshold, representing the highest umami intensity among the tested compounds.
To study the effects of the three peptides on the enhancement of umami taste by MSG, they were dissolved in a 0.35% MSG solution to prepare 1 mg/mL samples, and their regulatory effects on umami perception were systematically evaluated. Experimental data showed that the umami perception thresholds of the three peptides in aqueous solution were 0.11, 0.37, and 0.44 mg/mL, respectively, while in MSG solution, the thresholds significantly decreased to 0.09, 0.27, and 0.31 mg/mL. This indicates that all the synthesized peptides can effectively lower the umami perception threshold, with umami enhancement rates of 18.1%, 27%, and 22.7%. This proves that umami peptides have potential application value in the development of low-sodium seasonings and precise flavor regulation in food.
To further verify the taste functional properties of these peptides, this study used electronic tongue technology for analysis and measurement, with the results shown in
Table 9 and
Figure 18.
The results demonstrate that the main flavors of the three peptides are umami and sweetness, which is consistent with the previous sensory evaluation results presented in
Table 8. The ranking for umami was DDSMAATGL > DGEEDASM > DEEEVDI. In terms of sweetness, DDSMAATGL and DGEEDASM scored the same, both being higher than DEEEVDI.
Despite the valuable insights gained from this study, certain limitations should be acknowledged. First, the fermented sausage samples used were the result of a certain production process and of a certain geographical origin. Therefore, our results may not be easily translatable into other types of fermented sausages. Second, this research was limited to three selected umami peptides. Though these peptides produced significant findings, the limited number of studied peptides does not allow us to conclude that our results represent the broader spectrum of umami peptides in fermented sausage or in other food matrices.
4. Conclusions
In this study, we utilized metagenomic sequencing data of fermented sausages to explore and construct an integrated model for the prediction of potential umami peptides. The model integrates four sub-models: CNN, LSTM, Attention, and Transformer. Using the constructed model to predict and score the data, we selected the top 10 potential umami peptide sequences with the highest prediction scores. Notably, comprehensive SHAP (SHapley Additive exPlanations) analysis was conducted to elucidate the predictive mechanisms of the integrated model, revealing key amino acid positions and their directional contributions to umami perception.
For further validation, we constructed a Ramachandran plot to assess the structure of the umami receptor T1R1/T1R3, and the results showed that its structure is highly reliable. Then, we performed molecular docking analysis on these top 10 peptides to evaluate their binding ability and stability with the receptor. Based on the molecular docking results, we ultimately selected the three peptides with the lowest binding energy and most stable binding models as candidate umami peptides, namely, DDSMAATGL, DGEEDASM, and DEEEVDI.
The molecular docking results also indicated that amino acid residues Glu301, Arg277, Lys328, and His71 on the receptor play a key role in binding umami peptides through hydrogen bond formation. Further molecular dynamics simulations revealed that the complexes formed by these three peptides with the receptor have good structural stability and efficacy. Subsequently, we conducted sensory evaluations of these three synthetic peptides, all of which exhibited significant umami characteristics.
In summary, in this study, we successfully developed and validated an efficient umami peptide screening strategy based on metagenomic data and an integrated deep learning model. Compared to traditional methods, this approach presents significant advantages in terms of speed, cost-effectiveness, process simplification, and labor savings. This innovative research not only provides powerful computational tools and valuable technical references for the high-throughput discovery of functional peptides (such as umami peptides) but also greatly deepens the understanding of the molecular basis of umami generation through an in-depth analysis of molecular interaction mechanisms. The results of this study lay a solid foundation for the efficient mining of potential functional peptides from complex biological resources in the future, as well as promoting the application of umami peptides in food science and other fields, which has important theoretical significance and broad application prospects.


 ,
, 


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}