
Insights into Chemopreventive Effects of Rosmarinic Acid Against Aflatoxin B1-Induced Genotoxic Effects

 , , , ,
, , , ,  and
and 
Abstract

1. Introduction
2. Materials and Methods
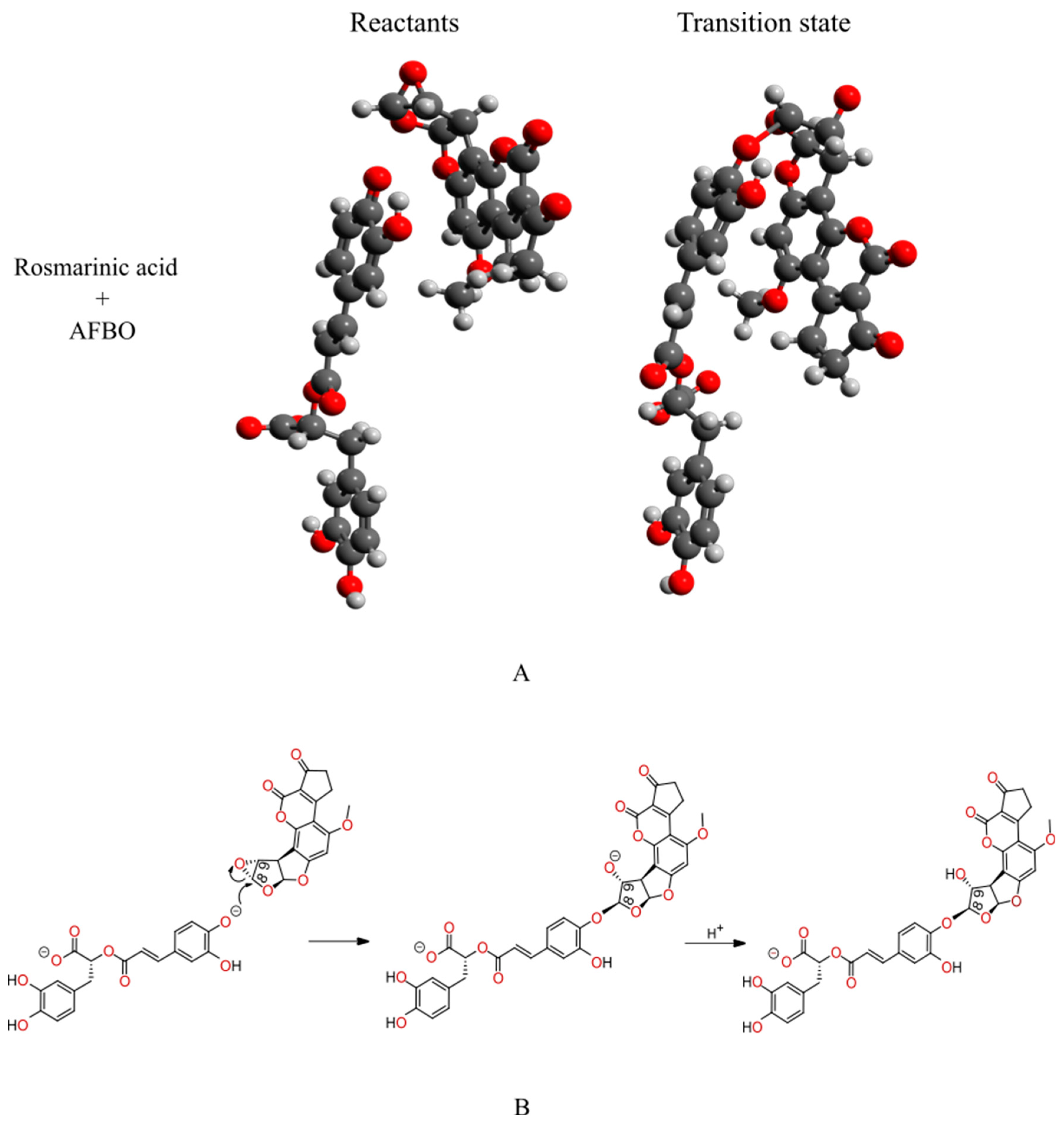
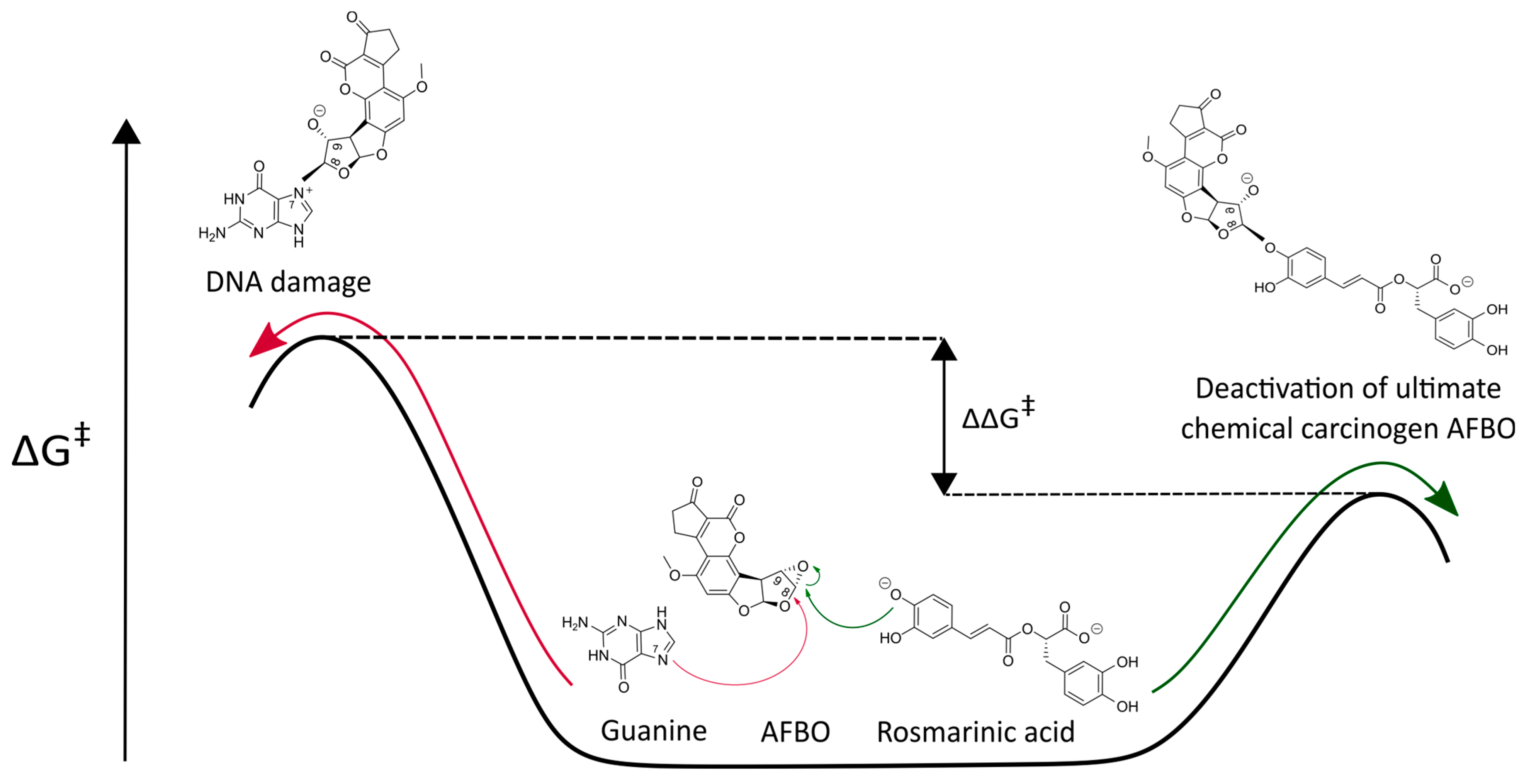
2.1. In Silico Investigation of Alkylation Reactions Between Rosmarinic Acid and AFBO—The Carcinogenic Metabolite of AFB1
2.2. Chemicals
2.3. Human Hepatoma HepG2 Cells
2.4. Determination of Cell Viability Using an MTT Assay
2.5. The Comet Assay
2.6. Flow Cytometric Analyses of Gamma-H2AX Formation, Histone p-H3-Positive Cells, Cell Proliferation, and Cell Cycle
3. Results and Discussion
3.1. Mechanistic Insights into the Alkylation Reaction Between Rosmarinic Acid and AFBO—The Carcinogenic Metabolite of AFB1
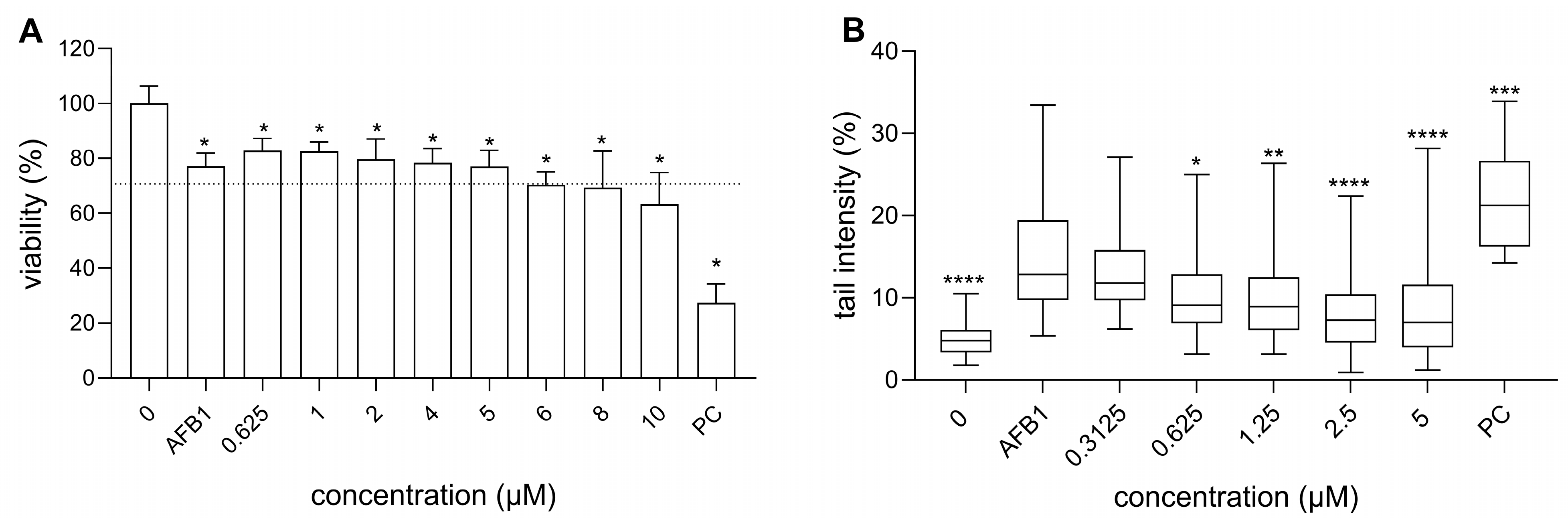
3.2. Cytotoxicity of RA, AFB1, and Their Binary Mixtures
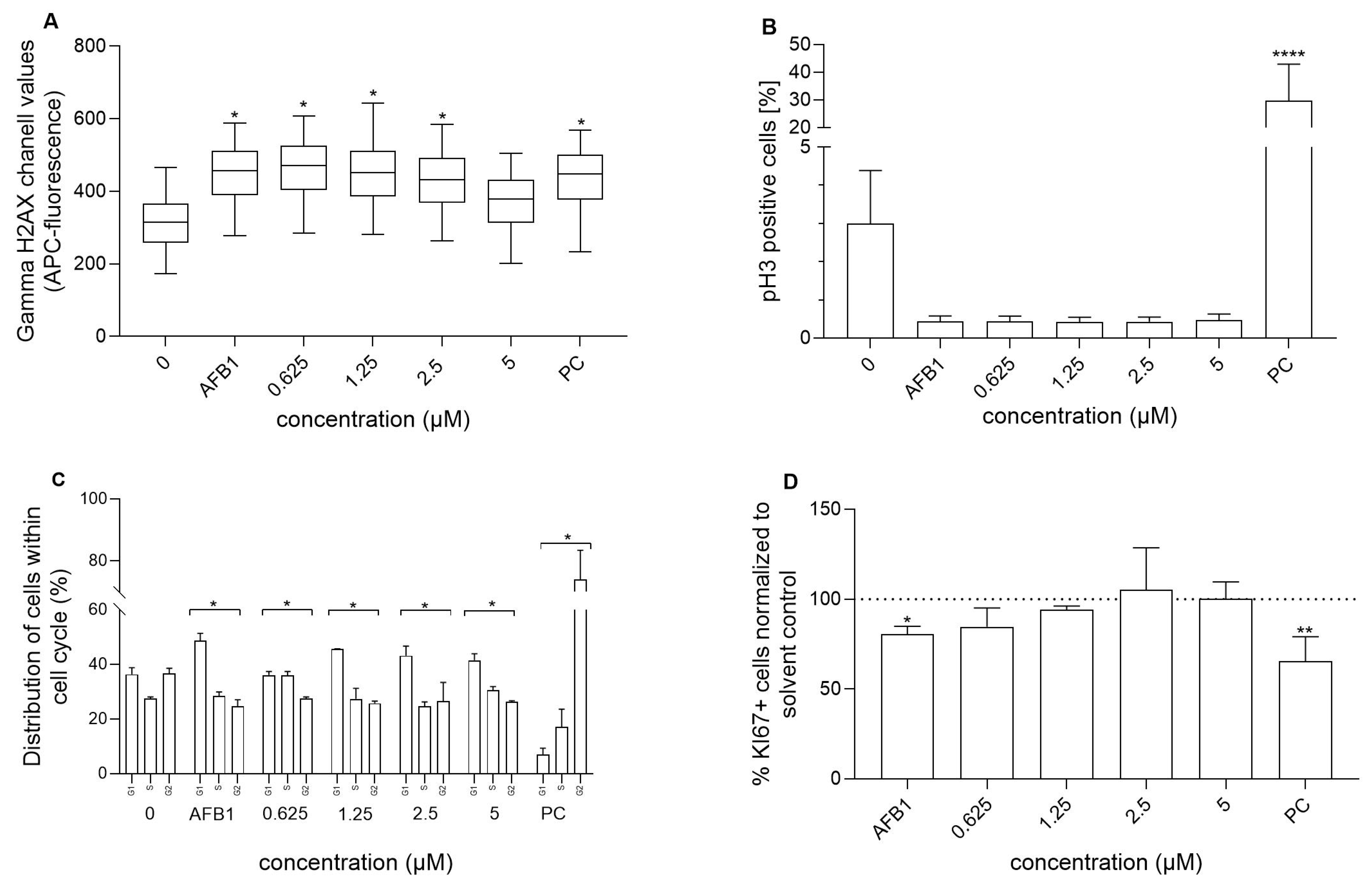
3.3. Genotoxicity of RA and AFB1, as Well as RA’s Chemoprotective Effects Against AFB1-Induced Genotoxicity
3.4. Influence of RA and Its Combination with AFB1 on Cell Cycle and Cell Proliferation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lešnik, S.; Furlan, V.; Bren, U. Rosemary (Rosmarinus officinalis L.): Extraction techniques, analytical methods and health-promoting biological effects. Phytochem. Rev. 2021, 20, 1273–1328. [Google Scholar] [CrossRef]
- Begum, A.; Sandhya, S.; Vinod, K.R.; Reddy, S.; Banji, D. An in-depth review on the medicinal flora Rosmarinus officinalis (Lamiaceae). Acta Sci. Pol. Technol. 2013, 12, 61–74. [Google Scholar]
- Pérez-Fons, L.; Garzón, M.a.T.; Micol, V. Relationship between the antioxidant capacity and effect of rosemary (Rosmarinus officinalis L.) polyphenols on membrane phospholipid order. J. Agric. Food Chem. 2010, 58, 161–171. [Google Scholar] [CrossRef]
- Žegura, B.; Dobnik, D.; Niderl, M.H.; Filipič, M. Antioxidant and antigenotoxic effects of rosemary (Rosmarinus officinalis L.) extracts in Salmonella typhimurium TA98 and HepG2 cells. Environ. Toxicol. Pharmacol. 2011, 32, 296–305. [Google Scholar] [CrossRef]
- Yu, M.-H.; Choi, J.-H.; Chae, I.-G.; Im, H.-G.; Yang, S.-A.; More, K.; Lee, I.-S.; Lee, J. Suppression of LPS-induced inflammatory activities by Rosmarinus officinalis L. Food Chem. 2013, 136, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Bakırel, T.; Bakırel, U.; Keleş, O.Ü.; Ülgen, S.G.; Yardibi, H. In vivo assessment of antidiabetic and antioxidant activities of rosemary (Rosmarinus officinalis) in alloxan-diabetic rabbits. J. Ethnopharmacol. 2008, 116, 64–73. [Google Scholar] [CrossRef]
- Bozin, B.; Mimica-Dukic, N.; Samojlik, I.; Jovin, E. Antimicrobial and antioxidant properties of rosemary and sage (Rosmarinus officinalis L. and Salvia officinalis L., Lamiaceae) essential oils. J. Agric. Food Chem. 2007, 55, 7879–7885. [Google Scholar] [CrossRef]
- Yesil-Celiktas, O.; Sevimli, C.; Bedir, E.; Vardar-Sukan, F. Inhibitory effects of rosemary extracts, carnosic acid and rosmarinic acid on the growth of various human cancer cell lines. Plant Foods Hum. Nutr. 2010, 65, 158–163. [Google Scholar] [CrossRef]
- Valdés, A.; García-Cañas, V.; Rocamora-Reverte, L.; Gómez-Martínez, Á.; Ferragut, J.A.; Cifuentes, A. Effect of rosemary polyphenols on human colon cancer cells: Transcriptomic profiling and functional enrichment analysis. Genes Nutr. 2013, 8, 43–60. [Google Scholar] [CrossRef]
- Huang, M.-T.; Ho, C.-T.; Wang, Z.Y.; Ferraro, T.; Lou, Y.-R.; Stauber, K.; Ma, W.; Georgiadis, C.; Laskin, J.D.; Conney, A.H. Inhibition of skin tumorigenesis by rosemary and its constituents carnosol and ursolic acid. Cancer Res. 1994, 54, 701–708. [Google Scholar]
- Wang, H.; Provan, G.J.; Helliwell, K. Determination of rosmarinic acid and caffeic acid in aromatic herbs by HPLC. Food Chem. 2004, 87, 307–311. [Google Scholar] [CrossRef]
- Adomako-Bonsu, A.G.; Chan, S.L.; Pratten, M.; Fry, J.R. Antioxidant activity of rosmarinic acid and its principal metabolites in chemical and cellular systems: Importance of physico-chemical characteristics. Toxicol. In Vitro 2017, 40, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yu, C.; Lu, Y.; Gu, Y.; Lu, J.; Xu, W.; Xuan, L.; Wang, Y. Pharmacokinetics, tissue distribution, metabolism, and excretion of depside salts from Salvia miltiorrhiza in rats. Drug Metab. Dispos. 2007, 35, 234–239. [Google Scholar] [CrossRef]
- Bhise, K.; Kashaw, S.K.; Sau, S.; Iyer, A.K. Nanostructured lipid carriers employing polyphenols as promising anticancer agents: Quality by design (QbD) approach. Int. J. Pharm. 2017, 526, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Marchev, A.S.; Vasileva, L.V.; Amirova, K.M.; Savova, M.S.; Koycheva, I.K.; Balcheva-Sivenova, Z.P.; Vasileva, S.M.; Georgiev, M.I. Rosmarinic acid-From bench to valuable applications in food industry. Trends Food Sci. Technol. 2021, 117, 182–193. [Google Scholar] [CrossRef]
- Taguchi, R.; Hatayama, K.; Takahashi, T.; Hayashi, T.; Sato, Y.; Sato, D.; Ohta, K.; Nakano, H.; Seki, C.; Endo, Y. Structure–activity relations of rosmarinic acid derivatives for the amyloid β aggregation inhibition and antioxidant properties. Eur. J. Med. Chem. 2017, 138, 1066–1075. [Google Scholar] [CrossRef]
- Yao, Y.; Mao, J.; Xu, S.; Zhao, L.; Long, L.; Chen, L.; Li, D.; Lu, S. Rosmarinic acid inhibits nicotine-induced C-reactive protein generation by inhibiting NLRP3 inflammasome activation in smooth muscle cells. J. Cell. Physiol. 2019, 234, 1758–1767. [Google Scholar] [CrossRef]
- Amaral, G.P.; Mizdal, C.R.; Stefanello, S.T.; Mendez, A.S.L.; Puntel, R.L.; de Campos, M.M.A.; Soares, F.A.A.; Fachinetto, R. Antibacterial and antioxidant effects of Rosmarinus officinalis L. extract and its fractions. J. Tradit. Med. Complement. 2019, 9, 383–392. [Google Scholar] [CrossRef]
- Radziejewska, I.; Supruniuk, K.; Nazaruk, J.; Karna, E.; Popławska, B.; Bielawska, A.; Galicka, A. Rosmarinic acid influences collagen, MMPs, TIMPs, glycosylation and MUC1 in CRL-1739 gastric cancer cell line. Biomed. Pharmacother. 2018, 107, 397–407. [Google Scholar] [CrossRef]
- Ma, Z.-J.; Yan, H.; Wang, Y.-J.; Yang, Y.; Li, X.-B.; Shi, A.-C.; Jing-Wen, X.; Yu-Bao, L.; Li, L.; Wang, X.-X. Proteomics analysis demonstrating rosmarinic acid suppresses cell growth by blocking the glycolytic pathway in human HepG2 cells. Biomed. Pharmacother. 2018, 105, 334–349. [Google Scholar] [CrossRef]
- Khalaf, A.A.; Hassanen, E.I.; Ibrahim, M.A.; Tohamy, A.F.; Aboseada, M.A.; Hassan, H.M.; Zaki, A.R. Rosmarinic acid attenuates chromium-induced hepatic and renal oxidative damage and DNA damage in rats. J. Biochem. Mol. Toxicol. 2020, 34, e22579. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.-J.; Lim, D.-S.; Kim, S.O.; Park, C.; Leem, S.-H.; Lee, H.; Kim, G.-Y.; Jeong, S.-J.; Choi, Y.H. Protection of oxidative stress-induced DNA damage and apoptosis by rosmarinic acid in murine myoblast C2C12 cells. Biotechnol. Bioprocess Eng. 2022, 27, 171–182. [Google Scholar] [CrossRef]
- Furtado, R.A.; De Araujo, F.R.R.; Resende, F.A.; Cunha, W.R.; Tavares, D.C. Protective effect of rosmarinic acid on V79 cells evaluated by the micronucleus and comet assays. J. Appl. Toxicol. 2010, 30, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Furtado, M.A.; de Almeida, L.C.F.; Furtado, R.A.; Cunha, W.R.; Tavares, D.C. Antimutagenicity of rosmarinic acid in Swiss mice evaluated by the micronucleus assay. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2008, 657, 150–154. [Google Scholar] [CrossRef]
- Wogan, G.N.; Hecht, S.S.; Felton, J.S.; Conney, A.H.; Loeb, L.A. Environmental and chemical carcinogenesis. Semin. Cancer Biol. 2004, 14, 473–486. [Google Scholar] [CrossRef]
- World Health Organization. Painting, Firefighting, and Shiftwork. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; World Health Organization: Geneva, Switzerland, 2012. [Google Scholar]
- Dai, Y.; Huang, K.; Zhang, B.; Zhu, L.; Xu, W. Aflatoxin B1-induced epigenetic alterations: An overview. Food Chem. Toxicol. 2017, 109, 683–689. [Google Scholar] [CrossRef]
- World Health Organization; Agency for Research on Cancer. Some naturally occurring substances: Food items and constituents, heterocyclic aromatic amines and mycotoxins. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 1993; Volume 56. [Google Scholar]
- Rushing, B.R.; Selim, M.I. Aflatoxin B1: A review on metabolism, toxicity, occurrence in food, occupational exposure, and detoxification methods. Food Chem. Toxicol. 2019, 124, 81–100. [Google Scholar] [CrossRef]
- Wang, J.-S.; Huang, T.; Su, J.; Liang, F.; Wei, Z.; Liang, Y.; Luo, H.; Kuang, S.-Y.; Qian, G.-S.; Sun, G. Hepatocellular carcinoma and aflatoxin exposure in Zhuqing village, Fusui county, People’s Republic of China. Cancer Epidemiol. Biomarkers Prev. 2001, 10, 143–146. [Google Scholar]
- Kew, M.C. Aflatoxins as a cause of hepatocellular carcinoma. J. Gastrointestin. Liver Dis. 2013, 22, 305–310. [Google Scholar]
- Kibugu, J.; Munga, L.; Mburu, D.; Maloba, F.; Auma, J.E.; Grace, D.; Lindahl, J.F. Dietary mycotoxins: An overview on toxicokinetics, toxicodynamics, toxicity, epidemiology, detection, and their mitigation with special emphasis on aflatoxicosis in humans and animals. Toxins 2024, 16, 483. [Google Scholar] [CrossRef]
- Furlan, V.; Tošović, J.; Bren, U. QM-CSA: A Novel Quantum Mechanics-Based Protocol for Evaluation of the Carcinogen-Scavenging Activity of Polyphenolic Compounds. Foods 2024, 13, 2708. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Bren, U.; Guengerich, F.P.; Mavri, J. Guanine alkylation by the potent carcinogen aflatoxin B1: Quantum chemical calculations. Chem. Res. Toxicol. 2007, 20, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Lajovic, A.; Nagy, L.D.; Guengerich, F.P.; Bren, U. Carcinogenesis of urethane: Simulation versus experiment. Chem. Res. Toxicol. 2015, 28, 691. [Google Scholar] [CrossRef]
- Furlan, V.; Bren, U. Protective Effects of [6]-Gingerol Against Chemical Carcinogens: Mechanistic Insights. Int. J. Mol. Sci. 2020, 21, 695. [Google Scholar] [CrossRef]
- Štern, A.; Furlan, V.; Novak, M.; Štampar, M.; Kolenc, Z.; Kores, K.; Filipič, M.; Bren, U.; Žegura, B. Chemoprotective Effects of Xanthohumol against the Carcinogenic Mycotoxin Aflatoxin B1. Foods 2021, 10, 1331. [Google Scholar] [CrossRef]
- MarvinSketch, Version 18.5.0; ChemAxon: Budapest, Hungary, 2019. Available online: https://www.chemaxon.com/products/marvin/marvinsketch/ (accessed on 15 May 2025).
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Novak, M.; Žegura, B.; Baebler, Š.; Štern, A.; Rotter, A.; Stare, K.; Filipič, M. Influence of selected anti-cancer drugs on the induction of DNA double-strand breaks and changes in gene expression in human hepatoma HepG2 cells. Environ. Sci. Pollut. Res. 2016, 23, 14751–14761. [Google Scholar] [CrossRef]
- Collins, A.; Møller, P.; Gajski, G.; Vodenková, S.; Abdulwahed, A.; Anderson, D.; Bankoglu, E.E.; Bonassi, S.; Boutet-Robinet, E.; Brunborg, G. Measuring DNA modifications with the comet assay: A compendium of protocols. Nat. Protoc. 2023, 18, 929–989. [Google Scholar] [CrossRef]
- Novak, M.; Žegura, B.; Nunić, J.; Gajski, G.; Gerić, M.; Garaj-Vrhovac, V.; Filipič, M. Assessment of the genotoxicity of the tyrosine kinase inhibitor imatinib mesylate in cultured fish and human cells. Mutat. Res.-Genet. Toxicol. Environ. Mutag. 2017, 814, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Štampar, M.; Breznik, B.; Filipič, M.; Žegura, B. Characterization of in vitro 3D cell model developed from human hepatocellular carcinoma (HepG2) cell line. Cells 2020, 9, 2557. [Google Scholar] [CrossRef] [PubMed]
- Štampar, M.; Žabkar, S.; Filipič, M.; Žegura, B. HepG2 spheroids as a biosensor-like cell-based system for (geno) toxicity assessment. Chemosphere 2022, 291, 132805. [Google Scholar] [CrossRef]
- Azqueta, A.; Stopper, H.; Zegura, B.; Dusinska, M.; Møller, P. Do cytotoxicity and cell death cause false positive results in the in vitro comet assay? Mutat. Res.-Genet. Toxicol. Environ. Mutag. 2022, 881, 503520. [Google Scholar] [CrossRef]
- Jin, B.; Liu, J.; Gao, D.; Xu, Y.; He, L.; Zang, Y.; Li, N.; Lin, D. Detailed studies on the anticancer action of rosmarinic acid in human Hep-G2 liver carcinoma cells: Evaluating its effects on cellular apoptosis, caspase activation and suppression of cell migration and invasion. J. BUON 2020, 25, 1383–1389. [Google Scholar]
- Ozgun, G.; Ozgun, E. The cytotoxic concentration of rosmarinic acid increases MG132-induced cytotoxicity, proteasome inhibition, autophagy, cellular stresses, and apoptosis in HepG2 cells. Hum. Exp. Toxicol. 2020, 39, 514–523. [Google Scholar] [CrossRef]
- Renzulli, C.; Galvano, F.; Pierdomenico, L.; Speroni, E.; Guerra, M.C. Effects of rosmarinic acid against aflatoxin B1 and ochratoxin-A-induced cell damage in a human hepatoma cell line (Hep G2). J. Appl. Toxicol. 2004, 24, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhu, Y.; Li, F.; Zhang, G.; Shi, J.; Ou, R.; Tong, Y.; Liu, Y.; Liu, L.; Lu, L. Spica prunellae and its marker compound rosmarinic acid induced the expression of efflux transporters through activation of Nrf2-mediated signaling pathway in HepG2 cells. J. Ethnopharmacol. 2016, 193, 1–11. [Google Scholar] [CrossRef]
- Helvacioglu, S.; Hamitoğlu, M.; Yildirim, E.; Korkut, Ş.V.; Aylin, Y.; Aydin, A. Protective Effects of Rosmarinic Acid and Epigallocatechin Gallate Against Doxorubicin-Induced Cytotoxicity and Genotoxicity in CHO-K1 Cells. Turk. J. Pharm. Sci. 2025, 21, 536. [Google Scholar]
- Psotova, J.; Svobodova, A.; Kolarova, H.; Walterova, D. Photoprotective properties of Prunella vulgaris and rosmarinic acid on human keratinocytes. J. Photochem. Photobiol. B Biol. 2006, 84, 167–174. [Google Scholar] [CrossRef]
- Lima, C.F.; Fernandes-Ferreira, M.; Pereira-Wilson, C. Phenolic compounds protect HepG2 cells from oxidative damage: Relevance of glutathione levels. Life Sci. 2006, 79, 2056–2068. [Google Scholar] [CrossRef]
- Vostálová, J.; Zdařilová, A.; Svobodová, A. Prunella vulgaris extract and rosmarinic acid prevent UVB-induced DNA damage and oxidative stress in HaCaT keratinocytes. Arch. Dermatol. Res. 2010, 302, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-R.; Li, Y.-y.; Wang, J.-Y.; Wang, H.-W.; Wang, H.-N.; Kang, X.-M.; Xu, W.-Q. Synthesis and characterization of a rosmarinic acid derivative that targets mitochondria and protects against radiation-induced damage in vitro. Radiat. Res. 2017, 188, 264–275. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, N.C.; Sarmento, M.S.; Nunes, E.A.; Porto, C.M.; Rosa, D.P.; Bona, S.R.; Rodrigues, G.; Marroni, N.P.; Pereira, P.; Picada, J.N. Rosmarinic acid as a protective agent against genotoxicity of ethanol in mice. Food Chem. Toxicol. 2012, 50, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Pereira, P.; Tysca, D.; Oliveira, P.; da Silva Brum, L.F.; Picada, J.N.; Ardenghi, P. Neurobehavioral and genotoxic aspects of rosmarinic acid. Pharmacol. Res. 2005, 52, 199–203. [Google Scholar] [CrossRef]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef]
- Rahmanian, N.; Shokrzadeh, M.; Eskandani, M. Recent advances in γH2AX biomarker-based genotoxicity assays: A marker of DNA damage and repair. DNA Repair 2021, 108, 103243. [Google Scholar] [CrossRef]
- Valdiglesias, V.; Giunta, S.; Fenech, M.; Neri, M.; Bonassi, S. γH2AX as a marker of DNA double strand breaks and genomic instability in human population studies. Mutat. Res. Rev. Mut. Res. 2013, 753, 24–40. [Google Scholar] [CrossRef]
- Watters, G.P.; Smart, D.J.; Harvey, J.S.; Austin, C.A. H2AX phosphorylation as a genotoxicity endpoint. Mutat. Res.-Genet. Toxicol. Environ. Mutag. 2009, 679, 50–58. [Google Scholar] [CrossRef]
- Braidy, N.; Matin, A.; Rossi, F.; Chinain, M.; Laurent, D.; Guillemin, G. Neuroprotective effects of rosmarinic acid on ciguatoxin in primary human neurons. Neurotox. Res. 2014, 25, 226–234. [Google Scholar] [CrossRef]
- Prigent, C.; Dimitrov, S. Phosphorylation of serine 10 in histone H3, what for? J. Cell Sci. 2003, 116, 3677–3685. [Google Scholar] [CrossRef] [PubMed]
- Khoury, L.; Zalko, D.; Audebert, M. Complementarity of phosphorylated histones H2AX and H3 quantification in different cell lines for genotoxicity screening. Arch. Toxicol. 2016, 90, 1983–1995. [Google Scholar] [CrossRef]
- Sharma, A.K.; Bhattacharya, S.; Khan, S.A.; Khade, B.; Gupta, S. Dynamic alteration in H3 serine 10 phosphorylation is G1-phase specific during ionization radiation induced DNA damage response in human cells. Mutat. Res.-Fundam. Mol. Mech. Mutag. 2015, 773, 83–91. [Google Scholar] [CrossRef]
- Liu, Y.; Du, M.; Zhang, G. Proapoptotic activity of aflatoxin B1 and sterigmatocystin in HepG2 cells. Toxicol. Rep. 2014, 1, 1076–1086. [Google Scholar] [CrossRef]
- Du, M.; Liu, Y.; Zhang, G. Interaction of aflatoxin B1 and fumonisin B1 in HepG2 cell apoptosis. Food Biosci. 2017, 20, 131–140. [Google Scholar] [CrossRef]
- Bahri, S.; Mies, F.; Ben Ali, R.; Mlika, M.; Jameleddine, S.; Mc Entee, K.; Shlyonsky, V. Rosmarinic acid potentiates carnosic acid induced apoptosis in lung fibroblasts. PLoS ONE 2017, 12, e0184368. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Shen, Y.; Zhu, L.; Yao, L.; Wang, X.; Zhang, A.; Li, J.; Wu, J.; Qin, L. Rosmarinic acid, the active component of Rubi Fructus, induces apoptosis of SGC-7901 and HepG2 cells through mitochondrial pathway and exerts anti-tumor effect. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2023, 396, 3743–3755. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-F.; Hong, C.; Klauck, S.M.; Lin, Y.-L.; Efferth, T. Molecular mechanisms of rosmarinic acid from Salvia miltiorrhiza in acute lymphoblastic leukemia cells. J. Ethnopharmacol. 2015, 176, 55–68. [Google Scholar] [CrossRef]
- Sun, X.; Kaufman, P.D. Ki-67: More than a proliferation marker. Chromosoma 2018, 127, 175–186. [Google Scholar] [CrossRef]
- Wang, L.; Yang, H.; Wang, C.; Shi, X.; Li, K. Rosmarinic acid inhibits proliferation and invasion of hepatocellular carcinoma cells SMMC 7721 via PI3K/AKT/mTOR signal pathway. Biomed. Pharmacother. 2019, 120, 109443. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, W.; Li, Z.; Chen, L.; Wen, C.; Ruan, Q.; Xu, Z.; Liu, R.; Xu, J.; Bai, Y. Rosmarinic acid decreases the malignancy of pancreatic cancer through inhibiting Gli1 signaling. Phytomedicine 2022, 95, 153861. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Yang, J.; Yang, Y.; Wang, X.; Chen, G.; Shi, A.; Lu, Y.; Jia, S.; Kang, X.; Lu, L. Rosmarinic acid exerts an anticancer effect on osteosarcoma cells by inhibiting DJ-1 via regulation of the PTEN-PI3K-Akt signaling pathway. Phytomedicine 2020, 68, 153186. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Ma, L.; Zhao, L.; Feng, W.; Zheng, X. Rosmarinic inhibits cell proliferation, invasion and migration via up-regulating miR-506 and suppressing MMP2/16 expression in pancreatic cancer. Biomed. Pharmacother. 2019, 115, 108878. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scavenger of AFBO | Activation Free Energies | ||
|---|---|---|---|
| a [kcal/mol] | b [kcal/mol] | [kcal/mol] | |
| RA | 15.13 | 14.86 | 15.1 [33] |
| Guanine | 16.99 | 16.88 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furlan, V.; Novak, M.; Štampar, M.; Štern, A.; Žegura, B.; Bren, U. Insights into Chemopreventive Effects of Rosmarinic Acid Against Aflatoxin B1-Induced Genotoxic Effects. Foods 2025, 14, 2111. https://doi.org/10.3390/foods14122111
Furlan V, Novak M, Štampar M, Štern A, Žegura B, Bren U. Insights into Chemopreventive Effects of Rosmarinic Acid Against Aflatoxin B1-Induced Genotoxic Effects. Foods. 2025; 14(12):2111. https://doi.org/10.3390/foods14122111
Chicago/Turabian StyleFurlan, Veronika, Matjaž Novak, Martina Štampar, Alja Štern, Bojana Žegura, and Urban Bren. 2025. "Insights into Chemopreventive Effects of Rosmarinic Acid Against Aflatoxin B1-Induced Genotoxic Effects" Foods 14, no. 12: 2111. https://doi.org/10.3390/foods14122111
APA StyleFurlan, V., Novak, M., Štampar, M., Štern, A., Žegura, B., & Bren, U. (2025). Insights into Chemopreventive Effects of Rosmarinic Acid Against Aflatoxin B1-Induced Genotoxic Effects. Foods, 14(12), 2111. https://doi.org/10.3390/foods14122111