1. Introduction
With the development of society and the growth of the population, the demand for meat consumption increases continuously. However, traditional meat production industries, such as livestock, poultry, and aquaculture, have reached their limits and brought about a series of unsustainable social problems, such as the decline in wild animal resources, the loss of biodiversity, environmental pollution, and ethical and health issues [
1,
2,
3]. Alternative protein or “artificial meat” has been considered one of the promising ways to solve these unsustainable issues. Since the first cultured beef was successfully grown in a lab [
4], the emergence of cellular agriculture, which is represented by cell-cultured meat, has provided a more advantageous solution for alternative protein production in recent years [
5,
6,
7]. Compared with other alternative protein production like plant-based proteins, cell-cultured meat is more likely acceptable to consumers duo to its utilization of cell and tissue engineering methods to create products that closely resemble natural meat in appearance, structure, nutrition, flavor, and taste [
8,
9].
As the largest vertebrate group, fish is an excellent model for exploring various biological processes [
10] and pathophysiological mechanisms [
11]. In addition, as a valuable source of high-quality protein, fish has great potential to meet the growing human demand for protein. As of now, nearly 1000 fish cell lines (URL:
https://www.cellosaurus.org/, accessed on 9 June 2025) have been successfully established, and these cell lines are widely used in the research fields of cytology, genetics, immunology, ecological toxicology, etc. [
12,
13,
14,
15]. However, compared with mammals, fish cell culture technology is still relatively lagging behind, and there is little need for translational applications. Recently, the pipeline of using muscle stem cells to produce cultured fish fillets was reported for the first time in large yellow croaker [
16], which sets a successful precedent for cell-cultured fish meat, but also promotes the emergence of a new field for fish cell culture translation research, i.e., “Cellular Aquaculture” [
17].
The production of cell-cultured meat still faces many challenges, such as the source and expansion of seed cell lines, low-cost serum-free culture systems, large-scale expansion, and efficient directional differentiation [
18,
19]. Among them, the selection of seed cells plays a crucial role, and its source directly determines the formulation of subsequent operation strategies. The use of MuSCs and AD-MSCs as seed cells is favored in cultured meat biotechnology, owing to their capacity for directed differentiation into myogenic and adipogenic lineages
in vitro. A representative application is the production of cell-cultured large yellow croaker meat, which integrates both MuSCs and AD-MSCs [
16]. However, fish have very rich diversity, and the survival and expansion of cells from different fish
in vitro requires the species-specific optimization of conditions [
20]. According to our experience, it is difficult for many fish MuSCs and AD-MSCs to be subcultured
in vitro and maintain stable proliferation and differentiation capabilities. For example, rainbow trout (
Oncorhynchus mykiss) [
21] and seabream (
Sparus aurata) [
22] muscle satellite cells are prone to spontaneous differentiation when cultured
in vitro, making it difficult to establish stable cell lines. We have also attempted to establish MuSC lines from certain other fish species, but all efforts have failed due to premature aging, rendering them unusable for production. Pluripotent stem cells (PSCs), such as ESCs and MSCs, are also considered an ideal cell source for cell-cultured meat production due to their infinite proliferation and excellent differentiation capabilities [
23]. However, the current reliance of PSCs on highly specialized culture media and intricate multi-stage differentiation processes poses significant challenges for scaling up cultivated meat production [
24].
Fish fin tissues have strong regenerative ability, and fin fibroblasts from fin show excellent proliferation ability
in vitro. A study has also shown that they can be induced to differentiate into different types of cells, such as muscle cells, adipocytes, and nerve cells [
25]. Therefore, fin fibroblasts also have the potential to serve as source cells for cell-cultured fish meat. Topmouth culter is an important economic fish which is widely distributed in the major water bodies of the middle and lower reaches of the Yangtze River in China. It is favored by consumers due to its large size, fast growth, tender and delicious meat, and rich nutritional value. Its artificial breeding technology is mature and has been widely promoted, forming an aquaculture industry worth billions of Yuan [
26]. However, the rich intermuscular spines of
Cyprinidae seriously affect their edible quality and economic value. Although intermuscular spines can be removed by means of gene editing technology, it may take around ten more years to establish intermuscular spine-free strains [
27]. Therefore, the production of meat through cell culture may be a more ideal way to obtain spineless and delicious fish meat.
Here, for the first time, we report the establishment and characterization of a Topmouth culter caudal fin cell line, TCCF2022, and further optimize a recipe by adding a small-molecule compound to maintain the characteristics of the cell line, including its pluripotency. Subsequently, we optimize the protocols for differentiating caudal fin cells into myotubes and adipocytes, featuring more defined components and clearer mechanisms compared to the myogenic and adipogenic induction approach reported previously [
25]. In general, we establish a reliable foundation for the future application of the TCCF2022 cell line in cell-cultured fish meat production.
2. Materials and Methods
2.1. Primary Culture
The primary caudal fin culture protocol was adapted from filefish [
25], with modifications to optimize. Topmouth culter fish juveniles, with a length of approximately 10 cm, were purchased from the local market in Hangzhou City, Zhejiang Province. The caudal fin tissue was collected and underwent a strict disinfection process. First, the caudal fin tissue was soaked in a 1:10,000 concentration of potassium permanganate solution for 30 min and was then washed 5 times with D-Hanks buffer (GENOM, GNM14175-5, Hangzhou, China) containing 2 × PS (Macklin, P917928, Shanghai, China) to remove excess potassium permanganate. Next, the fin tissue was disinfected by soaking in 75% alcohol for 15 s and rinsed 5 times with D-Hanks buffer to remove mucus and epithelium. Finally, the tissue was sliced into 1 mm blocks and attached to a six-well plate coated with L-type polylysine solution (Sangon, E607015, Shanghai, China) by the adhesion method. To promote the attachment growth of the tissue, the culture dish was first inverted and dried for 30 min, and then the sample blocks were digested for 10 min with 0.5 mL 0.25% trypsin–EDTA digestion solution (Cienry, CR25200, Huzhou, China). After trypsin treatment, 0.5 mL DMEM/F12 (Gibco, C11330500BT, Suzhou, China) containing 1 × PS and 10% fetal bovine serum (Gibco, 10099141C, Suzhou, China) was used to terminate digestion. Finally, 2 mL of DMEM/F12 (Gibco, C11330500BT, Suzhou, China) culture medium containing 20% fetal bovine serum, 5 ng/mL bFGF (Beyotime, P5453, purity > 96%, Shanghai, China), and 1 × PS were added to each well, and the plate was placed in a 5% CO
2 incubator at 27 °C for culture.
2.2. Subculture, Cryopreservation, and Recovery
When the primary cells had reached 90% confluency, the culture medium was carefully removed, and the cells were then rinsed twice with 1 mL of D-Hanks buffer. Following this, the cells were subjected to trypsin digestion in 0.5 mL 0.25% trypsin–EDTA solution. Once the digestion was complete, 0.5 mL of complete medium was added to promptly halt the digestion. The cells were then gently aspirated with a pipette and transferred to a 1.5 mL centrifuge tube, which then was subjected to centrifugation at 300× g for 3 min to remove the supernatant. The cells were resuspended in complete medium (DMEM/F12 medium containing 10% fetal bovine serum, 10 ng/mL bFGF, and 1 × PS) at a ratio of one to three for subculture. If the cells needed to be frozen, 1 mL of DMEM/F12 medium containing 20% FBS and 5% DMSO (GENOM, GNM10944-1, Hangzhou, China) was utilized for resuspension, and the cells were placed in a refrigerator at −80 °C for controlled cooling. Finally, the cells were carefully transferred to liquid nitrogen for long-term preservation.
For cell recovery, cryopreserved TCCF2022 cells in cryovials were thawed in a water bath at 37 °C and suspended in 5 mL of fresh DMEM/F12. After centrifugation at 300× g for 3 min, the cells were resuspended in 2 mL of DMEM/F12 and seeded into 6-well plates for culture at 27 °C.
2.3. Culture of Forskolin-Treated Cells
For Forskolin-treated cells, we used DMEM/F12 culture medium containing 10% fetal bovine serum, 10 ng/mL bFGF, 0.5μM Forskolin (Targetmol, T2939, purity 99.86%, Shanghai, China), and 1 × PS as conditioned medium, and the plate was placed in a 5% CO
2 incubator at 27 °C for culture. And the method of subculture was the same as in
Section 2.2.
2.4. Growth Curve Plotting
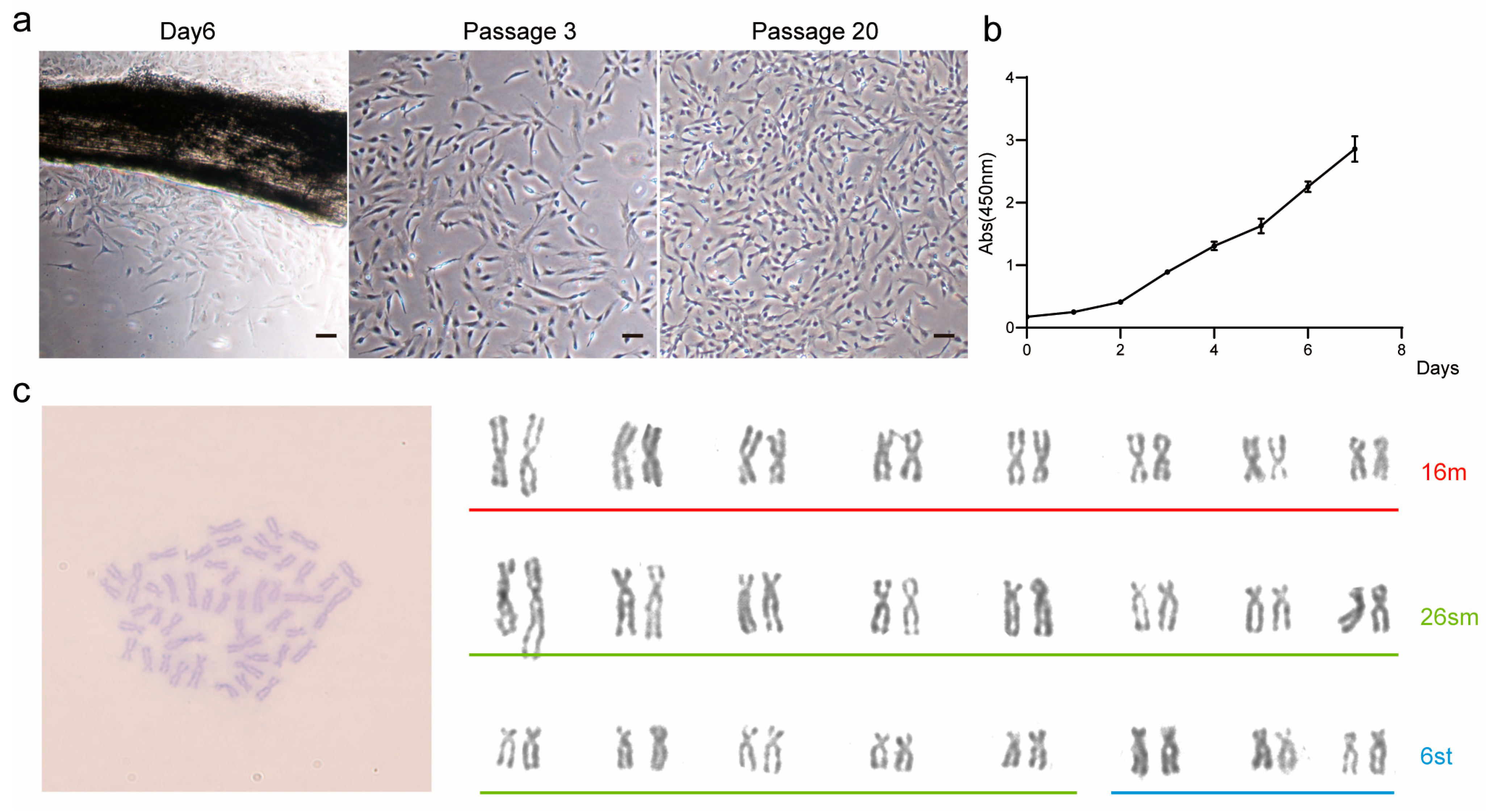
TCCF2022 cells were seeded into 96-well plates containing complete medium according to the standard of 4 × 103 cells per well, and cultured in an incubator at 27 °C under 5% CO2. After 1 h, when the cells successfully attached to the wall, the medium was removed and DMEM/F12 medium containing 100 UL of 10% hypersensitive cell proliferation assay (CCK-8) (Abbkine, BMU106, Wuhan, China) was added for 4 h of treatment. Cell growth was assessed by measuring absorbance at 450 nm. Subsequent tests were performed every 24 h using CCK8, and the actual absorbance was obtained by subtracting the absorbance of the blank control group. Each experimental group contained three biological replicates.
2.5. Karyotype Analysis
This method was adapted from the protocol adopted by Yucheng, L in 1983 [
28]. T25 cell culture dish was seeded with 10
6 cells, and when they had grown to 80% confluency, a 5 mL complete medium containing 1 ug/mL colchicine (MACKLIN, C804814, purity ≧ 98%, Shanghai, China) was added. After being incubated at 27 °C for 5 h, the medium was removed, and the cells were treated with 1 mL 0.25% trypsin–EDTA digestive solution for 5 min. Subsequently, 1 mL complete medium was added to stop the digestion, and the cells were gently blown with a pipette, transferred to a 15 mL centrifuge tube, and centrifuged at 300×
g for 3 min. The supernatant was removed, 1 mL D-Hanks was added for cleaning once, and the cells were centrifuged again to remove the supernatant. The cells were then treated with 4 mL 0.075 M KCl for 20 min for hypotonic treatment and prefixed with 1 mL Carnot fixing solution (ethanol/acetic acid = 3:1) for 5 min. After centrifugation at 500×
g for 5 min to remove the supernatant, 1 mL Carnot fixative was added for 30 min. The suspended cells were gently blown with a straw and centrifuged at 500×
g for 5 min to remove the supernatant. The fixation process was repeated once, and after centrifugation, 800 µL of fixative was removed. The cells were then resuspended with the remaining 200 µL fixative. They were dropped on a slide using the cold drop method, dried at 37 °C for 30 min, and stained with 5% Giemsa dye for 30 min. Excess dye was washed away, and the glass slides were dried and then sealed with water-neutral resin. The samples were observed and photographed using a 100× oil lens.
2.6. Alkaline Phosphatase Staining
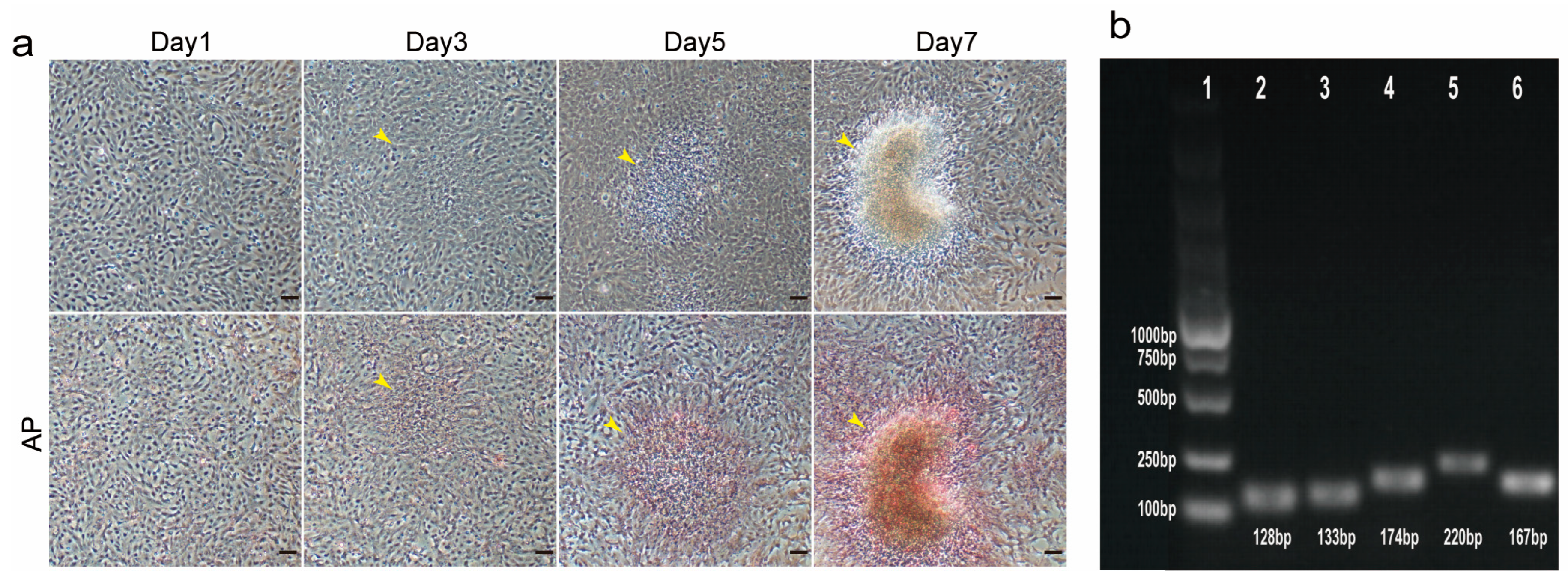
TCCF2022 cells were seeded into 12-well plates at a ratio of 2 × 105 cells per well, and cultured in an incubator containing 5% CO2 at 27 °C with 1 mL of complete medium. When the cells grew to 90% confluent, the culture was continued for 1, 3, 5, and 7 days. The medium was then removed, and the cells were rinsed twice with 1 mL PBS (Sangon Biotech, B548117-0500, Shanghai, China) solution and fixed at RT for 30 min with 0.5 mL 4% PFA. After removing 4% PFA, the cells were rinsed three times with 1 mL PBS solution and were then stained with an alkaline phosphatase staining kit (red) (Applygen, E1041-50, Beijing, China) for 1 h. After the dye was removed, the dye was rinsed three times with 1 mL PBS solution and finally photographed with an inverted phase contrast microscope.
2.7. RT-PCR
TCCF2022 cells were seeded into a 6-well plate at a ratio of 5 × 10
5 cells per well. After 3 days of cultivation, the medium was removed, and the cells were rinsed once with 1 mL PBS. Then, 1 mL of RNAiso plus (Takara, D9108A, Dalian, China) was added to the lysate cells and they were allowed to stand for 10 min; RNAiso plus was collected into a 1 mL centrifuge tube, 200 µL chloroform was added and the mixture was shaken violently for 15 s. The mixture was allowed to stand at room temperature for 10 min and then centrifuged at 12,000×
g at 4 °C for 10 min, and the supernatant was removed. Isopropyl alcohol was added in equal volume and the mixture was left at room temperature for 10 min; it was then centrifuged at 4 °C 12,000×
g for 10 min and the supernatant was removed. The mixture was rinsed once with 75% alcohol, centrifuged at 4 °C 12,000×
g for 5 min, and dried for 5 min after removing the alcohol, and the RNA was dissolved with DEPC water. The RNA was reverse-transcribed into first Strand cDNA using a PrimeScript™ II 1st Strand cDNA Synthesis Kit (TaKaRa, 6210A, Dalian, China). The amplification of the target fragment was performed using PrimeSTAR
® HS DNA Polymerase (TaKaRa, DR044A, Dalian, China). See
Supplementary Table S1 for the RT-PCR primers.
2.8. qRT-PCR
Total RNA extraction and cDNA synthesis were the same as the above RT-PCR procedure. CFX ConnectTM Optics Module (BIO-RAD) and TB green (TaKaRa, RR420A, Dalian, China) were used for qRT-PCR, with β-actin as the internal reference. Each gene group was analyzed with three biological replicates, and the relative expression levels were calculated by 2
−△△CT. See
Table S2 for the qRT-PCR primers.
2.9. RNA Sequencing and Transcriptome Analysis Sequencing
RNA HEanalysis protocols refer to our previous work [
16]. RNA libraries were constructed from the collected RNA samples utilizing the Illumina TruSeq™ RNA Library Preparation Kit and subjected to paired-end sequencing on the Illumina NovaSeq 6000 platform. Three biological replicates were analyzed per experimental group. Raw sequencing data underwent quality control processing using Fastp (v0.19.5), resulting in an average yield of ~50.5 million (SD ± 3.66 million) high-quality reads per biological replicate. De novo transcriptome assembly was conducted using Trinity software (v2.8.5). The functional annotation of the assembled transcripts was performed through BLAST+ (v2.9.0) alignment against multiple databases: NR, Swiss-Prot, Pfam, EggNOG, GO, and KEGG.
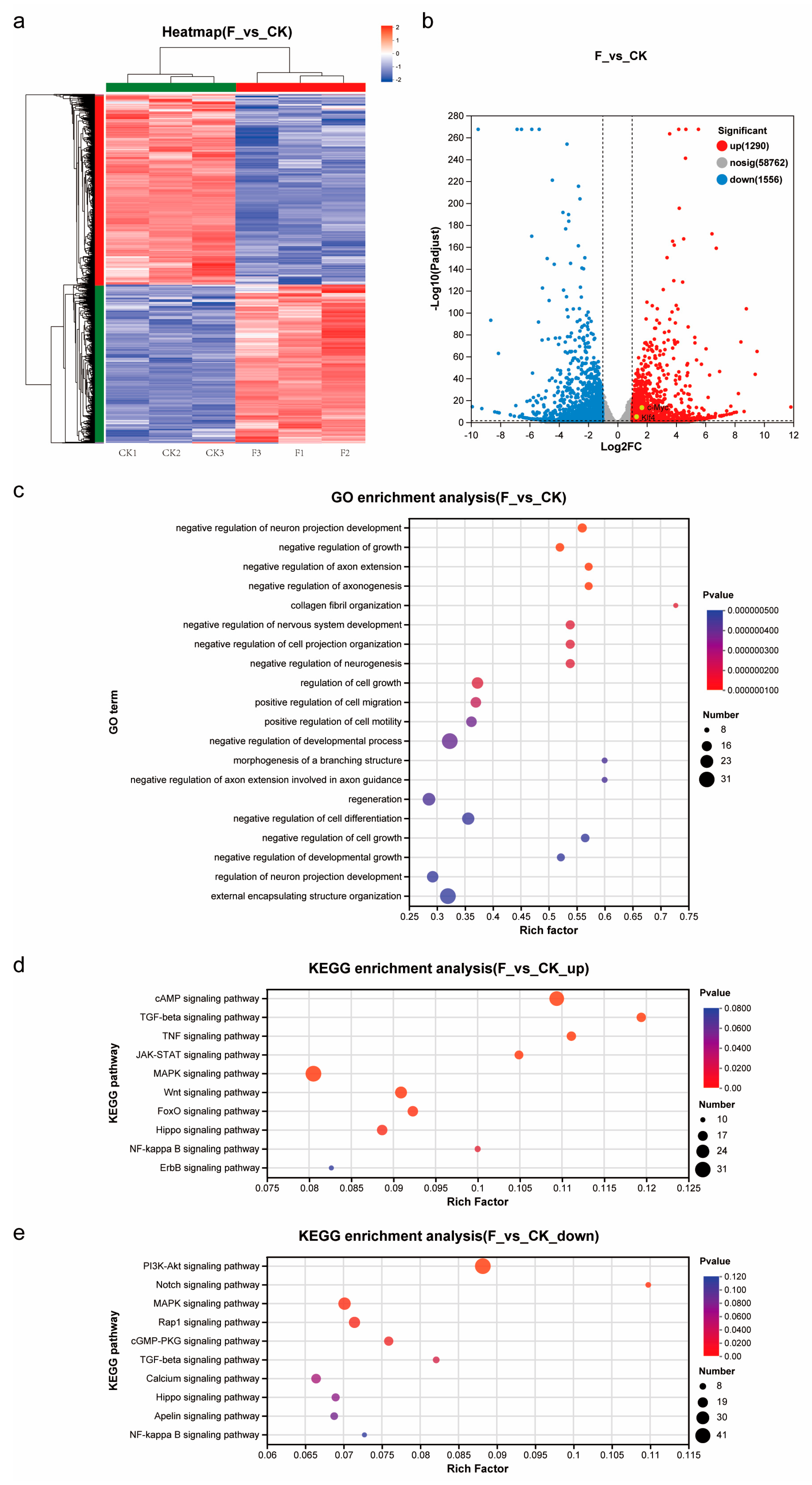
Transcript quantification was performed with RSEM (v1.3.3), with expression levels standardized to transcripts per million (TPM). Differential expression analysis was carried out using DESeq2 (v1.24.0), applying significance thresholds of |log2(fold change)| > 1 and adjusted p-value < 0.05, with results visualized through volcano plots. A heatmap depicting z-score normalized expression patterns of all differentially expressed genes between control (CK) and treated (F) groups was generated, implementing hierarchical clustering through the average linkage method with the Euclidean distance metric for both genes and samples.
Functional enrichment analysis was conducted through GO term classification using GOATools (v0.6.5) and KEGG pathway analysis with KOBAS (v2.1.1) for significantly differentially expressed genes.
2.10. Cryosection and HE Staining
The HE staining protocol refers to our previous work [
18]. The 3D spheroids were collected and fixed at 4 °C overnight using 4%PFA and 30% sucrose, and then they were encapsulated using O.C.T. Compound (Scigen, 4586, Guangzhou, China). A MICROM HM525 freezing microslicer was used to slice the sections at a shelf temperature of −20 °C, and the slice thickness was 10 μm. Then, the frozen slices were placed in distilled water for 2 min, stained with hematoxylin staining solution for 5 min, soaked in tap water to rinse off the excess staining solution for 10 min, washed again with distilled water, dehydrated with 95% ethanol for 5 s, dyed with eosin staining solution for 30 s, dehydrated with 95% ethanol for 2 min, and then dehydrated with fresh 95% ethanol for 2 min. Xylene transparent was added for 5 min and then replaced with fresh xylene transparent for 5 min, before being sealed with neutral gum.
2.11. Immunofluorescence and Histological Staining of Cryosections
Immunofluorescence and Histological staining protocols refer to our previous work [
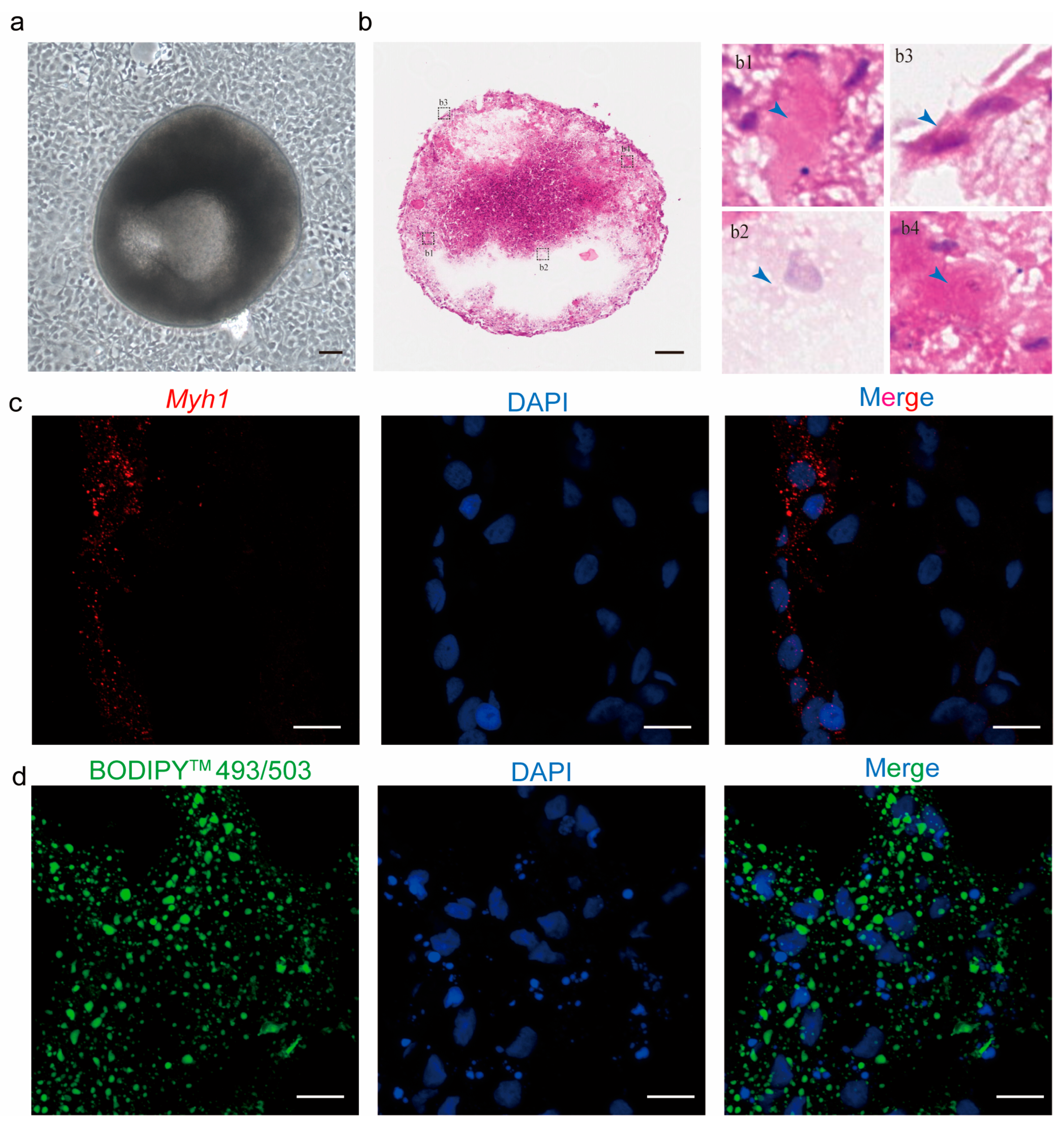
18]. Immunostaining was performed by a general protocol. The frozen sections were removed from −20 °C, thawed to room temperature, and then rinsed in PBS for 5 min. They were fixed with 4% PFA at RT for 10 min, followed by washing with PBS twice for 5 min each. Next, the sections were treated with 0.3% Triton X-100 (Sangon Biotech, A600198-0500, Shanghai, China) in PBS for 5 min and rinsed again with PBS twice for 5 min each. Finally, the sections were blocked at room temperature for 2 h using a blocking buffer containing 3% BSA-V (Solarbio, A8020, purity 97.00%, Beijing, China), 0.3% Triton X-100, and 2% denatured goat serum (Beyotime, C0265, Shanghai, China), incubated with the 1st antibody in blocking buffer at RT for 1 h, and finally counterstained with 1 μg/mL DAPI (Solarbio, C0060, Beijing, China). Myosin heavy chain (Bioss, bs-5885R, dilution 1:200, Beijing, China) for Myh1 was used as the 1st antibody. Goat antirabbit IgG secondary antibody, Alexa Fluor 647 (Beyotime, A0468, polyclonal, dilution 1:500, Shanghai, China) was used as the 2nd antibody.
To visualize lipid droplets, the sections underwent fixation with 4% PFA at RT for 30 min, followed by two washes in PBS for 5 min each, incubation with 4 μM BODIPY™ 493/503 (Thermo Fisher, D2191, Suzhou, China) lipid dye at RT for 30 min, and a final counterstaining with 1 μg/mL DAPI.
2.12. Myogenic and Adipogenic Induction with Small Molecular Chemical Treatment
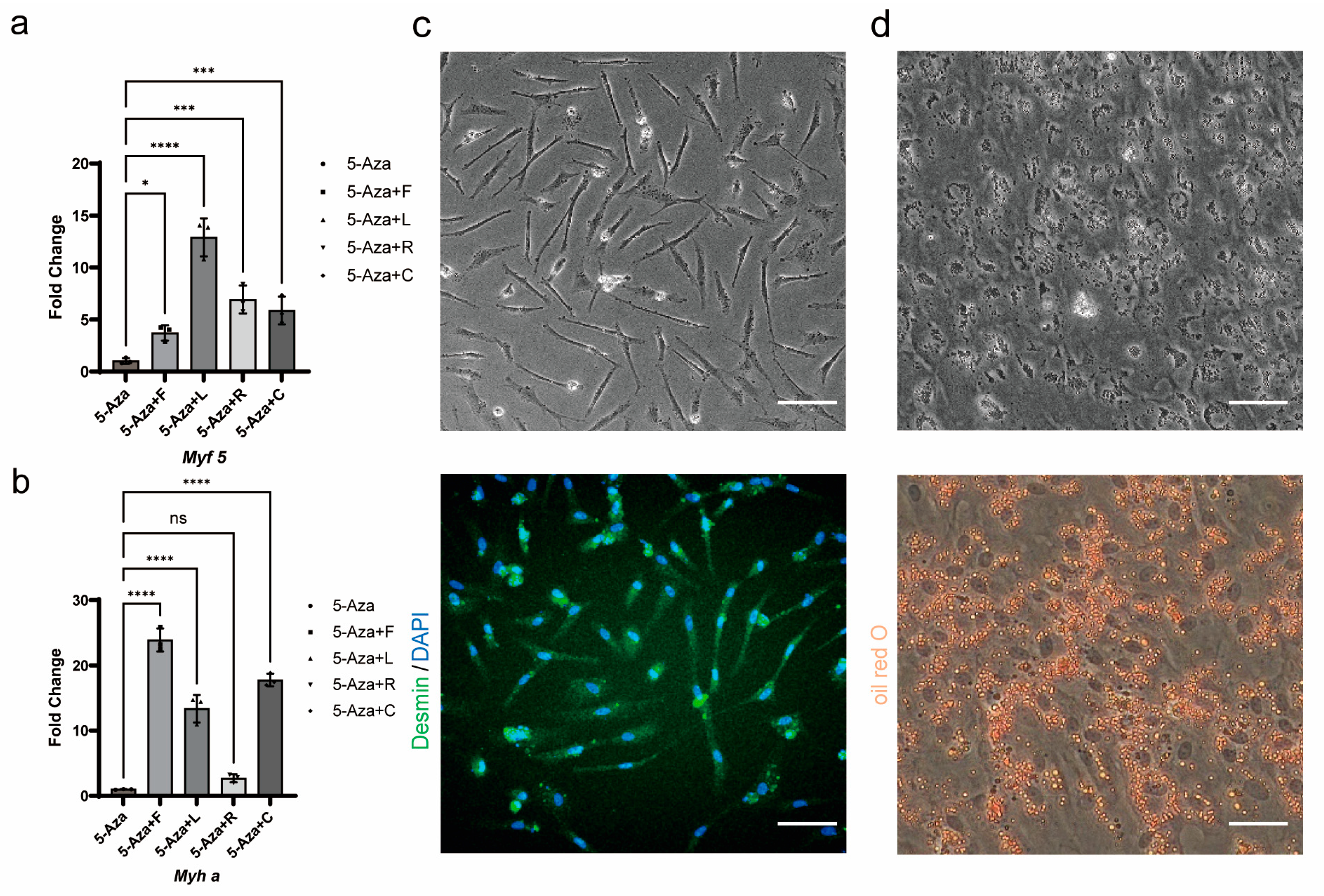
Myogenic induction: Myogenic induction was divided into two stages. In the first stage, Forskolin-treated cells were treated with DMEM/F12 medium containing 15 μM 5-Azacytidine (MCE, HY-10586, purity 99.91%, Shanghai, China), 10 μM Forskolin, 10 nM LY411575 (MCE, HY-50752, purity 98.68%, Shanghai, China), 5 μM RepSox (Targetmol, T6337, purity 99.62%, Shanghai, China), and 2 μM CHIR99021 (Targetmol, T2310, purity 99.29%, Shanghai, China) for 2 days. Subsequently, F12 medium (Biosharp, BL311A, Hefei, China) containing 2% fetal bovine serum was used for induction for 2 days in an incubator containing 5% CO2 at 27 °C to complete differentiation.
Adipogenic induction: Forskolin-treated cells were treated with DMEM/F12 medium containing 10% HS (horse serum, Biosharp, BL209A, Hefei, China), 10 μg/mL insulin (Targetmol, I189675, ≥27 USP units/mg, Shanghai, China), 0.5 μM IBMX (Targetmol, T1713, purity 99.86%, Shanghai, China), 0.25 μM dexamethasone (Targetmol, T0947L, purity 99.88%, Shanghai, China), and 1% Lipid Mixture (Peprotech, LM-200, Cranbury, NJ, USA) for 3 days in an incubator containing 5% CO2 at 27 °C.
2.13. Immunofluorescent and Histological Staining of Myogenic and Adipogenic Induction Cells
Immunofluorescence and Histological staining protocols refer to our previous work [
18]. Immunostaining was performed by a general protocol. Cells were fixed with 4% PFA (paraformaldehyde) at RT for 10 min, permeabilized with 0.3% Triton X-100 in PBS at RT for 10 min, blocked in blocking buffer for 60 min, incubated with the 1st antibody in blocking buffer at RT for 2 h or at 4 °C overnight, incubated with the 1st antibody in blocking buffer at RT for 1 h, and finally counterstained with 1 μg/mL DAPI. Desmin (Bioss, bs-1026R, dilution 1:200, Beijing, China) for myofiber/myotube was used as the 1st antibody. Goat antirabbit IgG secondary antibody, Alexa Fluor 488 (Beyotime, A0423, polyclonal, dilution 1:500, Shanghai, China) wasused as the 2nd antibody.
To observe lipid droplets in adipocytes, the cells were fixed with 4% PFA at RT for 30 min, washed twice with PBS (5 min each), incubated in 60% isopropanol for 2 min, and then stained with Oil Red O (Aladdin, O104972, Shanghai, China).
4. Discussion
Cellular agriculture technology is gradually becoming an innovative means to obtain high-quality alternative proteins, offering a sustainable solution for global population growth. Fish has always been an important source of animal protein, meeting people’s daily nutritional needs. However, traditional fisheries face challenges related to environmental and resource constraints. Therefore, the advancement of cell-cultured meat as an emerging industry, driven by stem cell technology, presents enormous potential. Stable, continuously expandable seed cells form the foundational cornerstone for scaling cell-cultured meat production. Edible meat in daily life is usually composed of muscle tissue, fat tissue, and connective tissue. Cell agriculture in mammals [
5] and birds [
7] usually uses MuSCs as seed cells. In fish, since fish fat contains long-chain omega-3 polyunsaturated fatty acids and can provide special flavor to fish, in order to improve the taste of cell-cultured meat and provide higher nutritional value, AD-MSCs and MuSCs are often used as seed cells for cell aquaculture. However, fish cell culture technologies remain underdeveloped, with stable, continuously passaged cell lines of MuSCs and AD-MSCs being difficult to establish across many fish species. The scarcity of reliable seed cell sources persists as a critical bottleneck. Notably, threadsail filefish (
Stephanolepis cirrhifer) caudal fin cells have been reported to generate myotubes and adipocytes
in vitro [
25], suggesting their potential as novel seed cells for cell-cultured meat that could circumvent species-specific limitations in conventional cell line development. Moreover, using fin tissue to obtain seed cells eliminates the need to sacrifice fish, thereby supporting biodiversity conservation and addressing potential ethical concerns associated with traditional methods.
Here, we report for the first time the establishment and characterization of the caudal fin cell line TCCF2022 from Topmouth culter (
Culter alburnus). In primary culture, epithelial-like and fibroblast-like cells migrated out from tissue blocks, with fibroblast-like cells gradually becoming dominant as passages progressed. These cells proliferated on average every 2–3 days at 27 °C, spontaneously aggregating to form 3D spheroids. Alkaline phosphatase staining showed positivity, and the cells exhibited the expression of pluripotency marker genes, including
Sox2,
Nanog,
Oct4, and
Klf4. This suggests that TCCF2022 cells possess strong pluripotency. Since the genome of Topmouth culter has not been well assembled and annotated, we first mined and annotated the unannotated genes
Sox2,
Nanog,
Oct4,
Klf4, and
Myha from its genome sequence and SRA database (see
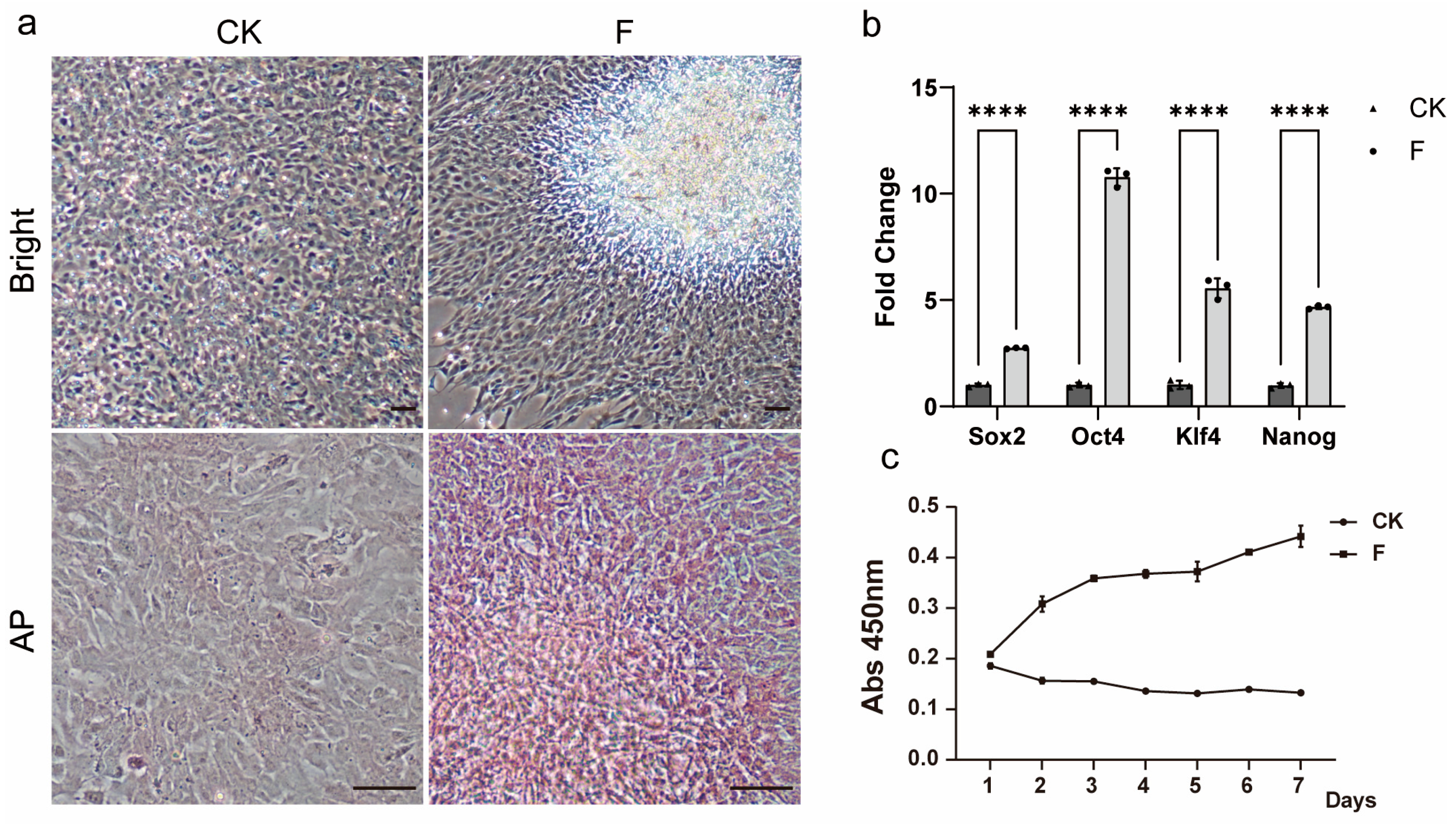
Supplementary Figure S1). However, with the increase in the number of transmission passages, the proliferation ability of the cells gradually decreased, up to about the 30th passage, when the cells even stopped growing. In an attempt to restore the cells’ ability to proliferate, we unexpectedly found that Forskolin as a single factor contributed to the recovery and maintenance of pluripotency, leading to the re-emergence of 3D growth in multi-passaged TCCF2022 cells. Other pluripotency features, such as
Sox2,
Oct4,
Nanog, and
Klf4 gene expressions, significantly increased.
Forskolin, a natural compound from plants, is an activator of intracellular adenylate cyclase, increasing intracellular cyclic adenosine monophosphate (cAMP) levels [
32], triggering a series of biochemical reactions, such as protein kinase A (PKA), influencing cell function and behavior, and is commonly used as one of the components for fibroblast reprogramming [
33,
34,
35]. Transcriptomic analysis revealed that the cAMP signal was indeed highly activated in Forskolin-treated TCCF2022 cells. However, whether Forskolin-treated TCCF2022 cells can be used as a seed cell source for artificial meat needs to be further determined.
We observed the emergence of myogenic and adipogenic lineage cells within the 3D spheroids, suggesting that Forskolin-treated TCCF2022 cells have the potential to differentiate into myotubes and adipocytes. But due to low efficiency and undesired cell populations with undefined identities, it is not currently possible to apply 3D organoid-like culture directly to the formation of cell-cultured fish meat. Therefore, we strategically directed our focus to first establishing the ways of transdifferentiation of caudal fin cells into myotubes and adipocytes in a 2D monolayer system. Although the differentiation of caudal fin cells into myogenic lineage cells and adipogenic lineage cells in a 2D monolayer system has already been reported, the proprietary composition of their myogenic induction medium is not clear or optimized for fish species, and the resulting cell identities also lack characterization [
25]. Moreover, the relevant induction efficiencies were not explicitly presented. Therefore, we attempted to develop a cost-effective, small-molecule cocktail with well-defined mechanisms, which could be beneficial to the cell-cultured fish meat research field.
In our optimized small-molecule cocktail (5LRCF), 5-Aza promotes myogenic differentiation by covalently binding to DNA methyltransferases (DNMTs), leading to the demethylation of the promoter regions of key myogenic genes (such as MyoD, Myogenin, and Myf5), which releases gene silencing and activates myogenic regulatory factors [
31,
36,
37]. However, our data reveals that treatment with 5-Aza alone only marginally enhances the myogenic capacity of Forskolin-treated TCCF2022 cells. Thus, we combined 5-Aza with complementary myogenic enhancers such as Forskolin, LY411575, RepSox, and CHIR99021. Forskolin was applied at optimized concentrations to enhance pluripotency-related gene expression, whereas LY411575 and RepSox have the ability to enhance the myogenic differentiation efficiency of MuSCs in large yellow croaker (
Larimichthys crocea) by the inhibition of Notch and TGF-β signaling, respectively [
16]. LY411575 inhibits Notch signaling, thereby mimicking the natural downregulation of this pathway that occurs when muscle stem cells transition from a stem cell fate to a differentiation state. RepSox inhibits the TGF-β signaling pathway, preventing the differentiation of muscle stem cells into myofibroblasts. The purpose of the addition of CHIR99021 is to activate the Wnt/β-catenin signaling pathway, upregulating the key membrane fusion proteins Myomaker and Myomerger, which drive myocyte membrane fusion, thereby promoting myotube formation [
38]. Finally, low-serum differentiation further promoted the formation of myotubes. Our combinatorial approaches successfully generated Desmin-positive multinucleated myotubes and lipid-laden adipocytes, enhancing differentiation efficiency remains a core focus of our current research. However, the molecular basis and mechanisms of Forskolin-treatment remain to be further investigated.
Although scaling up the production of cell-cultured fish meat using TCCF2022 cells still faces substantial challenges, including the development of serum-free culture media, the optimization of cellular immortalization strategies, the construction of suspension culture systems, and the determination of the optimal bioreactor configurations, the TCCF2022 cell line was successfully established from the caudal fin of Topmouth culter and demonstrated robust proliferative capacity and multipotent differentiation potential, endowing it with significant value as seed cells for cell-cultured fish meat production. Furthermore, this method is effective and has robust manipulability to maintain its pluripotency and myogenic/adipogenic induction, offering another option for producing cell-cultured meat from economically important fish species, where MuSCs and AD-MSCs lines are difficult to established.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}