
AOPxSVM: A Support Vector Machine for Identifying Antioxidant Peptides Using a Block Substitution Matrix and Amino Acid Composition, Transformation, and Distribution Embeddings

 and
and
Abstract
1. Introduction
2. Materials and Methods
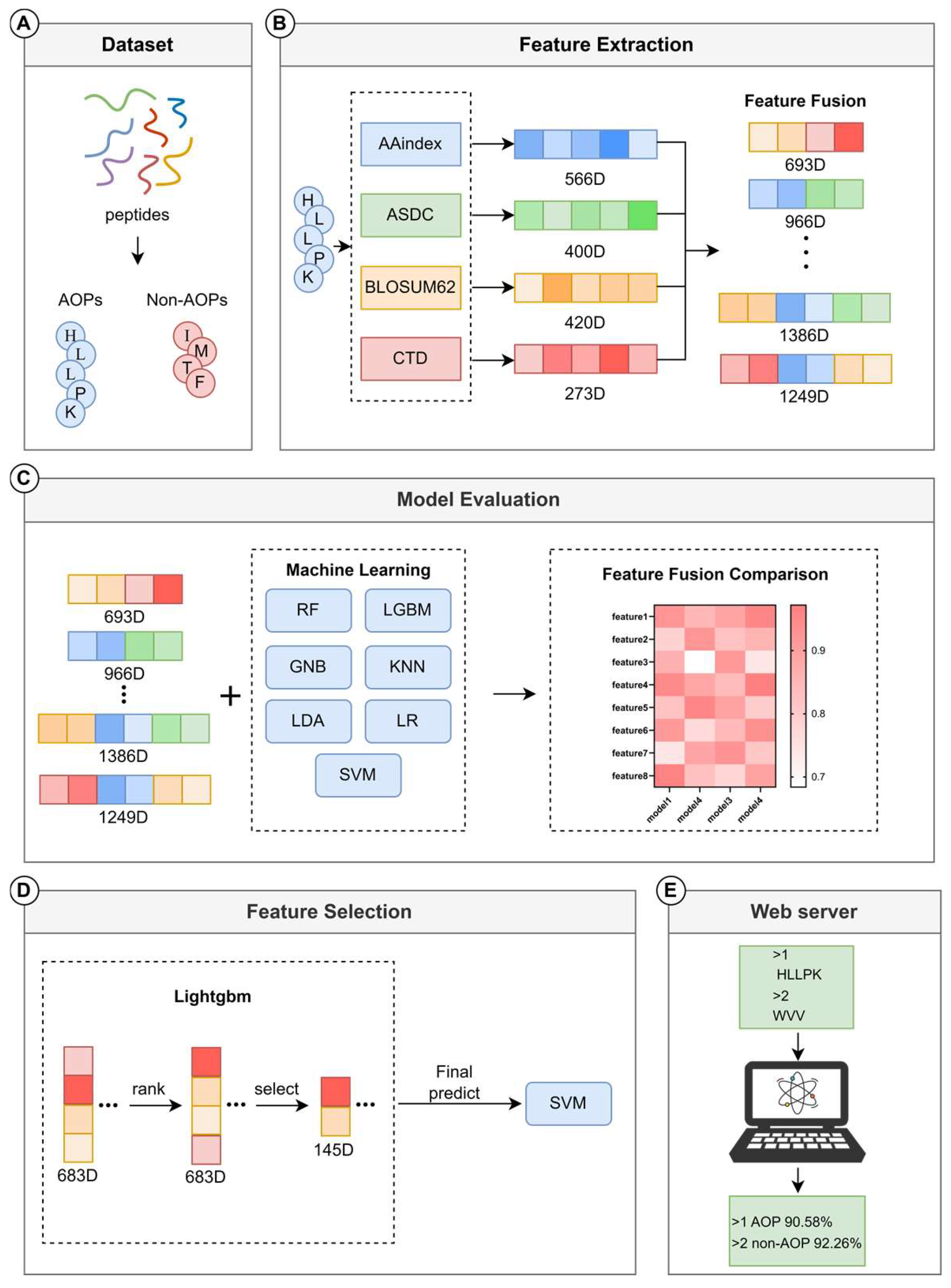
2.1. Benchmark Dataset
2.2. Feature Extraction
2.2.1. Physicochemical Property Feature
- (1)
- Amino Acid Index (AAindex)
- (2)
- Composition Transformation and Distribution (CTD)
2.2.2. Sequence Fingerprinting
- (1)
- Adaptive Skip Dipeptide Composition (ASDC)
2.2.3. Sequence Evolution Features
- (1)
- Block Substitution Matrix 62 (BLOSUM62)
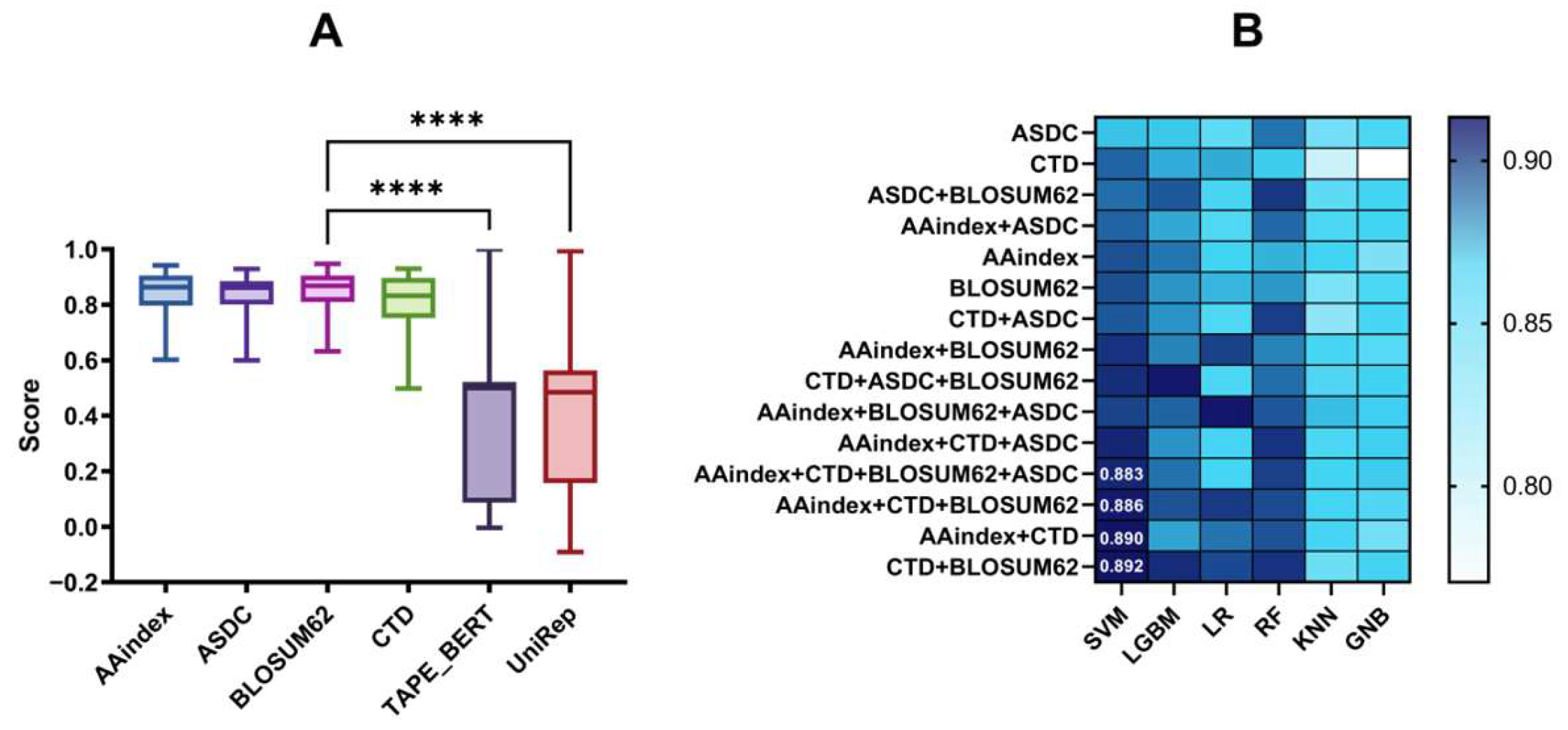
2.2.4. Deep Learning-Based Embedded Features
- (1)
- TAPE_BERT
- (2)
- UniRep
2.3. Machine Learning Methods
2.4. Feature Selection Methods
2.5. Model Evaluation Metrics
2.6. Friedman Test
3. Results
3.1. Selection of Baseline Models with Different Features and Fusion Features
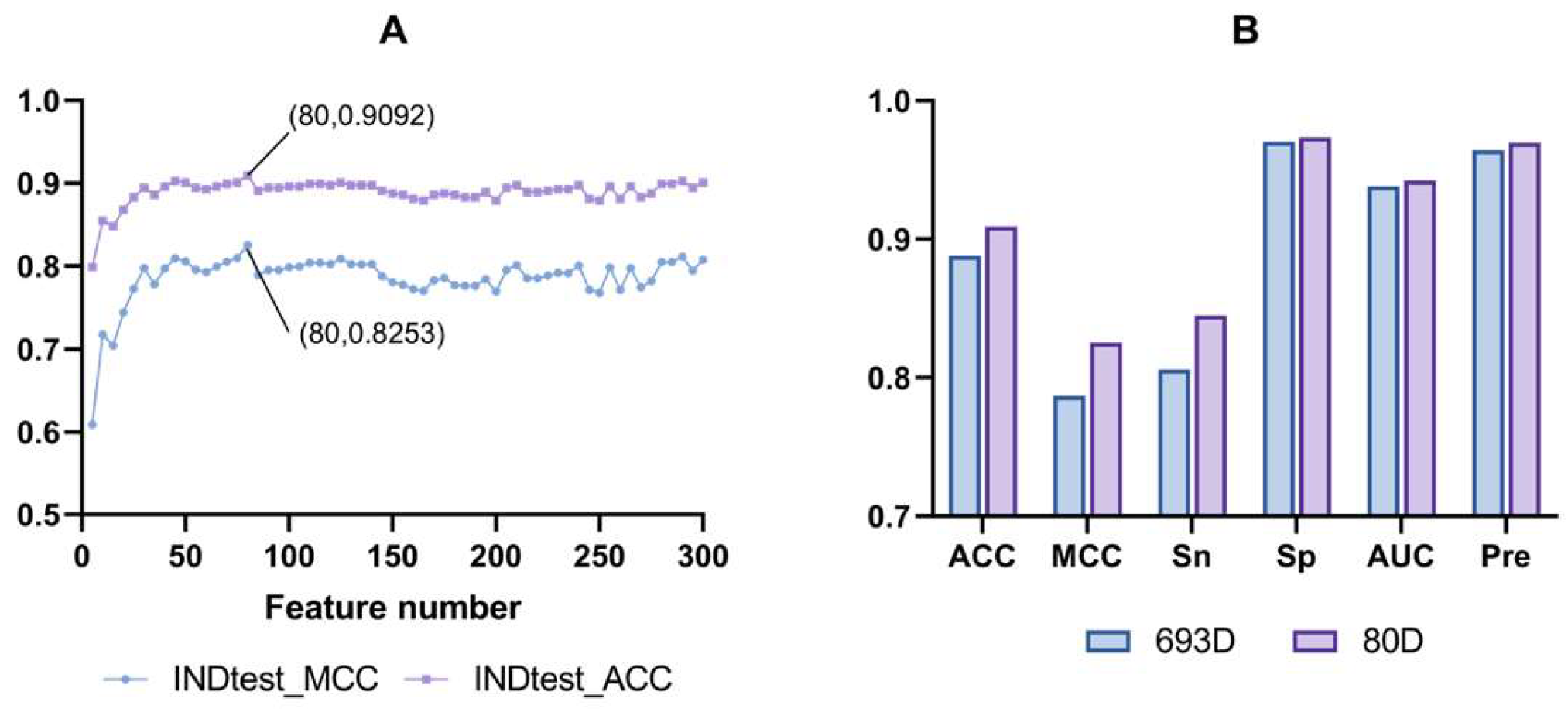
3.2. Feature Selection Optimization
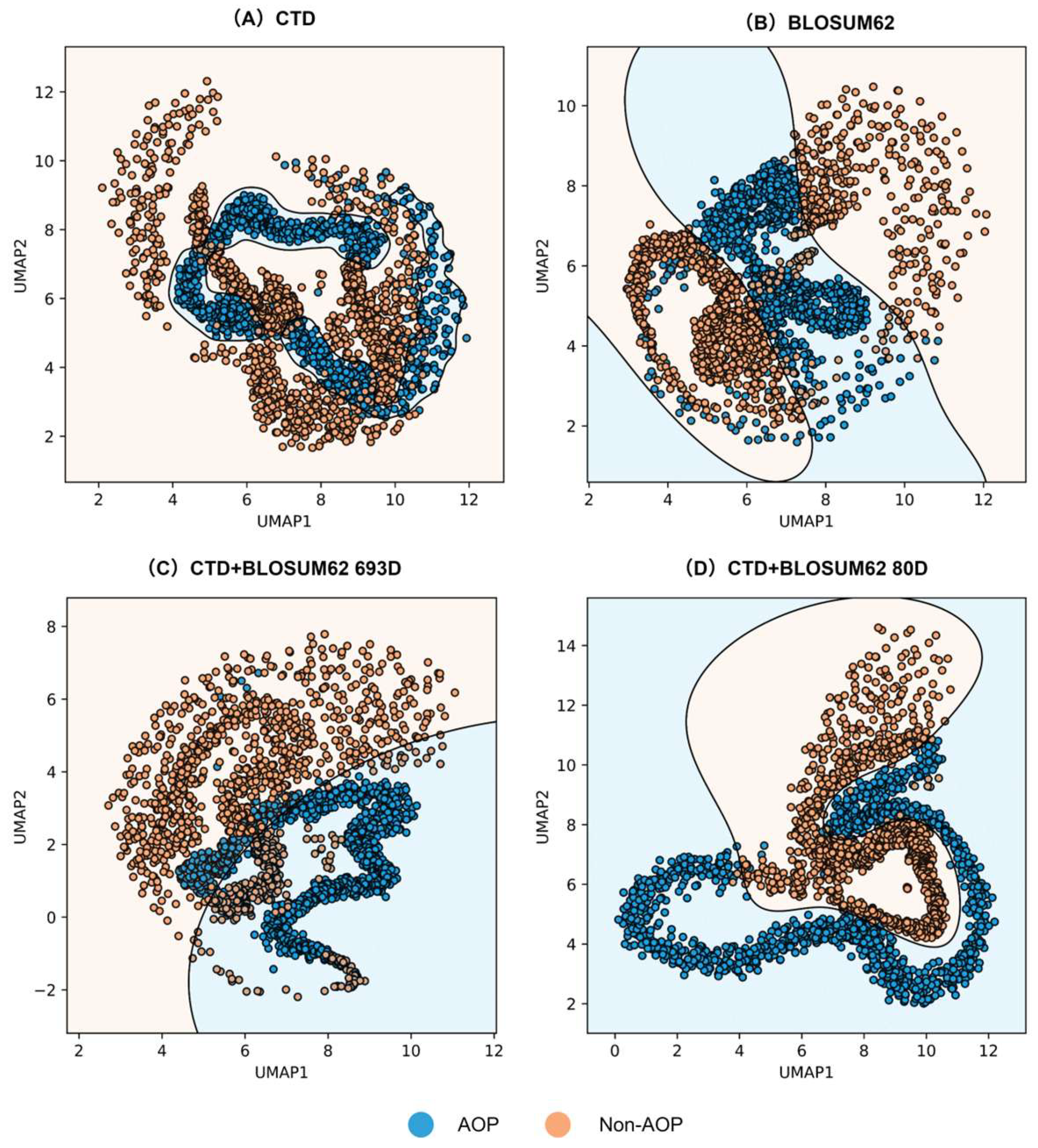
3.3. Feature Visualization
3.4. Comparison with Existing Methods
3.5. Web Server Development
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Min, B.; Ahn, D. Mechanism of lipid peroxidation in meat and meat products—A review. Food Sci. Biotechnol. 2005, 14, 152–163. [Google Scholar]
- Lorenzo, J.M.; Gómez, M. Shelf life of fresh foal meat under MAP, overwrap and vacuum packaging conditions. Meat Sci. 2012, 92, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Brecht, J.K. Physiology of Lightly Processed Fruits and Vegetables. Hortscience 1995, 30, 18–22. [Google Scholar] [CrossRef]
- Sabbatino, F.; Conti, V.; Liguori, L.; Polcaro, G.; Corbi, G.; Manzo, V.; Tortora, V.; Carlomagno, C.; Vecchione, C.; Filippelli, A. Molecules and mechanisms to overcome oxidative stress inducing cardiovascular disease in cancer patients. Life 2021, 11, 105. [Google Scholar] [CrossRef]
- Rock, C.L.; Jacob, R.A.; Bowen, P.E. Update on the biological characteristics of the antioxidant micronutrients: Vitamin C, vitamin E, and the carotenoids. J. Am. Diet. Assoc. 1996, 96, 693–702. [Google Scholar] [CrossRef]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118. [Google Scholar] [CrossRef]
- Rao, A.; Bharani, M.; Pallavi, V. Role of antioxidants and free radicals in health and disease. Adv. Pharmacol. Toxicol. 2006, 7, 29–38. [Google Scholar]
- Stefanis, L.; Burke, R.E.; Greene, L.A. Apoptosis in neurodegenerative disorders. Curr. Opin. Neurol. 1997, 10, 299–305. [Google Scholar] [CrossRef]
- Wang, R.; Jiang, Y.; Jin, J.; Yin, C.; Yu, H.; Wang, F.; Feng, J.; Su, R.; Nakai, K.; Zou, Q. DeepBIO: An automated and interpretable deep-learning platform for high-throughput biological sequence prediction, functional annotation and visualization analysis. Nucleic Acids Res. 2023, 51, 3017–3029. [Google Scholar] [CrossRef]
- World Health Organization. Diet, Nutrition and the Prevention of Chronic Diseases: Report of a Joint WHO/FAO Expert Consultation; WHO Technical Report Series 916; World Health Organization: Geneva, Switzerland, 2002; pp. 1–149. [Google Scholar]
- Ito, N.; Fukushima, S.; Tsuda, H. Carcinogenicity and modification of the carcinogenic response by BHA, BHT, and other antioxidants. CRC Crit. Rev. Toxicol. 1985, 15, 109–150. [Google Scholar] [CrossRef]
- López-García, G.; Dublan-García, O.; Arizmendi-Cotero, D.; Gómez Oliván, L.M. Antioxidant and antimicrobial peptides derived from food proteins. Molecules 2022, 27, 1343. [Google Scholar] [CrossRef] [PubMed]
- Sohaib, M.; Anjum, F.M.; Sahar, A.; Arshad, M.S.; Rahman, U.U.; Imran, A.; Hussain, S. Antioxidant proteins and peptides to enhance the oxidative stability of meat and meat products: A comprehensive review. Int. J. Food Prop. 2017, 20, 2581–2593. [Google Scholar] [CrossRef]
- Ahmadi-Vavsari, F.; Farmani, J.; Dehestani, A. Recombinant production of a bioactive peptide from spotless smooth-hound (Mustelus griseus) muscle and characterization of its antioxidant activity. Mol. Biol. Rep. 2019, 46, 2599–2608. [Google Scholar] [CrossRef]
- Chen, N.; Yang, H.; Sun, Y.; Niu, J.; Liu, S. Purification and identification of antioxidant peptides from walnut (Juglans regia L.) protein hydrolysates. Peptides 2012, 38, 344–349. [Google Scholar] [CrossRef]
- Qin, D.; Bo, W.; Zheng, X.; Hao, Y.; Li, B.; Zheng, J.; Liang, G. DFBP: A comprehensive database of food-derived bioactive peptides for peptidomics research. Bioinformatics 2022, 38, 3275–3280. [Google Scholar] [CrossRef]
- Wei, L.; He, W.; Malik, A.; Su, R.; Cui, L.; Manavalan, B. Computational prediction and interpretation of cell-specific replication origin sites from multiple eukaryotes by exploiting stacking framework. Brief. Bioinform. 2021, 22, bbaa275. [Google Scholar] [CrossRef]
- Olsen, T.H.; Yesiltas, B.; Marin, F.I.; Pertseva, M.; García-Moreno, P.J.; Gregersen, S.; Overgaard, M.T.; Jacobsen, C.; Lund, O.; Hansen, E.B. AnOxPePred: Using deep learning for the prediction of antioxidative properties of peptides. Sci. Rep. 2020, 10, 21471. [Google Scholar] [CrossRef]
- Shen, Y.; Liu, C.; Chi, K.; Gao, Q.; Bai, X.; Xu, Y.; Guo, N. Development of a machine learning-based predictor for identifying and discovering antioxidant peptides based on a new strategy. Food Control 2022, 131, 108439. [Google Scholar] [CrossRef]
- Xiao, C.; Zhou, Z.; She, J.; Yin, J.; Cui, F.; Zhang, Z. PEL-PVP: Application of plant vacuolar protein discriminator based on PEFT ESM-2 and bilayer LSTM in an unbalanced dataset. Int. J. Biol. Macromol. 2024, 277, 134317. [Google Scholar] [CrossRef]
- Qin, D.; Jiao, L.; Wang, R.; Zhao, Y.; Hao, Y.; Liang, G. Prediction of antioxidant peptides using a quantitative structure− activity relationship predictor (AnOxPP) based on bidirectional long short-term memory neural network and interpretable amino acid descriptors. Comput. Biol. Med. 2023, 154, 106591. [Google Scholar] [CrossRef]
- Chen, J.; Zou, Q.; Li, J. DeepM6ASeq-EL: Prediction of Human N6-Methyladenosine (m6A) Sites with LSTM and Ensemble Learning. Front. Comput. Sci. 2022, 16, 162302. [Google Scholar] [CrossRef]
- Du, Z.; Ding, X.; Xu, Y.; Li, Y. UniDL4BioPep: A universal deep learning architecture for binary classification in peptide bioactivity. Brief. Bioinform. 2023, 24, bbad135. [Google Scholar] [CrossRef] [PubMed]
- Li, W.X.; Liu, X.J.; Liu, Y.F.; Zheng, Z.J. High-Accuracy Identification and Structure-Activity Analysis of Antioxidant Peptides via Deep Learning and Quantum Chemistry. J. Chem. Inf. Model. 2025, 65, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Ren, X.; Luo, X.; Wang, Z.; Li, Z.; Luo, X.; Shen, J.; Li, Y.; Yuan, D.; Nussinov, R.J.N.C. A foundation model identifies broad-spectrum antimicrobial peptides against drug-resistant bacterial infection. Nat. Commun. 2024, 15, 7538. [Google Scholar] [CrossRef]
- Zhou, W.Y.; Liu, Y.F.; Li, Y.X.; Kong, S.Q.; Wang, W.L.; Ding, B.Y.; Han, J.Y.; Mou, C.Z.; Gao, X.; Liu, J.T. TriNet: A tri-fusion neural network for the prediction of anticancer and antimicrobial peptides. Patterns 2023, 4, 100702. [Google Scholar] [CrossRef]
- Zhang, J.H.; Zhang, Z.H.; Pu, L.R.; Tang, J.J.; Guo, F. AIEpred: An Ensemble Predictive Model of Classifier Chain to Identify Anti-Inflammatory Peptides. IEEE-ACM Trans. Comput. Biol. Bioinform. 2021, 18, 1831–1840. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, R.; Feng, J.; Jin, J.; Liang, S.; Li, Z.; Yu, Y.; Ma, A.; Su, R.; Zou, Q. Explainable deep hypergraph learning modeling the peptide secondary structure prediction. Adv. Sci. 2023, 10, 2206151. [Google Scholar] [CrossRef]
- Li, H.; Liu, B. BioSeq-Diabolo: Biological sequence similarity analysis using Diabolo. PLoS Comput. Biol. 2023, 19, e1011214. [Google Scholar] [CrossRef]
- Kawashima, S.; Pokarowski, P.; Pokarowska, M.; Kolinski, A.; Katayama, T.; Kanehisa, M. AAindex: Amino acid index database, progress report 2008. Nucleic Acids Res. 2007, 36, D202–D205. [Google Scholar] [CrossRef]
- Li, Z.-R.; Lin, H.H.; Han, L.; Jiang, L.; Chen, X.; Chen, Y.Z. PROFEAT: A web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence. Nucleic Acids Res. 2006, 34, W32–W37. [Google Scholar] [CrossRef]
- Dubchak, I.; Muchnik, I.; Mayor, C.; Dralyuk, I.; Kim, S.H. Recognition of a protein fold in the context of the SCOP classification. Proteins Struct. Funct. Bioinform. 1999, 35, 401–407. [Google Scholar] [CrossRef]
- Zou, X.; Ren, L.; Cai, P.; Zhang, Y.; Ding, H.; Deng, K.; Yu, X.; Lin, H.; Huang, C. Accurately identifying hemagglutinin using sequence information and machine learning methods. Front. Med. 2023, 10, 1281880. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Yuan, S.S.; Li, J.; Huang, C.B.; Lin, H.; Liao, B. A First Computational Frame for Recognizing Heparin-Binding Protein. Diagnostics 2023, 13, 2465. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Tang, J.; Zou, Q. SkipCPP-Pred: An improved and promising sequence-based predictor for predicting cell-penetrating peptides. BMC Genom. 2017, 18, 742. [Google Scholar] [CrossRef]
- Wei, L.; Zhou, C.; Chen, H.; Song, J.; Su, R. ACPred-FL: A sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics 2018, 34, 4007–4016. [Google Scholar] [CrossRef]
- Eddy, S.R. Where did the BLOSUM62 alignment score matrix come from? Nat. Biotechnol. 2004, 22, 1035–1036. [Google Scholar] [CrossRef]
- Asgari, E.; McHardy, A.C.; Mofrad, M.R. Probabilistic variable-length segmentation of protein sequences for discriminative motif discovery (DiMotif) and sequence embedding (ProtVecX). Sci. Rep. 2019, 9, 3577. [Google Scholar] [CrossRef]
- Li, Y.; Wei, X.; Yang, Q.; Xiong, A.; Li, X.; Zou, Q.; Cui, F.; Zhang, Z. msBERT-Promoter: A multi-scale ensemble predictor based on BERT pre-trained model for the two-stage prediction of DNA promoters and their strengths. BMC Biol. 2024, 22, 126. [Google Scholar] [CrossRef]
- Joshi, M.; Singh, B.K. Deep Learning Techniques for Brain Lesion Classification Using Various MRI (from 2010 to 2022): Review and Challenges. Medinformatics 2024, 1–21. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Alley, E.C.; Khimulya, G.; Biswas, S.; AlQuraishi, M.; Church, G.M. Unified rational protein engineering with sequence-based deep representation learning. Nat. Methods 2019, 16, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Suzek, B.E.; Wang, Y.; Huang, H.; McGarvey, P.B.; Wu, C.H.; Consortium, U. UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015, 31, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, C.; Chen, R.; Cao, D.; Zeng, X.J.E.S.w.A. Geometric Deep Learning for Drug Discovery. Expert Syst. Appl. 2024, 240, 122498. [Google Scholar] [CrossRef]
- Noble, W.S. What is a support vector machine? Nat. Biotechnol. 2006, 24, 1565–1567. [Google Scholar] [CrossRef]
- Wang, Y.; Zhai, Y.; Ding, Y.; Zou, Q. SBSM-Pro: Support Bio-sequence Machine for Proteins. Sci. China Inf. Sci. 2024, 67, 144–159. [Google Scholar] [CrossRef]
- Meher, P.K.; Hati, S.; Sahu, T.K.; Pradhan, U.; Gupta, A.; Rath, S.N. SVM-Root: Identification of Root-Associated Proteins in Plants by Employing the Support Vector Machine with Sequence-Derived Features. Curr. Bioinform. 2024, 19, 91–102. [Google Scholar] [CrossRef]
- Li, H.; Pang, Y.; Liu, B. BioSeq-BLM: A platform for analyzing DNA, RNA, and protein sequences based on biological language models. Nucleic Acids Res. 2021, 49, e129. [Google Scholar] [CrossRef]
- Ke, G.L.; Meng, Q.; Finley, T.; Wang, T.F.; Chen, W.; Ma, W.D.; Ye, Q.W.; Liu, T.Y. LightGBM: A Highly Efficient Gradient Boosting Decision Tree. Adv. Neural 2017, 30, 3149–3157. [Google Scholar]
- Jiang, J.; Li, J.; Li, J.; Pei, H.; Li, M.; Zou, Q.; Lv, Z. A machine learning method to identify umami peptide sequences by using multiplicative LSTM embedded features. Foods 2023, 12, 1498. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Ru, X.; Li, L.; Zou, Q. Incorporating Distance-Based Top-n-gram and Random Forest to Identify Electron Transport Proteins. J. Proteome Res. 2019, 18, 2931–2939. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.; Hart, P. Nearest neighbor pattern classification. IEEE Trans. Inf. Theory 1967, 13, 21–27. [Google Scholar] [CrossRef]
- Boopathi, V.; Subramaniyam, S.; Malik, A.; Lee, G.; Manavalan, B.; Yang, D.-C. mACPpred: A support vector machine-based meta-predictor for identification of anticancer peptides. Int. J. Mol. Sci. 2019, 20, 1964. [Google Scholar] [CrossRef] [PubMed]
- Zulfiqar, H.; Guo, Z.; Ahmad, R.M.; Ahmed, Z.; Cai, P.; Chen, X.; Zhang, Y.; Lin, H.; Shi, Z. Deep-STP: A deep learning-based approach to predict snake toxin proteins by using word embeddings. Front. Med. 2023, 10, 1291352. [Google Scholar] [CrossRef]
- Lv, Z.; Cui, F.; Zou, Q.; Zhang, L.; Xu, L. Anticancer peptides prediction with deep representation learning features. Brief. Bioinform. 2021, 22, bbab008. [Google Scholar] [CrossRef]
- Kumar, M.; Rath, N.K.; Swain, A.; Rath, S.K. Feature selection and classification of microarray data using MapReduce based ANOVA and K-nearest neighbor. Procedia Comput. Sci. 2015, 54, 301–310. [Google Scholar] [CrossRef]
- Battiti, R. Using mutual information for selecting features in supervised neural net learning. IEEE Trans. Neural Netw. 1994, 5, 537–550. [Google Scholar] [CrossRef]
- Zhu, H.; Hao, H.; Yu, L. Identification of microbe–disease signed associations via multi-scale variational graph autoencoder based on signed message propagation. BMC Biol. 2024, 22, 172. [Google Scholar] [CrossRef]
- Huang, Z.; Guo, X.; Qin, J.; Gao, L.; Ju, F.; Zhao, C.; Yu, L. Accurate RNA velocity estimation based on multibatch network reveals complex lineage in batch scRNA-seq data. BMC Biol. 2024, 22, 290. [Google Scholar] [CrossRef]
- Guo, X.; Huang, Z.; Ju, F.; Zhao, C.; Yu, L. Highly Accurate Estimation of Cell Type Abundance in Bulk Tissues Based on Single-Cell Reference and Domain Adaptive Matching. Adv. Sci. 2024, 11, 2306329. [Google Scholar] [CrossRef]
- Zhang, H.Q.; Arif, M.; Thafar, M.A.; Albaradei, S.; Cai, P.; Zhang, Y.; Tang, H.; Lin, H. PMPred-AE: A computational model for the detection and interpretation of pathological myopia based on artificial intelligence. Front. Med. 2025, 12, 1529335. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ai, C.; Yang, H.; Dong, R.; Tang, J.; Zheng, S.; Guo, F. RetroCaptioner: Beyond attention in end-to-end retrosynthesis transformer via contrastively captioned learnable graph representation. Bioinformatics 2024, 40, btae561. [Google Scholar] [CrossRef] [PubMed]
- Ai, C.; Yang, H.; Liu, X.; Dong, R.; Ding, Y.; Guo, F. MTMol-GPT: De novo multi-target molecular generation with transformer-based generative adversarial imitation learning. PLoS Comput. Biol. 2024, 20, e1012229. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Lv, H.; Shao, J.; Chen, S.; Liu, B. TPpred-SC: Multi-functional therapeutic peptideprediction based on multi-label supervisedcontrastive learning. Sci. China Inf. Sci. 2024, 67, 212105. [Google Scholar] [CrossRef]
- Mubango, E.; Fu, Z.; Dou, P.; Tan, Y.; Luo, Y.; Chen, L.; Wu, K.; Hong, H. Dual function antioxidant and anti-inflammatory fish maw peptides: Isolation and structure-activity analysis via tandem molecular docking and quantum chemical calculation. Food Chem. 2025, 465, 141970. [Google Scholar] [CrossRef]
- Cao, C.; Ding, B.; Li, Q.; Kwok, D.; Wu, J.; Long, Q. Power analysis of transcriptome-wide association study: Implications for practical protocol choice. PLoS Genet. 2021, 17, e1009405. [Google Scholar] [CrossRef]
- Cao, C.; He, J.; Mak, L.; Perera, D.; Kwok, D.; Wang, J.; Li, M.; Mourier, T.; Gavriliuc, S.; Greenberg, M. Reconstruction of microbial haplotypes by integration of statistical and physical linkage in scaffolding. Mol. Biol. Evol. 2021, 38, 2660–2672. [Google Scholar] [CrossRef]
- Cao, C.; Wang, J.; Kwok, D.; Cui, F.; Zhang, Z.; Zhao, D.; Li, M.J.; Zou, Q. webTWAS: A resource for disease candidate susceptibility genes identified by transcriptome-wide association study. Nucleic Acids Res. 2022, 50, D1123–D1130. [Google Scholar] [CrossRef]
- Zhou, Z.; Xiao, C.; Yin, J.; She, J.; Duan, H.; Liu, C.; Fu, X.; Cui, F.; Qi, Q.; Zhang, Z. PSAC-6mA: 6mA site identifier using self-attention capsule network based on sequence-positioning. Comput. Biol. Med. 2024, 171, 108129. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Dataset | AOPP.test01 | AOPP.test2023 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Model | Val_ACC | ACC | MCC | Sn | Sp | AUC | Pre | F1 | ACC | MCC | Sn | Sp | Pre | F1 |
| AOPP | 0.8969 | 0.9043 | 0.8181 | 0.8284 | 0.9802 | 0.9043 | 0.9767 | 0.8965 | 0.9267 | 0.8595 | 0.8667 | 0.9867 | 0.9848 | 0.9220 |
| AnOxPP | —— | —— | —— | —— | —— | —— | —— | —— | 0.8800 | 0.7610 | 0.9060 | 0.8530 | 0.8610 | 0.8829 |
| AnOxPePred a | —— | —— | —— | —— | —— | —— | —— | —— | 0.7530 | 0.4330 | 0.8100 | 0.6270 | 0.8260 | 0.8179 |
| UniDL4BioPep | —— | —— | —— | —— | —— | —— | —— | —— | 0.5800 | 0.1633 | 0.6800 | 0.4800 | 0.5667 | 0.6182 |
| SBSM-Pro | —— | 0.7888 | 0.5786 | 0.7591 | 0.8185 | —— | 0.8070 | 0.7823 | 0.7333 | 0.4668 | 0.7200 | 0.7467 | 0.7397 | 0.7297 |
| AOPxSVM * | 0.9056 | 0.9092 | 0.8253 | 0.8449 | 0.9736 | 0.9423 | 0.9697 | 0.9030 | 0.9333 | 0.8670 | 0.9200 | 0.9467 | 0.9452 | 0.9324 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Wang, H.; Yu, Q.; Cai, J.; Jiang, L.; Luo, X.; Zou, Q.; Lv, Z. AOPxSVM: A Support Vector Machine for Identifying Antioxidant Peptides Using a Block Substitution Matrix and Amino Acid Composition, Transformation, and Distribution Embeddings. Foods 2025, 14, 2014. https://doi.org/10.3390/foods14122014
Li R, Wang H, Yu Q, Cai J, Jiang L, Luo X, Zou Q, Lv Z. AOPxSVM: A Support Vector Machine for Identifying Antioxidant Peptides Using a Block Substitution Matrix and Amino Acid Composition, Transformation, and Distribution Embeddings. Foods. 2025; 14(12):2014. https://doi.org/10.3390/foods14122014
Chicago/Turabian StyleLi, Rujun, Haotian Wang, Qiunan Yu, Jing Cai, Liangzhen Jiang, Ximei Luo, Quan Zou, and Zhibin Lv. 2025. "AOPxSVM: A Support Vector Machine for Identifying Antioxidant Peptides Using a Block Substitution Matrix and Amino Acid Composition, Transformation, and Distribution Embeddings" Foods 14, no. 12: 2014. https://doi.org/10.3390/foods14122014
APA StyleLi, R., Wang, H., Yu, Q., Cai, J., Jiang, L., Luo, X., Zou, Q., & Lv, Z. (2025). AOPxSVM: A Support Vector Machine for Identifying Antioxidant Peptides Using a Block Substitution Matrix and Amino Acid Composition, Transformation, and Distribution Embeddings. Foods, 14(12), 2014. https://doi.org/10.3390/foods14122014