
High-Throughput Determination of Multiclass Chemical Hazards in Poultry Muscles and Eggs Using UPLC–MS/MS

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Instruments and Equipment
2.3. Sample Preparation
2.4. Instrumental Conditions
2.5. Preparation of Standards and Working Solutions
2.6. Quality Assurance
3. Results and Discussion
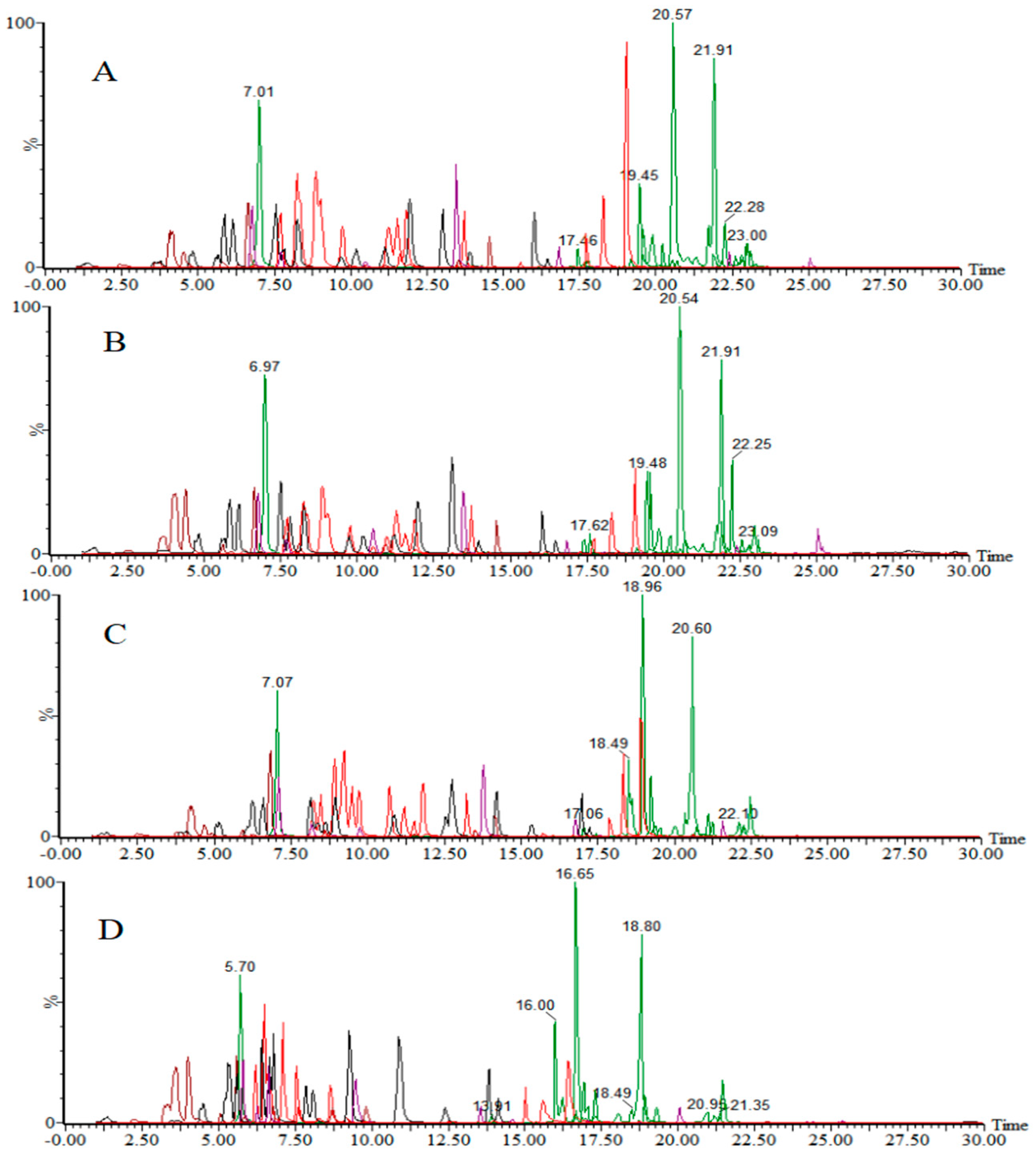
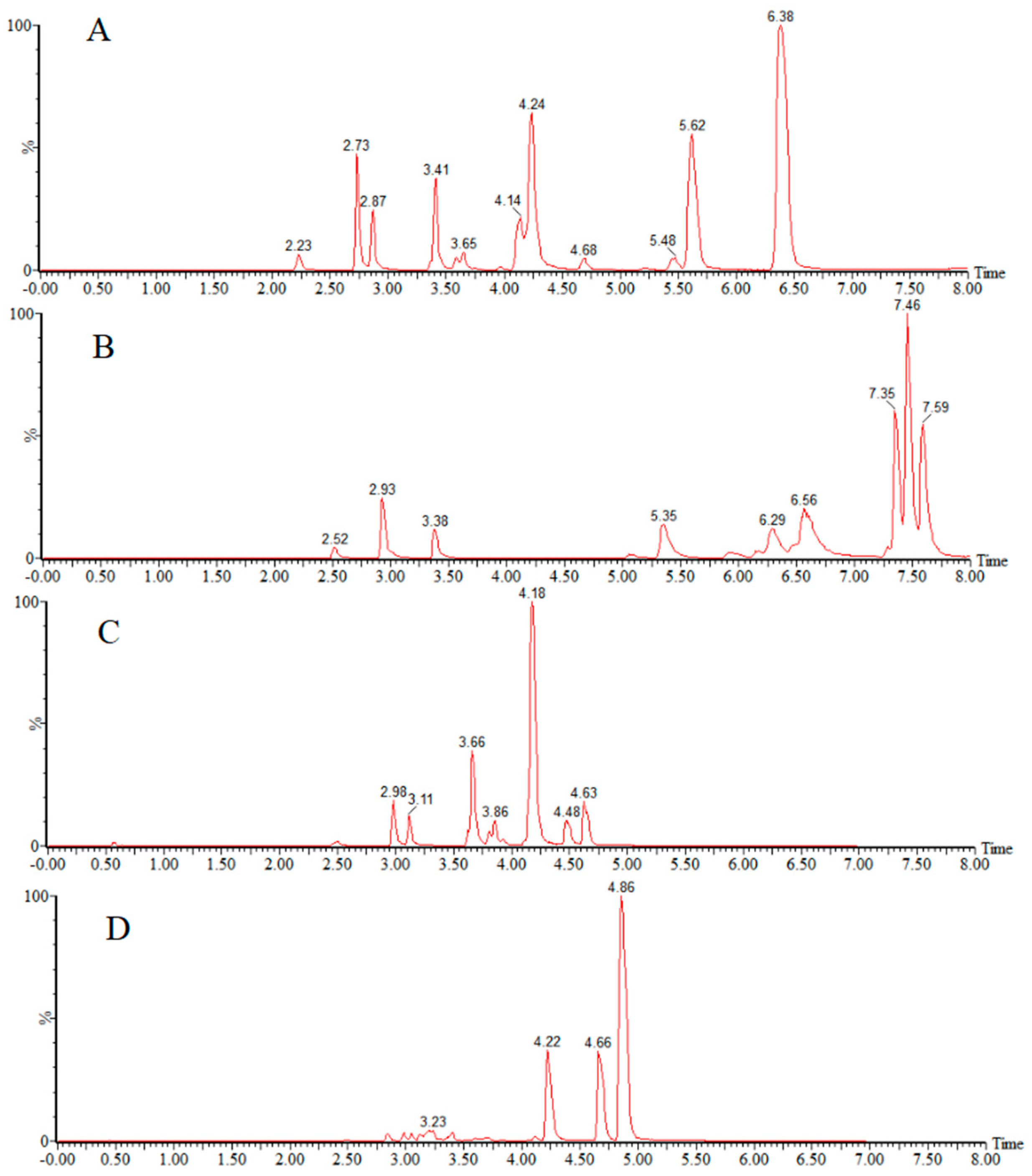
3.1. Optimization of UPLC–MS/MS
3.1.1. Mass Spectrometry Parameters
3.1.2. Chromatographic Conditions
3.2. Optimization of Sample Pretreatment
3.2.1. Extraction Solvents
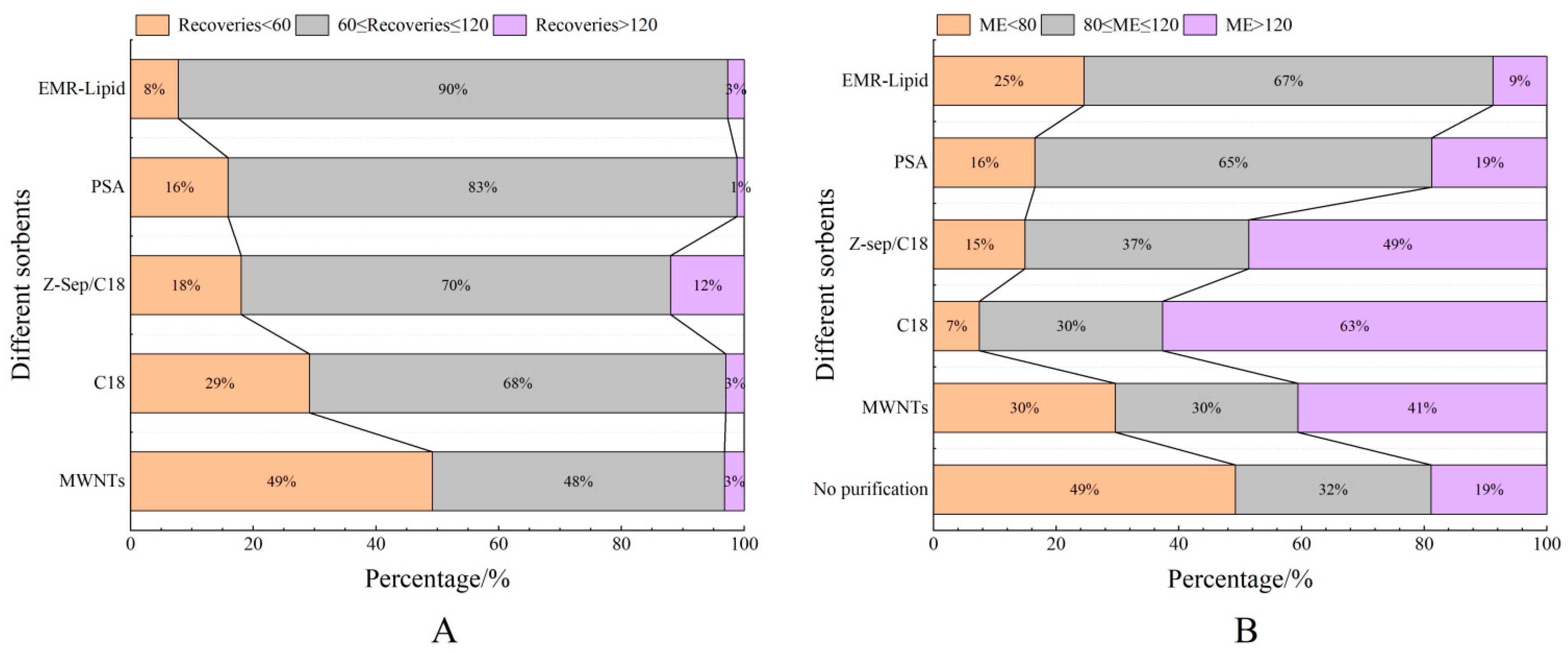
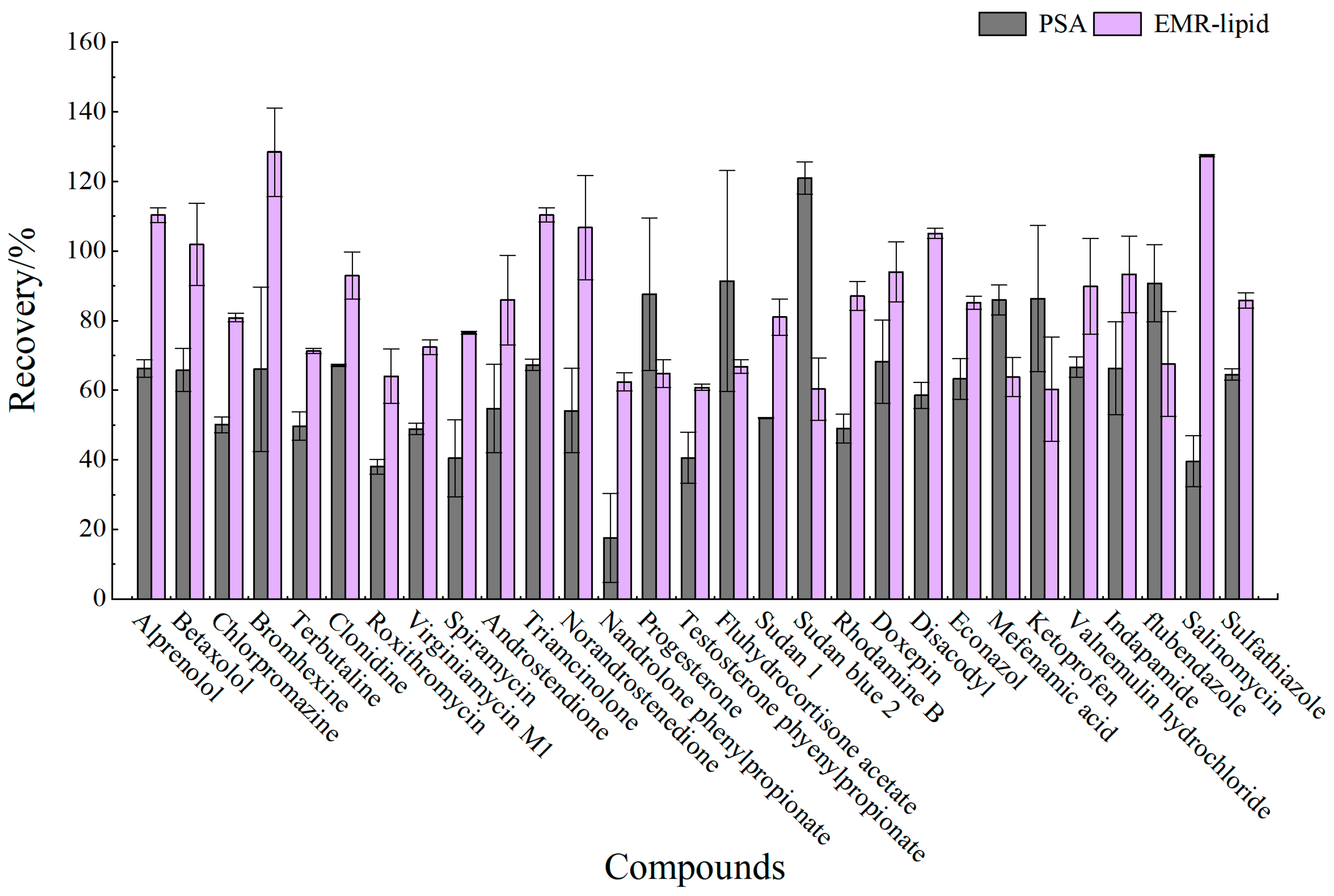
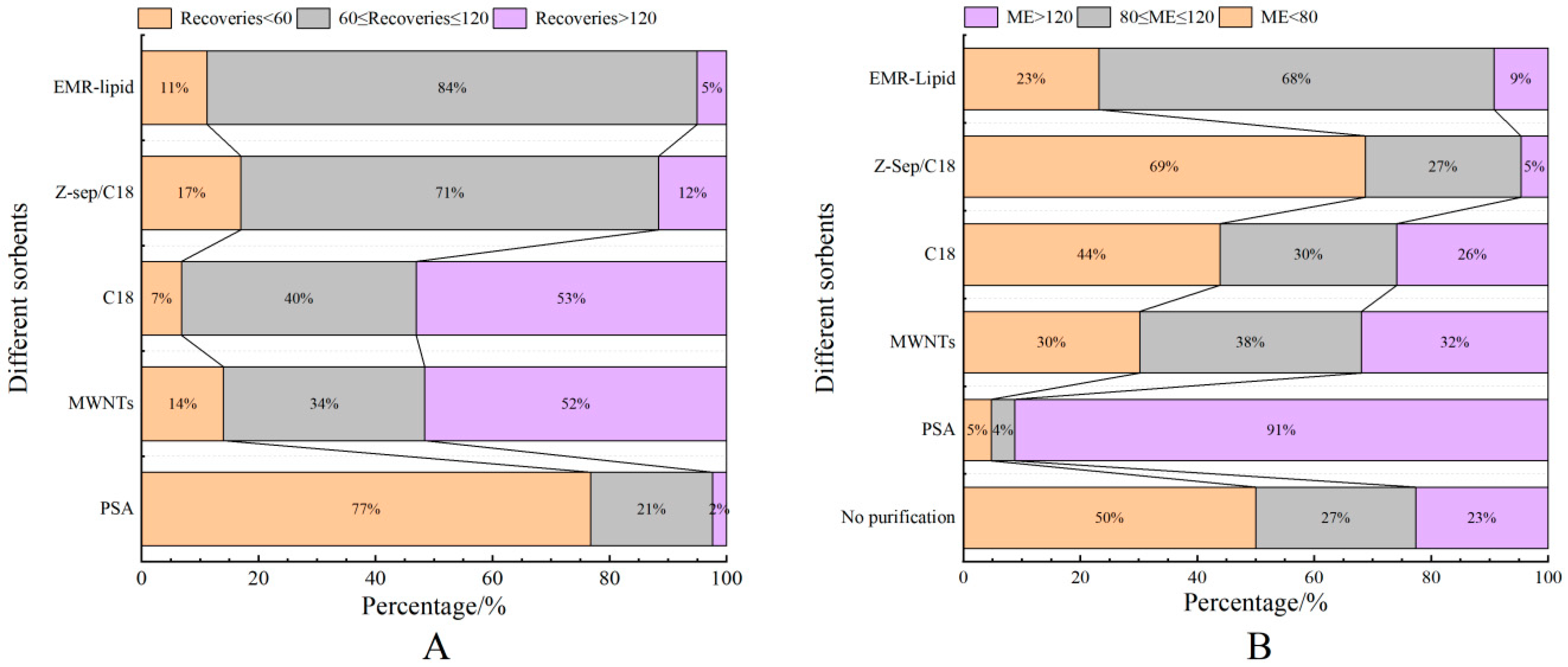
3.2.2. Purification Materials
3.3. Method Validation
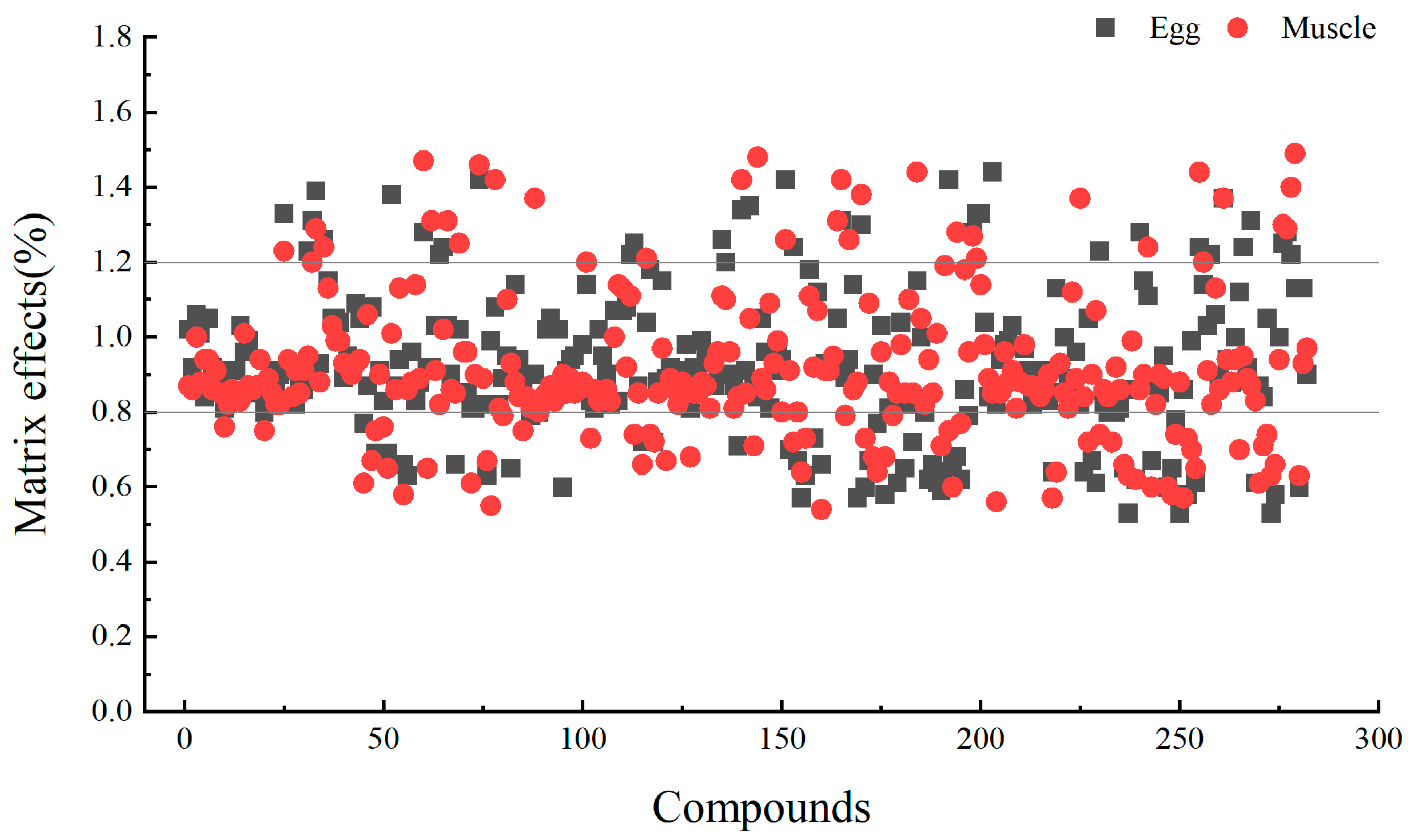
3.3.1. Matrix Effect
3.3.2. Linear Range and Method Limit of Quantification (LOQ)
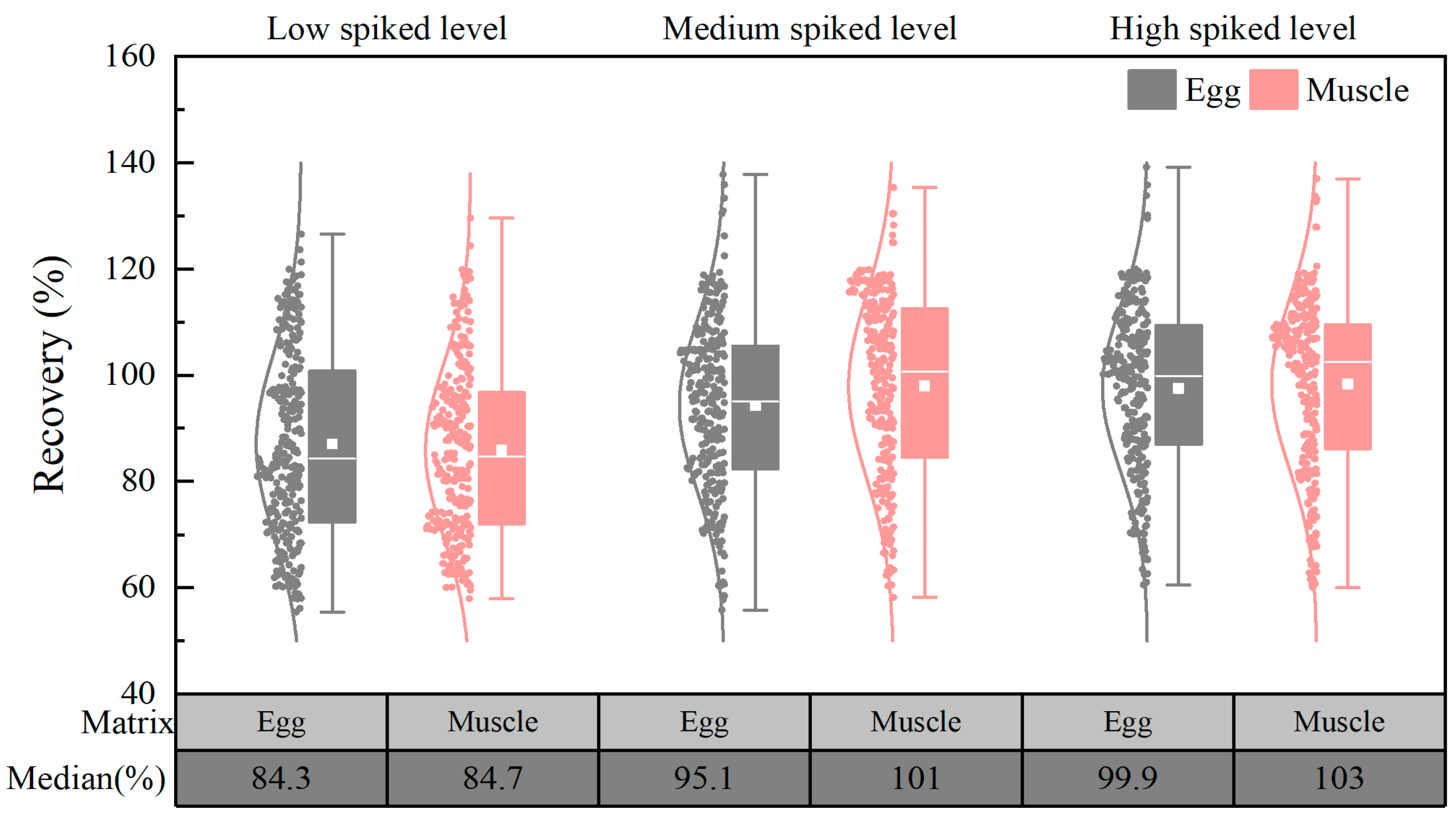
3.3.3. Spiking Recoveries and Precisions
3.4. Application
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- China Economic Net. Agricultural Economy Shows Steady and Positive Development in 2024. Available online: http://www.ce.cn/xwzx/gnsz/gdxw/202501/17/t20250117_39269913.shtml (accessed on 10 February 2025).
- Getahun, M.; Abebe, R.B.; Sendekie, A.K.; Woldeyohanis, A.E.; Kasahun, A.E. Evaluation of antibiotics residues in milk and meat using different analytical methods. Int. J. Anal. Chem. 2023, 2023, 4380261. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ying, G.-G.; Deng, W.-J. Antibiotic residues in food: Extraction, analysis, and human health concerns. J. Agric. Food Chem. 2019, 67, 7569–7586. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, E.; Chaurand, P.; Raghavan, V. Visualizing the distribution of strawberry plant metabolites at different maturity stages by MALDI-TOF imaging mass spectrometry. Food Chem. 2021, 345, 128838. [Google Scholar] [CrossRef] [PubMed]
- Manav, Ö.G.; Dinç-Zor, Ş.; Alpdoğan, G. Optimization of a modified QuEChERS method by means of experimental design for multiresidue determination of pesticides in milk and dairy products by GC-MS. Microchem. J. 2019, 144, 124–129. [Google Scholar] [CrossRef]
- Lozano, A.; Hernando, M.D.; Uclés, S.; Hakme, E.; Fernández-Alba, A.R. Identification and measurement of veterinary drug residues in beehive products. Food Chem. 2019, 274, 61–70. [Google Scholar] [CrossRef]
- Kang, J.; Park, S.J.; Park, H.C.; Hossain, M.A.; Kim, M.A.; Son, S.W.; Lim, C.M.; Kim, T.W.; Cho, B.H. Multiresidue screening of veterinary drugs in meat, milk, egg, and fish using liquid chromatography coupled with ion trap time-of-flight mass spectrometry. Appl. Biochem. Biotechnol. 2017, 182, 635–652. [Google Scholar] [CrossRef]
- Desmarchelier, A.; Fan, K.; Minh Tien, M.; Savoy, M.C.; Tarres, A.; Fuger, D.; Goyon, A.; Bessaire, T.; Mottier, P. Determination of 105 antibiotic, anti-inflammatory, antiparasitic agents and tranquilizers by LC-MS/MS based on an acidic QuEChERS-like extraction. Food Addit. Contam. Part A 2018, 35, 646–660. [Google Scholar] [CrossRef]
- Savoy, M.C.; Woo, P.M.; Ulrich, P.; Tarres, A.; Mottier, P.; Desmarchelier, A. Determination of 14 aminoglycosides by LC-MS/MS using molecularly imprinted polymer solid phase extraction for clean-up. Food Addit. Contam. Part A 2018, 35, 674–685. [Google Scholar] [CrossRef]
- Yu, X.; Wu, X.; Xie, Y.; Tong, K.; Wang, M.; Li, J.; Fan, C.; Chen, H. Development and validation of a method for determination of 43 antimicrobial drugs in western-style pork products by UPLC-MS/MS with the aid of experimental design. Molecules 2022, 27, 8206. [Google Scholar] [CrossRef]
- Mookantsa, S.O.S.; Dube, S.; Nindi, M.M. Multiclass determination of 87 mixed veterinary drugs, pesticides and mycotoxin residues in beef muscle samples by ionic liquid-based dispersive liquid-liquid microextraction and liquid chromatography tandem mass spectrometry. Foods 2025, 14, 720. [Google Scholar] [CrossRef]
- Fan, Z.; Li, Y.; Fan, X.; Wang, P.; Yang, R.; Xie, C. Simultaneous determination of three active forms of vitamin B12 in situ produced during fermentation by LC-MS/MS. Foods 2025, 14, 309. [Google Scholar] [CrossRef] [PubMed]
- Badawy, M.E.I.; El-Nouby, M.A.M.; Kimani, P.K.; Lim, L.W.; Rabea, E.I. A review of the modern principles and applications of solid-phase extraction techniques in chromatographic analysis. Anal. Sci. 2022, 38, 1457–1487. [Google Scholar] [CrossRef] [PubMed]
- Piatkowska, M.; Jedziniak, P.; Zmudzki, J. Multiresidue method for the simultaneous determination of veterinary medicinal products, feed additives and illegal dyes in eggs using liquid chromatography-tandem mass spectrometry. Food Chem. 2016, 197, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Xie, K.; Lee, K. Veterinary drug residues in animal-derived foods: Sample preparation and analytical methods. Foods 2021, 10, 127767. [Google Scholar] [CrossRef]
- Li, X.; Yu, H.; Peng, R.; Gan, P. Determination of 19 sulfonamides residues in pork samples by combining QuEChERS with dispersive liquid-liquid microextraction followed by UHPLC-MS/MS. J. Sep. Sci. 2017, 40, 1377–1384. [Google Scholar] [CrossRef]
- Zhou, J.; Xu, J.J.; Cong, J.M.; Cai, Z.X.; Zhang, J.S.; Wang, J.L.; Ren, Y.P. Optimization for quick, easy, cheap, effective, rugged and safe extraction of mycotoxins and veterinary drugs by response surface methodology for application to egg and milk. J. Chromatogr. A 2018, 1532, 20–29. [Google Scholar] [CrossRef]
- Peters, R.J.; Bolck, Y.J.; Rutgers, P.; Stolker, A.A.; Nielen, M.W. Multi-residue screening of veterinary drugs in egg, fish and meat using high-resolution liquid chromatography accurate mass time-of-flight mass spectrometry. J. Chromatogr. A 2009, 1216, 8206–8216. [Google Scholar] [CrossRef]
- Chen, D.; Tao, Y.; Liu, Z.; Liu, Z.; Wang, Y.; Huang, L.; Yuan, Z. Development of a liquid chromatography-tandem mass spectrometry (LC-MS/MS) method for the quantification of glucocorticoid residues in edible tissues of swine, cattle, sheep, and chicken. Food Addit. Contam. Part A 2010, 27, 1363–1371. [Google Scholar] [CrossRef]
- Jin, Q.; Xu, Q.; Zhao, Z.; Si, W.; Bai, B.; Chen, L.; Zhou, C. Simultaneous determination of six acidic herbicides and metabolites in plant origin matrices by QuEChERS-UPLC-MS/MS. Molecules 2025, 30, 852. [Google Scholar] [CrossRef]
- Huang, X.; Feng, R.; Hu, Q.; Mao, X.; Zhou, H. Contamination status and health risk assessment of 73 mycotoxins in four edible and medicinal plants using an optimized QuEChERS pretreatment coupled with LC-MS/MS. Toxins 2025, 17, 52. [Google Scholar] [CrossRef]
- Xu, X.; Xu, X.; Han, M.; Qiu, S.; Hou, X. Development of a modified QuEChERS method based on magnetic multiwalled carbon nanotubes for the simultaneous determination of veterinary drugs, pesticides and mycotoxins in eggs by UPLC-MS/MS. Food Chem. 2019, 276, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Rizzetti, T.M.; de Souza, M.P.; Prestes, O.D.; Adaime, M.B.; Zanella, R. Optimization of sample preparation by central composite design for multi-class determination of veterinary drugs in bovine muscle, kidney and liver by ultra-high-performance liquid chromatographic-tandem mass spectrometry. Food Chem. 2018, 246, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Liu, X.; Kong, F.; Tang, L.; Wang, Q.; Li, W.; Xu, W.; Wen, S.; Chen, L.; Li, Y. Multi-residue determination of 325 pesticides in chicken eggs with EMR-Lipid clean-up by UHPLC-MS/MS and GC-MS/MS. Chromatographia 2020, 83, 593–599. [Google Scholar] [CrossRef]
- GB/T 27417-2017; Conformity assessment—Guidance on Validation and Verification of Chemical Analytical Methods. Standardization Administration of China: Beijing, China, 2017.
- Zhu, X.; Wang, M.; Hou, F.; He, Y.; Zhu, Y.; Dai, H.; Huang, M.; Yang, Y.; Wu, L. Multi-residue analysis of pesticides and veterinary drugs by ultra-performance liquid chromatography-tandem mass spectrometry using a modified QuEChERS method. Microchem. J. 2025, 208, 112384. [Google Scholar] [CrossRef]
- Tao, Y.; Chen, D.; Yu, G.; Yu, H.; Pan, Y.; Wang, Y.; Huang, L.; Yuan, Z. Simultaneous determination of lincomycin and spectinomycin residues in animal tissues by gas chromatography-nitrogen phosphorus detection and gas chromatography-mass spectrometry with accelerated solvent extraction. Food Addit. Contam. Part A 2011, 28, 145–154. [Google Scholar] [CrossRef]
- Ahammed Shabeer, T.P.; Girame, R.; Utture, S.; Oulkar, D.; Banerjee, K.; Ajay, D.; Arimboor, R.; Menon, K.R.K. Optimization of multi-residue method for targeted screening and quantitation of 243 pesticide residues in cardamom (Elettaria cardamomum) by gas chromatography tandem mass spectrometry (GC-MS/MS) analysis. Chemosphere 2018, 193, 447–453. [Google Scholar] [CrossRef]
- Sun, D.; Jin, Y.; Zhao, Q.; Tang, C.; Li, Y.; Wang, H.; Qin, Y.; Zhang, J. Modified EMR-lipid method combined with HPLC-MS/MS to determine folates in egg yolks from laying hens supplemented with different amounts of folic acid. Food Chem. 2021, 337, 127767. [Google Scholar] [CrossRef]
- Long, Y.; Huang, Y.; Zhu, M.; Ma, Y.; Gan, B.; Wang, Y.; Yu, Q.; Xie, J.; Chen, Y. Development of QuEChERS clean-up based on EMR-lipid for simultaneous analysis of 9 mycotoxins, acrylamide and 5-hydroxymethylfurfural in biscuit by UHPLC-MS/MS. Food Chem. 2023, 409, 135265. [Google Scholar] [CrossRef]
- Chu, N.; Shu, X.; Yuan, L.; Zhang, X.; Tang, M.; Yang, J.; Li, D.; Wu, S. Determination of 52 hidden chemical pesticides in biopesticide products by GC-MS/MS and LC-MS/MS. J. Environ. Sci. Health B 2022, 57, 504–515. [Google Scholar] [CrossRef]
- Lin, Z.; Lin, Y.; Lin, J.; Zhang, Y.; Fang, S. Trace analysis of fenbutatin oxide in soil and plant- and animal-derived foods using modified QuEChERS coupled with HPLC-MS/MS. ACS Omega 2021, 6, 10260–10265. [Google Scholar] [CrossRef]
- Wu, Y.L.; Chen, R.X.; Xue, Y.; Yang, T.; Zhao, J.; Zhu, Y. Simultaneous determination of amantadine, rimantadine and memantine in chicken muscle using multi-walled carbon nanotubes as a reversed-dispersive solid phase extraction sorbent. J. Chromatogr. B 2014, 965, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Guo, Y.; Hu, X.; Yu, Y.; Chen, J.; Su, J.; Lian, W.; Wu, X.; Meng, X. Simultaneous detection of four pesticides in agricultural products by a modified QuEChERS method and LC-MS/MS. J. Environ. Sci. Health B 2023, 58, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Ly, T.K.; Ho, T.D.; Behra, P.; Nhu-Trang, T.T. Determination of 400 pesticide residues in green tea leaves by UPLC-MS/MS and GC-MS/MS combined with QuEChERS extraction and mixed-mode SPE clean-up method. Food Chem. 2020, 326, 126928. [Google Scholar] [CrossRef]
- Areo, O.M.; Olowoyo, J.O.; Sethoga, L.S.; Adebo, O.A.; Njobeh, P.B. Determination of pesticide residues in rooibos (Aspalathus linearis) teas in South Africa. Toxicol. Rep. 2022, 9, 852–857. [Google Scholar] [CrossRef]
- Sandín-España, P.; Mateo-Miranda, M.; López-Goti, C.; Seris-Barrallo, E.; Alonso-Prados, J.L. Analysis of pesticide residues by QuEChERS method and LC-MS/MS for a new extrapolation of maximum residue levels in persimmon minor crop. Molecules 2022, 27, 126928. [Google Scholar] [CrossRef]
- Zhang, L.; Jia, Q.; Liao, G.; Qian, Y.; Qiu, J. Multi-residue determination of 244 chemical contaminants in chicken eggs by liquid chromatography-tandem mass spectrometry after effective lipid clean-up. Agriculture 2022, 12, 869. [Google Scholar] [CrossRef]
- de Souza, Y.P.; de Oliveira, A.C.; Bastos, L.H.P.; Sartori, A.V.; Spisso, B.F. Development and validation of a miniaturized two-step neutral and acidified acetonitrile extraction method for simultaneous determination of pesticides, veterinary drugs, and mycotoxins in eggs by UHPLC-MS/MS. Food Anal. Methods 2025, 14, 720. [Google Scholar] [CrossRef]
- Luo, P.; Liu, X.; Kong, F.; Chen, L.; Wang, Q.; Li, W.; Wen, S.; Tang, L.; Li, Y. Simultaneous determination of 169 veterinary drugs in chicken eggs with EMR-Lipid clean-up using ultra-high performance liquid chromatography tandem mass spectrometry. Anal. Methods 2019, 11, 1657–1662. [Google Scholar] [CrossRef]
- Wang, C.; Li, X.; Yu, F.; Wang, Y.; Ye, D.; Hu, X.; Zhou, L.; Du, J.; Xia, X. Multi-class analysis of veterinary drugs in eggs using dispersive-solid phase extraction and ultra-high performance liquid chromatography-tandem mass spectrometry. Food Chem. 2021, 334, 127598. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, W.; Guo, W.; Li, Y.; Jiang, R.; Li, H.; Wang, S.; Li, Z. Simultaneous screening and analysis of 155 veterinary drugs in livestock foods using ultra-high performance liquid chromatography tandem quadrupole linear-ion-trap mass spectrometry. Food Chem. 2022, 393, 133260. [Google Scholar] [CrossRef]
- Chen, D.; Yu, J.; Tao, Y.; Pan, Y.; Xie, S.; Huang, L.; Peng, D.; Wang, X.; Wang, Y.; Liu, Z.; et al. Qualitative screening of veterinary anti-microbial agents in tissues, milk, and eggs of food-producing animals using liquid chromatography coupled with tandem mass spectrometry. J. Chromatogr. B 2016, 1017–1018, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, H.F.; Sharkawy, A.A. Hormonal residues in chicken and cattle meat: A risk threat the present and future consumer health. Food Chem. Toxicol. 2023, 182, 114172. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrix | Compound | MDL(μg/kg) | Detection Technique | Column | Time Cycle (min) | Pretreatment | Recovery (%) | References |
|---|---|---|---|---|---|---|---|---|
| Eggs | 244 chemical contaminants | (LOQ) ≤ 5 | LC–MS/M | Zorbax Eclipse XDB-C18 | 25.1 | Extraction with 5% formic acid in acetonitrile; purification: EMR-lipid | 51.33–118.22 | [38] |
| Eggs | 285 compounds (pesticides, veterinary drugs, and mycotoxins) | LOQ: 0.1–50 | UHPLC–MS/MS | BEH C18 | 25 | Extraction with 1% formic acid in acetonitrile | Satisfactory recoveries (70 to 120) | [39] |
| Chicken eggs | 169 veterinary drugs | LOD: 0.01–3.81 | UHPLC–MS/MS | Eclipse Plus C18 | 24 | Extraction with ethyl formate–acetonitrile; purification: EMR-lipid dSPE | 57–124 | [40] |
| Eggs | 78 veterinary drugs | LOQ: 0.1–1 | UHPLC–MS/MS | Acquity UPLC BEH C18 | 9.5 | Extraction with acetonitrile containing 15% formic acid (v/v); purification by MCX sorbent (mixed-mode cation-eXchange sorbent) | 70.5–119.2 | [41] |
| Eggs | 74 veterinary drugs | LOQ: 0.1–17.3 | UPLC–MS/MS | Shimpack XR-ODS III column | 16 | Extraction with 1% acetic acid in acetonitrile; clean-up with Fe3O4-MWCNTs (magnetic multi-walled carbon nanotubes) | 60.5–114.6 | [22] |
| Livestock foods | 155 veterinary drugs | LOD: 0.5–5 | UHPLC–QTRAP–MS | Eclipse Plus C18 | 22 | Extraction with acetonitrile/water/formic acid mixture (80:19.8:0.2); SPE (PRiME HLB) | 46.4–120 | [42] |
| Animal-derived food | 120 drugs | LOD: 0.5–3.0 | LC–MS/MS | Hypersil Gold C18 | 60 | Extraction with 90% ACN (v/v); SPE (HLB) | / | [43] |
| Chicken muscle eggs | 280 chemical hazards | LOD: 0.05–10; LOQ: 0.1–20 | UHPLC–MS/MS | ACQUITY UPLC BEH C18 | 40 | Extraction with 1% acetic acid in 85% acetonitrile; Purification: EMR-lipid | 55.4–139 | This study |
| Compounds | Poultry Eggs (n = 144) | Poultry Muscle (n = 67) | ||||||
|---|---|---|---|---|---|---|---|---|
| Number of Detected | Maximum (μg/kg) | Minimum (μg/kg) | Average (μg/kg) * | Number of Detected | Maximum (μg/kg) | Minimum (μg/kg) | Average (μg/kg) * | |
| Trimethoprim | 6 | 292 | 1.75 | 86.4 | _ | _ | _ | _ |
| Sulfadiazine | 3 | 638 | 0.67 | 401 | 2 | 1.4 | 1.19 | 1.29 |
| Sulfamethizole | _ | _ | _ | _ | 2 | 5.77 | 1.33 | _ |
| Sulfamonomethoxine | 5 | 0.97 | 0.62 | 0.86 | 3 | 2.18 | 1.81 | 2.06 |
| Sulfathinoxaline | 1 | 15.9 | _ | _ | 1 | 1.69 | _ | _ |
| Sulfadimidine | _ | _ | _ | _ | 2 | 8.37 | 2.15 | _ |
| Sulfaphenazole | _ | _ | _ | _ | 1 | 1.16 | _ | _ |
| Sulfameter | 1 | 3.95 | _ | _ | _ | _ | _ | _ |
| Sulfisomidine | 1 | 0.62 | _ | _ | _ | _ | _ | _ |
| Nalidixic acid | 2 | 1.09 | 0.48 | _ | 3 | 21.4 | 8.65 | 16.8 |
| Oxolinic acid | _ | _ | _ | _ | 4 | 2.07 | 0.58 | 0.87 |
| Ofloxacin | 1 | 0.82 | _ | _ | _ | _ | _ | _ |
| Enrofloxacin | 5 | 1.96 | 0.35 | 0.65 | _ | _ | _ | _ |
| Pazufloxacin | 1 | 1.77 | _ | _ | _ | _ | _ | _ |
| Ciprofloxacin | 1 | 2.92 | _ | _ | _ | _ | _ | _ |
| Salinomycin | _ | _ | _ | _ | 2 | 0.77 | 0.77 | _ |
| Maduramicin | 3 | 0.65 | 0.33 | 0.49 | _ | _ | _ | _ |
| Guanabenz | 2 | 13 | 2 | _ | 3 | 21.9 | 1.37 | 14.4 |
| Nicarbazin | 3 | 17.7 | 0.14 | 6.96 | 5 | 6.64 | 1.2 | 3.92 |
| Diclazuril | 8 | 8.56 | 0.46 | 3.15 | 5 | 55 | 1.22 | 16.3 |
| Toltrazuril | _ | _ | _ | _ | 1 | 333 | _ | _ |
| Azithromycin | _ | _ | _ | _ | 2 | 56.1 | 24.5 | _ |
| Metronidazole | 1 | 19.1 | _ | _ | _ | _ | _ | _ |
| Ronidazole | 1 | 2.13 | _ | _ | _ | _ | _ | _ |
| Flubendazole | 7 | 0.56 | 0.11 | 0.28 | _ | _ | _ | _ |
| Albendazole sulfoxide | 2 | 1.79 | 0.88 | _ | _ | _ | _ | _ |
| Fenbendazole sulfone | 1 | 2.28 | _ | _ | _ | _ | _ | _ |
| Florfenicol | 3 | 115 | 3 | 59.4 | _ | _ | _ | _ |
| Griseofulvin | _ | _ | _ | _ | 1 | 0.59 | _ | _ |
| Isoxsuprine | 1 | 0.91 | _ | _ | 1 | 0.11 | _ | _ |
| Chlorpromazine | 2 | 0.33 | 0.24 | _ | _ | _ | _ | _ |
| Cortisone | _ | _ | _ | _ | 34 | 23.4 | 1.02 | 7.62 |
| Hydrocortisone | _ | _ | _ | _ | 8 | 39.6 | 8.52 | 22.7 |
| Betamethasone | _ | _ | _ | _ | 2 | 0.91 | 0.55 | _ |
| Progesterone | 89 | 211 | 3.53 | 54.5 | 13 | 38.6 | 2.22 | 9.4 |
| Androstendione | 29 | 7.65 | 0.11 | 3.42 | _ | _ | _ | _ |
| Corticosterone | _ | _ | _ | _ | 33 | 8.08 | 0.79 | 3.15 |
| Testosterone | _ | _ | _ | _ | 1 | 2.86 | _ | _ |
| Basic violet 1 | _ | _ | _ | _ | 1 | 0.59 | _ | _ |
| Rhodamine B | 2 | 0.75 | 0.12 | _ | _ | _ | _ | _ |
| 4-formylaminoantipyrine | 1 | 2.2 | _ | _ | _ | _ | _ | _ |
| Chlorpheniramine | 4 | 1.62 | 0.72 | 1.17 | _ | _ | _ | _ |
| Clomipramine | 1 | 0.36 | _ | _ | _ | _ | _ | _ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, R.; Chen, L.; Du, M.; Guo, Q.; Zhong, C.; Zhang, J.; Yu, X. High-Throughput Determination of Multiclass Chemical Hazards in Poultry Muscles and Eggs Using UPLC–MS/MS. Foods 2025, 14, 1660. https://doi.org/10.3390/foods14101660
Chen R, Chen L, Du M, Guo Q, Zhong C, Zhang J, Yu X. High-Throughput Determination of Multiclass Chemical Hazards in Poultry Muscles and Eggs Using UPLC–MS/MS. Foods. 2025; 14(10):1660. https://doi.org/10.3390/foods14101660
Chicago/Turabian StyleChen, Rong, Lan Chen, Mingyue Du, Qiaozhen Guo, Ciping Zhong, Jing Zhang, and Xiaoqin Yu. 2025. "High-Throughput Determination of Multiclass Chemical Hazards in Poultry Muscles and Eggs Using UPLC–MS/MS" Foods 14, no. 10: 1660. https://doi.org/10.3390/foods14101660
APA StyleChen, R., Chen, L., Du, M., Guo, Q., Zhong, C., Zhang, J., & Yu, X. (2025). High-Throughput Determination of Multiclass Chemical Hazards in Poultry Muscles and Eggs Using UPLC–MS/MS. Foods, 14(10), 1660. https://doi.org/10.3390/foods14101660