
Rosemarinic Acid-Induced Destabilization of Aβ Peptides: Insights from Molecular Dynamics Simulations

 ,
, 
 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Molecular Docking Analysis
2.2. MD Analysis
2.3. Analytical Methods of Silico Simulation
2.4. MM-PBSA Analysis
2.5. Free Energy Landscape (FEL)
3. Results and Discussions
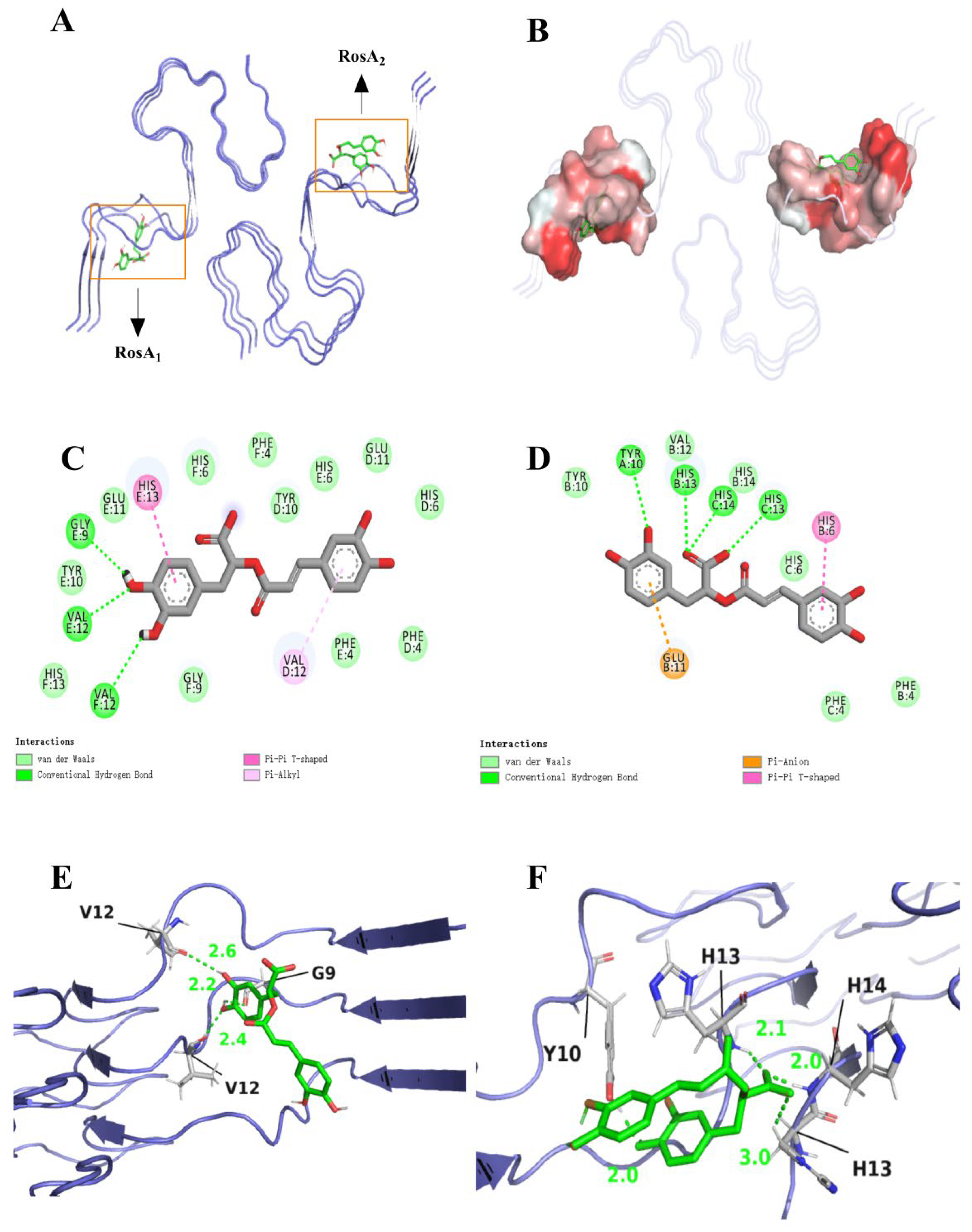
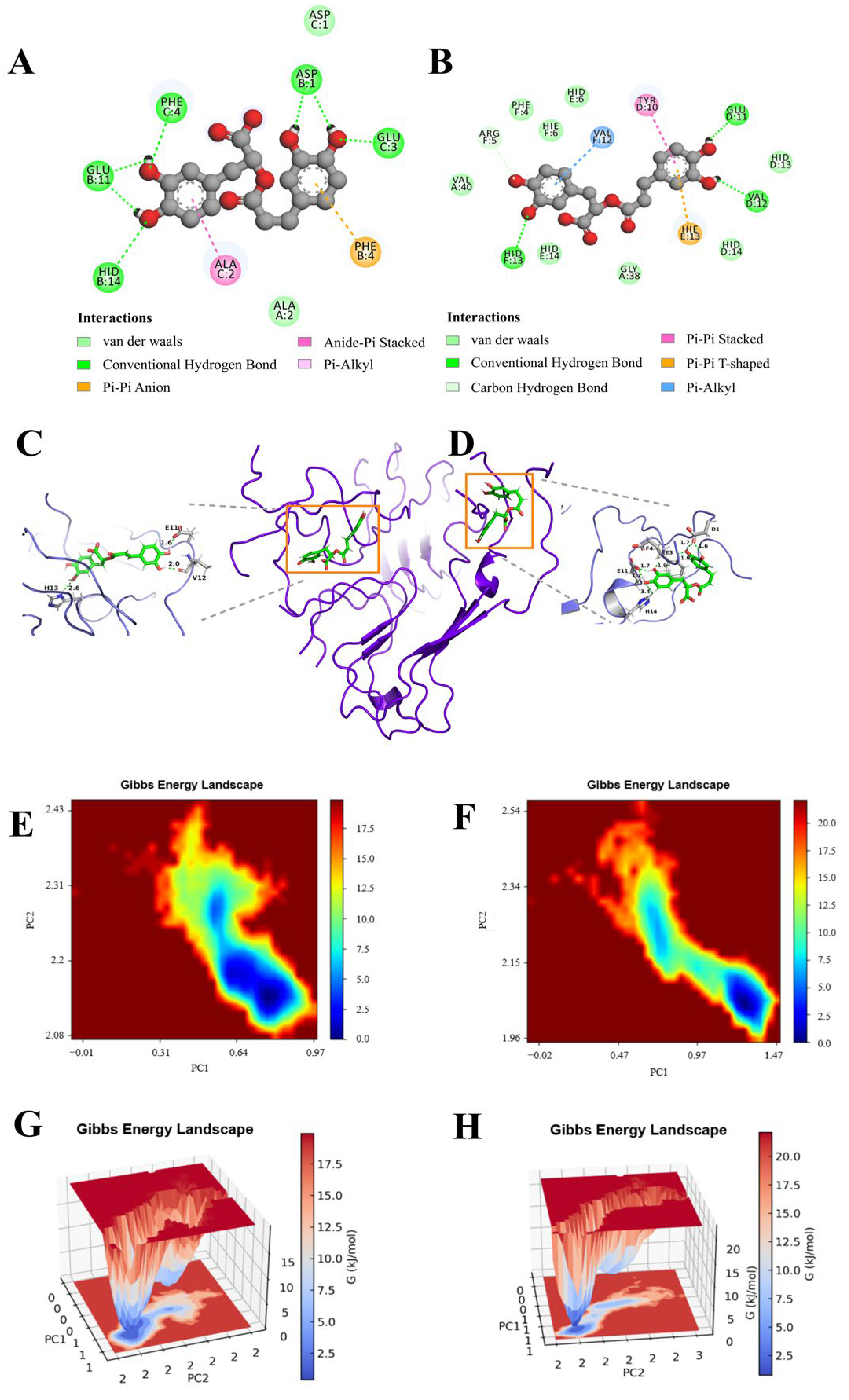
3.1. Molecular Interaction and Binding Mode
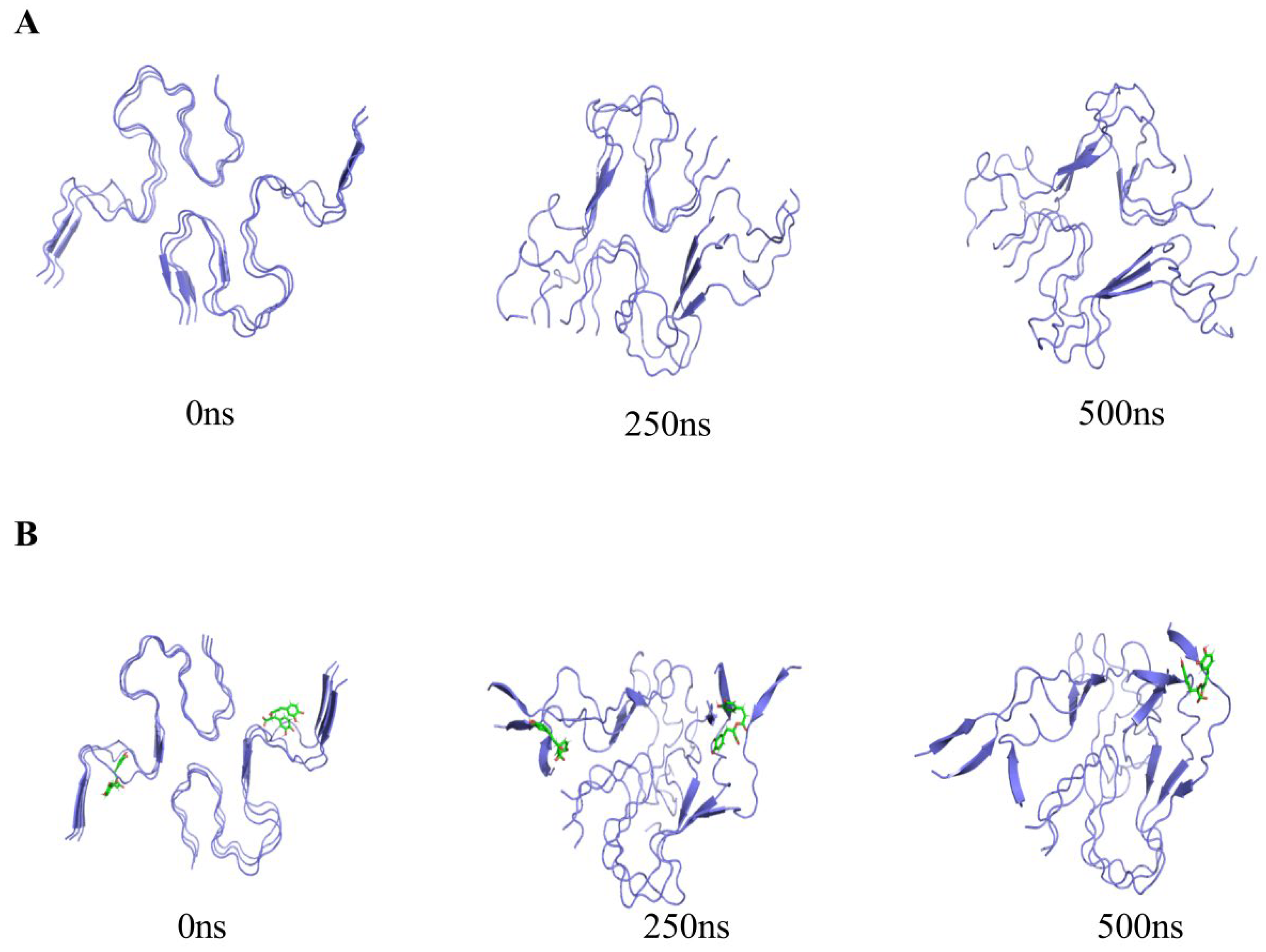
3.2. Trajectory Processing and Inspection
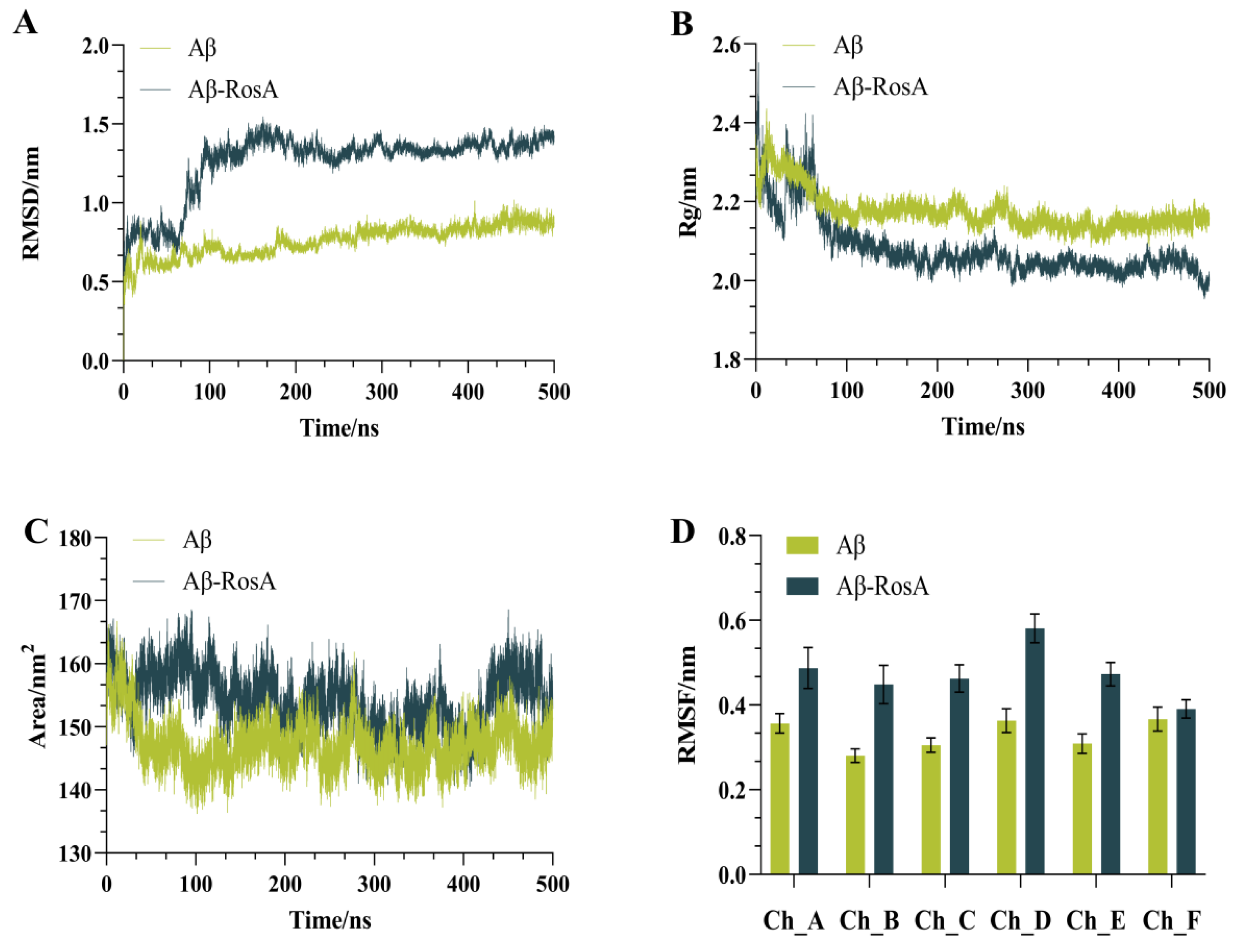
3.3. Stability Analysis
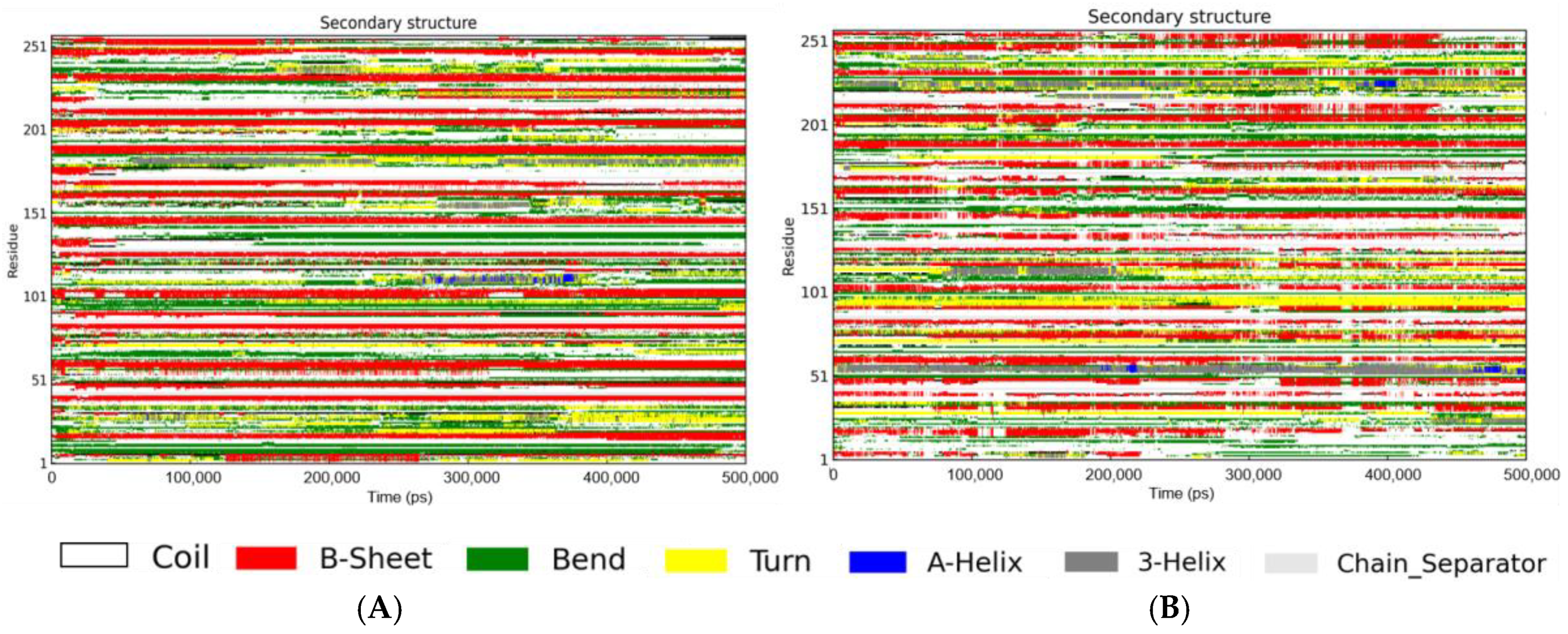
3.4. Secondary Structure in the Presence of RosA
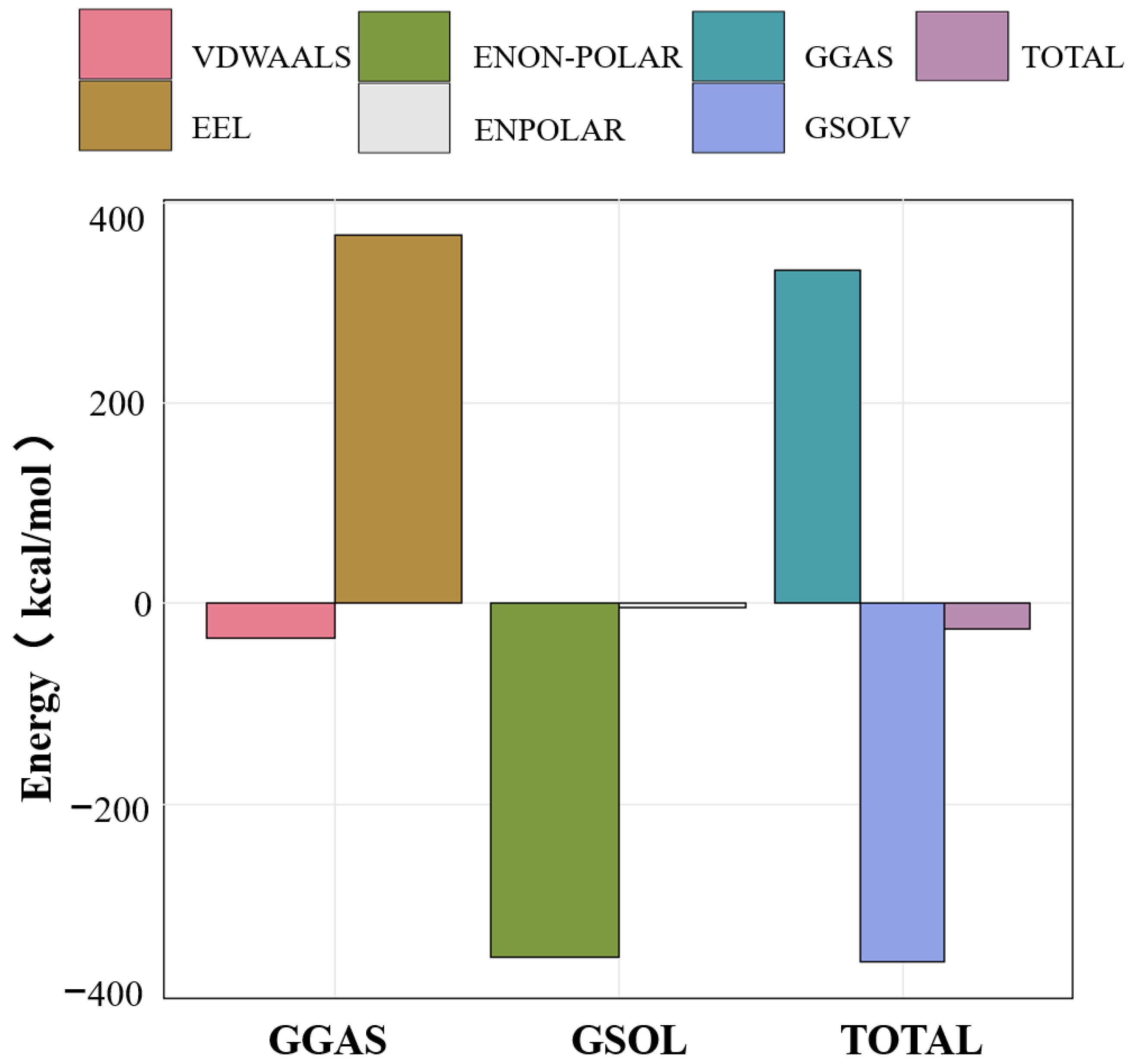
3.5. MMPBSA Binding Free Energy Analysis
3.6. Conformational Changes and Force Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, F.; Chen, W.; Dabbour, M.; Kumah Mintah, B.; Xu, H.; Pan, J.; Dai, C.; Ma, H.; He, R. Preparation of housefly (Musca domestica) larvae protein hydrolysates: Influence of dual-sweeping-frequency ultrasound-assisted enzymatic hydrolysis on yield, antioxidative activity, functional and structural attributes. Food Chem. 2024, 440, 138253. [Google Scholar] [CrossRef]
- Khabiya, R.; Karati, D.; Dwivedi, S.; Dwivedi, A.; Mukherjee, S. The promising role of bioactive congeners present in Cassytha filiformis in Alzheimer’s disease: An explicative review. Brain Disord. 2024, 13, 100125. [Google Scholar] [CrossRef]
- 2022 Alzheimer’s disease facts and figures. Alzheimers Dement. 2022, 18, 700–789. [CrossRef]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Rauk, A. The chemistry of Alzheimer’s disease. Chem. Soc. Rev. 2009, 38, 2698–2715. [Google Scholar] [CrossRef]
- Hayne, D.J.; Lim, S.; Donnelly, P.S. Metal complexes designed to bind to amyloid-β for the diagnosis and treatment of Alzheimer’s disease. Chem. Soc. Rev. 2014, 43, 6701–6715. [Google Scholar] [CrossRef]
- Giuffrida, M.L.; Caraci, F.; Pignataro, B.; Cataldo, S.; De Bona, P.; Bruno, V.; Molinaro, G.; Pappalardo, G.; Messina, A.; Palmigiano, A.; et al. β-amyloid monomers are neuroprotective. J. Neurosci. 2009, 29, 10582–10587. [Google Scholar] [CrossRef]
- Matuszyk, M.M.; Garwood, C.J.; Ferraiuolo, L.; Simpson, J.E.; Staniforth, R.A.; Wharton, S.B. Biological and methodological complexities of beta-amyloid peptide: Implications for Alzheimer’s disease research. J. Neurochem. 2022, 160, 434–453. [Google Scholar] [CrossRef] [PubMed]
- Chopra, B.; Dhingra, A.K. Natural products: A lead for drug discovery and development. Phytother. Res. 2021, 35, 4660–4702. [Google Scholar] [CrossRef]
- Praticò, D.; Delanty, N. Oxidative injury in diseases of the central nervous system: Focus on Alzheimer’s disease. Am. J. Med. 2000, 109, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Rottkamp, C.A.; Nunomura, A.; Raina, A.K.; Sayre, L.M.; Perry, G.; Smith, M.A. Oxidative Stress, Antioxidants, and Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2000, 14, S62–S66. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Rottkamp, C.A.; Nunomura, A.; Raina, A.K.; Perry, G. Oxidative stress in Alzheimer’s disease. Biochim. Biophys. Acta 2000, 1502, 139–144. [Google Scholar] [CrossRef]
- Yatin, S.M.; Yatin, M.; Aulick, T.; Ain, K.B.; Butterfield, D.A. Alzheimer’s amyloid β-peptide associated free radicals increase rat embryonic neuronal polyamine uptake and ornithine decarboxylase activity: Protective effect of vitamin E. Neurosci. Lett. 1999, 263, 17–20. [Google Scholar] [CrossRef]
- Perrig, W.J.; Perrig, P.; Stähelin, H.B. The relation between antioxidants and memory performance in the old and very old. J. Am. Geriatr. Soc. 1997, 45, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Schaffner, W.; Hertel, C. Phytoestrogen kaempferol (3,4′,5,7-tetrahydroxyflavone) protects PC12 and T47D cells from β-amyloid-induced toxicity. J. Neurosci. Res. 1999, 57, 399–404. [Google Scholar] [CrossRef]
- Kihara, T.; Shimohama, S.; Sawada, H.; Kimura, J.; Kume, T.; Kochiyama, H.; Maeda, T.; Akaike, A. Nicotinic receptor stimulation protects neurons against β-amyloid toxicity. Ann. Neurol. 1997, 42, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Mountaki, C.; Dafnis, I.; Panagopoulou, E.A.; Vasilakopoulou, P.B.; Karvelas, M.; Chiou, A.; Karathanos, V.T.; Chroni, A. Mechanistic insight into the capacity of natural polar phenolic compounds to abolish Alzheimer’s disease-associated pathogenic effects of apoE4 forms. Free Radic. Biol. Med. 2021, 171, 284–301. [Google Scholar] [CrossRef]
- Mohammad-Beigi, H.; Aliakbari, F.; Sahin, C.; Lomax, C.; Tawfike, A.; Schafer, N.P.; Amiri-Nowdijeh, A.; Eskandari, H.; Møller, I.M.; Hosseini-Mazinani, M.; et al. Oleuropein derivatives from olive fruit extracts reduce α-synuclein fibrillation and oligomer toxicity. J. Biol. Chem. 2019, 294, 4215–4232. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.; Abdullah, Y.; Benner, J.; Eberle, D.; Gehlen, K.; Hücherig, S.; Janiak, V.; Kim, K.H.; Sander, M.; Weitzel, C.; et al. Evolution of rosmarinic acid biosynthesis. Phytochemistry 2009, 70, 1663–1679. [Google Scholar] [CrossRef]
- Nunes, S.; Madureira, A.R.; Campos, D.; Sarmento, B.; Gomes, A.M.; Pintado, M.; Reis, F. Therapeutic and nutraceutical potential of rosmarinic acid—Cytoprotective properties and pharmacokinetic profile. Crit. Rev. Food Sci. Nutr. 2017, 57, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, D.B.; Castro, I.; Lopes-Rodrigues, V.; Pereira, J.M.; Barros, L.; Ferreira, I.; Xavier, C.P.R.; Vasconcelos, M.H. Melissa officinalis L. ethanolic extract inhibits the growth of a lung cancer cell line by interfering with the cell cycle and inducing apoptosis. Food Funct. 2018, 9, 3134–3142. [Google Scholar] [CrossRef]
- Noor, S.; Mohammad, T.; Rub, M.A.; Raza, A.; Azum, N.; Yadav, D.K.; Hassan, M.I.; Asiri, A.M. Biomedical features and therapeutic potential of rosmarinic acid. Arch. Pharm. Res. 2022, 45, 205–228. [Google Scholar] [CrossRef]
- Rollo, J.L.; Banihashemi, N.; Vafaee, F.; Crawford, J.W.; Kuncic, Z.; Holsinger, R.M. Unraveling the mechanistic complexity of Alzheimer’s disease through systems biology. Alzheimers Dement. 2016, 12, 708–718. [Google Scholar] [CrossRef]
- Vieira, T.F.; Sousa, S.F. Comparing AutoDock and Vina in Ligand/Decoy Discrimination for Virtual Screening. Appl. Sci. 2019, 9, 4538. [Google Scholar] [CrossRef]
- Tavan, M.; Hanachi, P.; de la Luz Cádiz-Gurrea, M.; Segura Carretero, A.; Mirjalili, M.H. Natural Phenolic Compounds with Neuroprotective Effects. Neurochem. Res. 2024, 49, 306–326. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Pan, F.; Wang, O.; Zhao, L.; Zhao, L. Antibacterial Effect of Sesame Protein-Derived Peptides against Escherichia coli and Staphylococcus aureus: In Silico and In Vitro Analysis. Nutrients 2024, 16, 175. [Google Scholar] [CrossRef] [PubMed]
- Zargari, F.; Lotfi, M.; Shahraki, O.; Nikfarjam, Z.; Shahraki, J. Flavonoids as potent allosteric inhibitors of protein tyrosine phosphatase 1B: Molecular dynamics simulation and free energy calculation. J. Biomol. Struct. Dyn. 2018, 36, 4126–4142. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Pan, F.; Zhao, L.; Zhao, S.; Yi, J.; Cai, S. Interfering effects on the bioactivities of several key proteins of COVID-19/variants in diabetes by compounds from Lianqiao leaves: In silico and in vitro analyses. Int. J. Biol. Macromol. 2022, 207, 715–729. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yin, B.; Wang, W.; Sun, H. Effects of Disulfide Bonds on Binding of Inhibitors to β-Amyloid Cleaving Enzyme 1 Decoded by Multiple Replica Accelerated Molecular Dynamics Simulations. ACS Chem. Neurosci. 2020, 11, 1811–1826. [Google Scholar] [CrossRef]
- Zarezade, V.; Rezaei, H.; Shakerinezhad, G.; Safavi, A.; Nazeri, Z.; Veisi, A.; Azadbakht, O.; Hatami, M.; Sabaghan, M.; Shajirat, Z. The identification of novel inhibitors of human angiotensin-converting enzyme 2 and main protease of Sars-Cov-2: A combination of in silico methods for treatment of COVID-19. J. Mol. Struct. 2021, 1237, 130409. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yu, N.; Zhao, X.; Quan, W.; Shu, M. 3D-QSAR, molecular docking, and molecular dynamics analysis of dihydrodiazaindolone derivatives as PARP-1 inhibitors. J. Mol. Model. 2023, 29, 131. [Google Scholar] [CrossRef] [PubMed]
- Reena Kumari, V.K.P.D.; Vikram, D. Promising antivirals for PLpro of SARS-CoV-2 using virtual screening, molecular docking, dynamics, and MMPBSA. J. Biomol. Struct. Dyn. 2023, 41, 4650–4666. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.Y.; Wang, L.; Ge, H.; Lu, X.L.; Pei, Z.; Gu, Q.; Xu, J. Salvianolic acid A, a polyphenolic derivative from Salvia miltiorrhiza bunge, as a multifunctional agent for the treatment of Alzheimer’s disease. Mol. Divers. 2013, 17, 515–524. [Google Scholar] [CrossRef]
- Dong, Q.; Cui, Z.; Wu, X.; Li, L.; Lu, F.; Liu, F. Natural flavonoid hesperetin blocks amyloid β-protein fibrillogenesis, depolymerizes preformed fibrils and alleviates cytotoxicity caused by amyloids. Food Funct. 2024, 15, 4233–4245. [Google Scholar] [CrossRef]
- Ono, K.; Li, L.; Takamura, Y.; Yoshiike, Y.; Zhu, L.; Han, F.; Mao, X.; Ikeda, T.; Takasaki, J.-i.; Nishijo, H.; et al. Phenolic Compounds Prevent Amyloid β-Protein Oligomerization and Synaptic Dysfunction by Site-specific Binding. J. Biol. Chem. 2012, 287, 14631–14643. [Google Scholar] [CrossRef]
- El Khatabi, K.; El-Mernissi, R.; Aanouz, I.; Ajana, M.A.; Lakhlifi, T.; Khan, A.; Wei, D.-Q.; Bouachrine, M. Identification of novel acetylcholinesterase inhibitors through 3D-QSAR, molecular docking, and molecular dynamics simulation targeting Alzheimer’s disease. J. Mol. Model. 2021, 27, 302. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.G.; Okumura, H. Promotion and Inhibition of Amyloid-β Peptide Aggregation: Molecular Dynamics Studies. Int. J. Mol. Sci. 2021, 22, 1859. [Google Scholar] [CrossRef] [PubMed]
- Lakey-Beitia, J.; Kumar, D.J.; Hegde, M.L.; Rao, K.S. Carotenoids as Novel Therapeutic Molecules Against Neurodegenerative Disorders: Chemistry and Molecular Docking Analysis. Int. J. Mol. Sci. 2019, 20, 5553. [Google Scholar] [CrossRef] [PubMed]
- Ouassaf, M.; Belaidi, S.; Chtita, S.; Lanez, T.; Abul Qais, F.; Md Amiruddin, H. Combined molecular docking and dynamics simulations studies of natural compounds as potent inhibitors against SARS-CoV-2 main protease. J. Biomol. Struct. Dyn. 2022, 40, 11264–11273. [Google Scholar] [CrossRef]
- Bhattacharya, K.; Bhattacharjee, A.; Chakraborty, M.; Das, D. Molecular Dynamics and Binding Energetics of Fluspirilene with BACE1: Implications for Alzheimer’s Disease Intervention. Pept. Sci. 2024, 116, e24349. [Google Scholar] [CrossRef]
- Shahwan, M.; Khan, M.S.; Husain, F.M.; Shamsi, A. Understanding binding between donepezil and human ferritin: Molecular docking and molecular dynamics simulation approach. J. Biomol. Struct. Dyn. 2022, 40, 3871–3879. [Google Scholar] [CrossRef] [PubMed]
- Wälti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Böckmann, A.; Güntert, P.; Meier, B.H.; Riek, R. Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. USA 2016, 113, E4976–E4984. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.J.; Garst, J.E.; Linnabary, R.D.; Channell, R.B. Perilla ketone: A potent lung toxin from the mint plant, Perilla frutescens Britton. Science 1977, 197, 573–574. [Google Scholar] [CrossRef]
- Fawzi, N.L.; Kohlstedt, K.L.; Okabe, Y.; Head-Gordon, T. Protofibril assemblies of the arctic, Dutch, and Flemish mutants of the Alzheimer’s Aβ1–40 peptide. Biophys. J. 2008, 94, 2007–2016. [Google Scholar] [CrossRef]
- Tallei, T.E.; Tumilaar, S.G.; Niode, N.J.; Fatimawali; Kepel, B.J.; Idroes, R.; Effendi, Y.; Sakib, S.A.; Emran, T.B. Potential of Plant Bioactive Compounds as SARS-CoV-2 Main Protease (Mpro) and Spike (S) Glycoprotein Inhibitors: A Molecular Docking Study. Scientifica 2020, 2020, 6307457. [Google Scholar] [CrossRef] [PubMed]
- Cosconati, S.; Forli, S.; Perryman, A.L.; Harris, R.; Goodsell, D.S.; Olson, A.J. Virtual Screening with AutoDock: Theory and Practice. Expert Opin. Drug Discov. 2010, 5, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Thakur, M.; Singh, S.K.; Sharma, L.K.; Chandra, K. Exploring the effect of nsSNPs in human YPEL3 gene in cellular senescence. Sci. Rep. 2020, 10, 15301. [Google Scholar] [CrossRef]
- Aris, P.; Mohamadzadeh, M.; Zarei, M.; Xia, X. Computational Design of Novel Griseofulvin Derivatives Demonstrating Potential Antibacterial Activity: Insights from Molecular Docking and Molecular Dynamics Simulation. Int. J. Mol. Sci. 2024, 25, 1039. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Pan, F.; Zhu, Z.; Yang, Z.; Wang, O.; Li, Q.; Zhao, L.; Zhao, L. Construction of a QSAR Model Based on Flavonoids and Screening of Natural Pancreatic Lipase Inhibitors. Nutrients 2023, 15, 3489. [Google Scholar] [CrossRef]
- Bhardwaj, P.; Biswas, G.P.; Mahata, N.; Ghanta, S.; Bhunia, B. Exploration of binding mechanism of triclosan towards cancer markers using molecular docking and molecular dynamics. Chemosphere 2022, 293, 133550. [Google Scholar] [CrossRef] [PubMed]
- Anjum, F.; Shahwan, M.; Alhumaydhi, F.A.; Sharaf, S.E.; Al Abdulmonem, W.; Shafie, A.; Bilgrami, A.L.; Shamsi, A.; Md Ashraf, G. Mechanistic insight into the binding between Ferritin and Serotonin: Possible implications in neurodegenerative diseases. J. Mol. Liq. 2022, 351, 118618. [Google Scholar] [CrossRef]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Curcumin has potent anti-amyloidogenic effects for Alzheimer’s β-amyloid fibrils in vitro. J. Neurosci. Res. 2004, 75, 742–750. [Google Scholar] [CrossRef]
- Noguchi-Shinohara, M.; Hamaguchi, T.; Sakai, K.; Komatsu, J.; Iwasa, K.; Horimoto, M.; Nakamura, H.; Yamada, M.; Ono, K. Effects of Melissa officinalis Extract Containing Rosmarinic Acid on Cognition in Older Adults Without Dementia: A Randomized Controlled Trial. J. Alzheimers Dis. 2023, 91, 805–814. [Google Scholar] [CrossRef]
- Singh, K.; Kaur, A.; Goyal, D.; Goyal, B. Mechanistic insights into the mitigation of Aβ aggregation and protofibril destabilization by a D-enantiomeric decapeptide rk10. Phys. Chem. Chem. Phys. 2022, 24, 21975–21994. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, J.; Zhang, H.-X. Effect of Quercetin on the Protein-Substrate Interactions in SIRT6: Insight from MD simulations. J. Mol. Graph. Model. 2024, 130, 108778. [Google Scholar] [CrossRef] [PubMed]
- Al-Khafaji, K.; Taskin Tok, T. Molecular dynamics simulation, free energy landscape and binding free energy computations in exploration the anti-invasive activity of amygdalin against metastasis. Comput. Methods Programs Biomed. 2020, 195, 105660. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, A.; Yankner, B.A. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc. Natl. Acad. Sci. USA 1994, 91, 12243–12247. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Secondary Structure Component | Aβ Peptide | Aβ Peptide System |
|---|---|---|
| a Helix | 1 | 3 |
| b β-Sheet | 27 | 22 |
| Coil | 42 | 45 |
| Bend | 21 | 18 |
| Turn | 7 | 9 |
| Chain_separator | 2 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, L.; Jiang, W.; Zhu, Z.; Pan, F.; Xing, X.; Zhou, F.; Zhao, L. Rosemarinic Acid-Induced Destabilization of Aβ Peptides: Insights from Molecular Dynamics Simulations. Foods 2024, 13, 4170. https://doi.org/10.3390/foods13244170
Zhao L, Jiang W, Zhu Z, Pan F, Xing X, Zhou F, Zhao L. Rosemarinic Acid-Induced Destabilization of Aβ Peptides: Insights from Molecular Dynamics Simulations. Foods. 2024; 13(24):4170. https://doi.org/10.3390/foods13244170
Chicago/Turabian StyleZhao, Liang, Weiye Jiang, Zehui Zhu, Fei Pan, Xin Xing, Feng Zhou, and Lei Zhao. 2024. "Rosemarinic Acid-Induced Destabilization of Aβ Peptides: Insights from Molecular Dynamics Simulations" Foods 13, no. 24: 4170. https://doi.org/10.3390/foods13244170
APA StyleZhao, L., Jiang, W., Zhu, Z., Pan, F., Xing, X., Zhou, F., & Zhao, L. (2024). Rosemarinic Acid-Induced Destabilization of Aβ Peptides: Insights from Molecular Dynamics Simulations. Foods, 13(24), 4170. https://doi.org/10.3390/foods13244170