
Genomic and Transcriptomic Analyses Reveal Multiple Strategies for Vibrio parahaemolyticus to Tolerate Sub-Lethal Concentrations of Three Antibiotics
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. The Characterization of Genome Features of the V. parahaemolyticus Isolates
2.1.1. V. parahaemolyticus Strains and Culture Conditions
2.1.2. Genomic DNA Preparation, Sequencing, Assembly, and Annotation
2.1.3. Comparative Genome Analysis
2.2. Phylogenetic Relationship of the V. parahaemolyticus Isolates
2.2.1. Serotyping and Multi-Locus Sequence Typing (MLST) Analyses
2.2.2. Phylogenetic Tree Analysis
2.3. Antibiotic Resistance of V. parahaemolyticus Isolates
2.3.1. Determination of Minimum Inhibitory Concentrations (MICs) of Antibiotics
2.3.2. Antibiotic Stress Conditions
2.4. Tolerance Mechanisms of the V. parahaemolyticus Isolates to Sub-LCs of Three Antibiotics
2.4.1. Cell Membrane Permeability (CMP) and Fluidity (CMF) and Cell Surface Hydrophobicity (CSH) Analysis
2.4.2. Scanning Electron Microscope (SEM) Assay
2.4.3. Illumina RNA Sequencing
2.5. Data Analysis
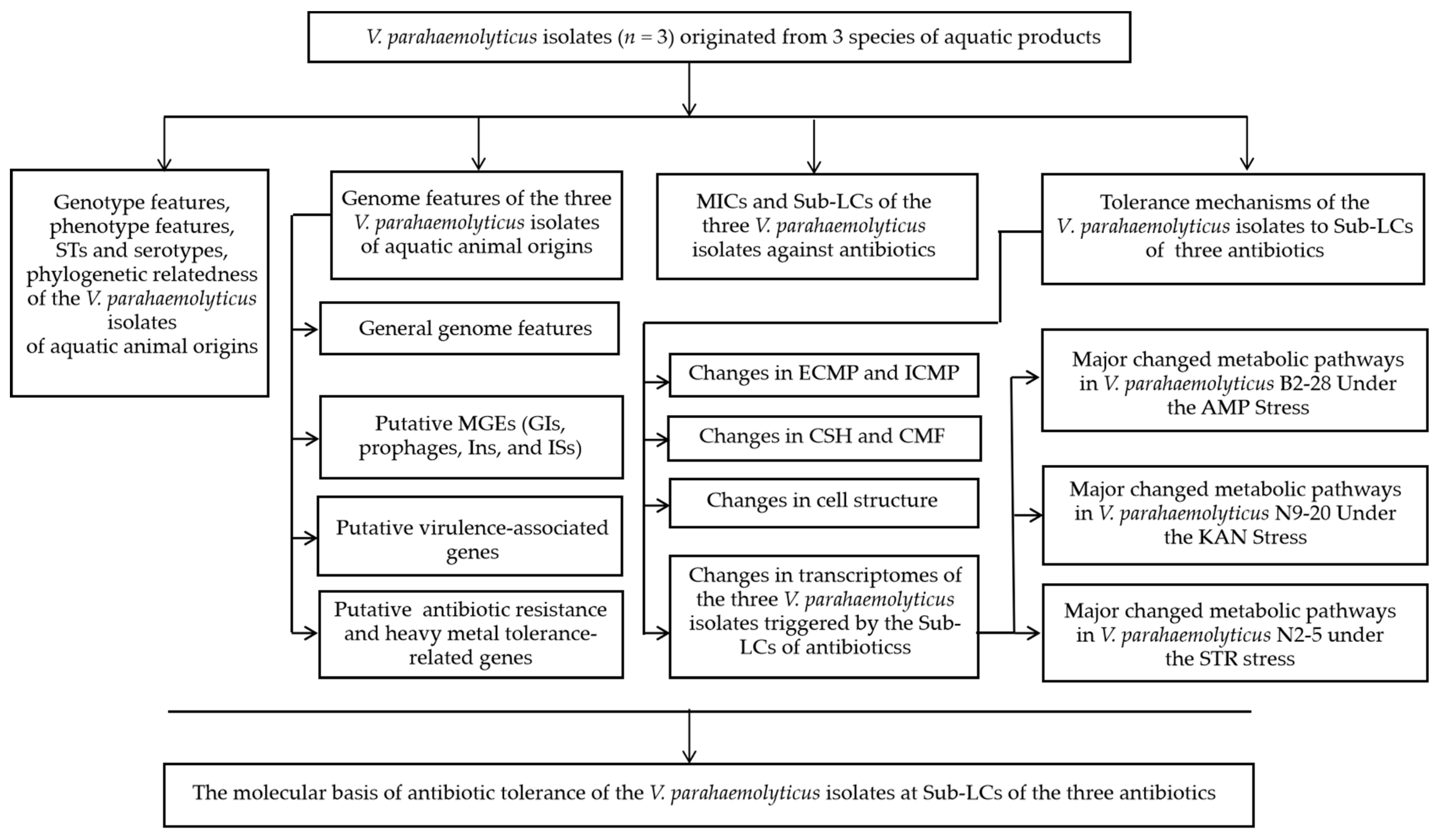
3. Results and Discussion
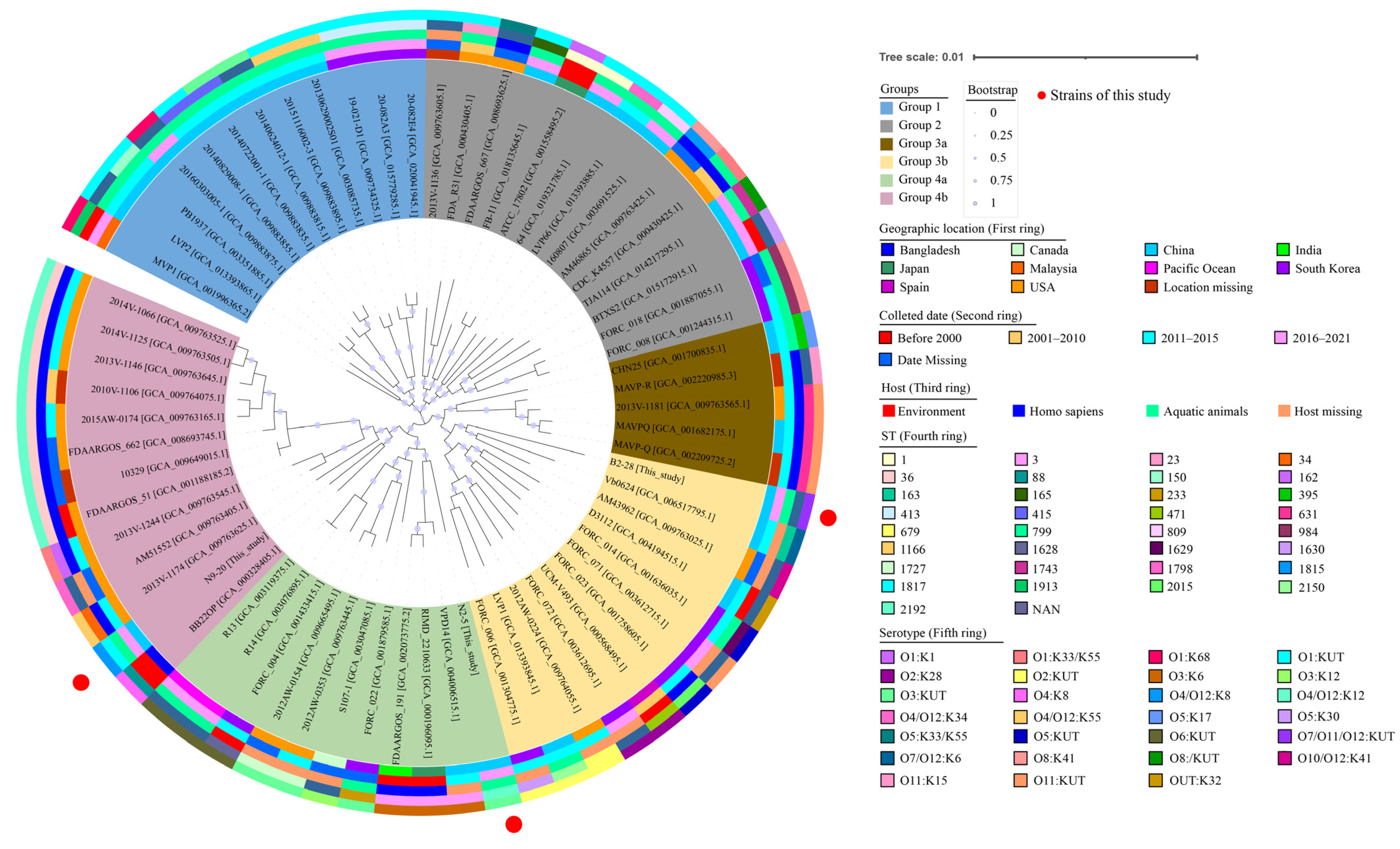
3.1. The Genotypes, Phenotypes, Sequence Types (STs), and Phylogenetic Relatedness of the V. parahaemolyticus Isolates
3.1.1. Genotypes and Phenotypes
3.1.2. Sequence Types
3.1.3. Phylogenetic Relatedness
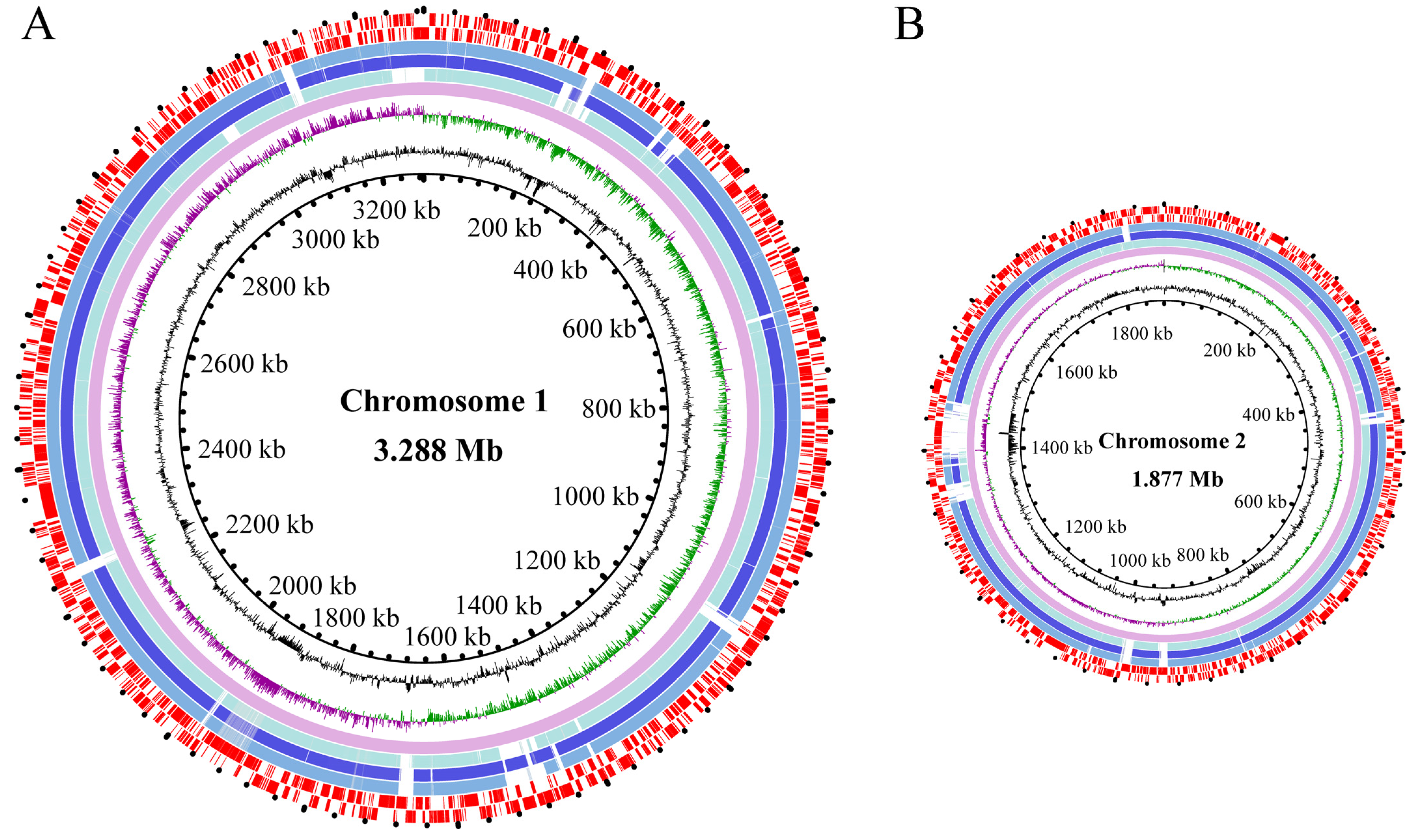
3.2. The Genome Features of the Three V. parahaemolyticus Isolates of Aquatic Animal Origins
3.2.1. General Genome Features
3.2.2. Putative MGEs
Genomic Islands
Prophages
Integrons
Insertion Sequences
3.2.3. Putative Virulence-Associated Genes
3.2.4. Heavy Metal Resistance- and Antibiotic Resistance-Associated Genes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotic and Heavy Metal | Resistance Gene | V. parahaemolyticus Isolate | Reference |
|---|---|---|---|
| Antibiotic | |||
| Beta-lactamases | blaCARB-17 | B2-28, N9-20, N2-5 | [76] |
| blaCARB-21 | N2-5 | [76] | |
| Elfamycin | tuf | B2-28, N9-20, N2-5 | [69] |
| Fluoroquinolone | crp | B2-28, N9-20, N2-5 | [70] |
| mfd | B2-28 | [77] | |
| gyrA | B2-28 | [78] | |
| Fosfomycin | uhpT | B2-28, N9-20, N2-5 | [72] |
| Peptide, rifamycin | rpoB | B2-28, N9-20, N2-5 | [71] |
| Tetracycline | tet (34) | B2-28, N9-20, N2-5 | [79] |
| tet (35) | B2-28, N9-20, N2-5 | [79] | |
| Heavy metal | |||
| Ni, Co | fecE | B2-28, N9-20, N2-5 | [80] |
| Fe, Cu, Mn | pfr | B2-28, N9-20, N2-5 | [81] |
| Co, Ni, Fe | rcnR/yohL | N2-5 | [81] |
| Cd, Hg | PA0320 | N2-5 | [81] |
3.3. The MICs and Sublethal Concentrations (Sub-LCs) of the Three V. parahaemolyticus Isolates against Antibiotics
3.4. The Tolerance Mechanisms of V. parahaemolyticus Isolates from Aquatic Animals to the Sub-LCs of the Three Antibiotics
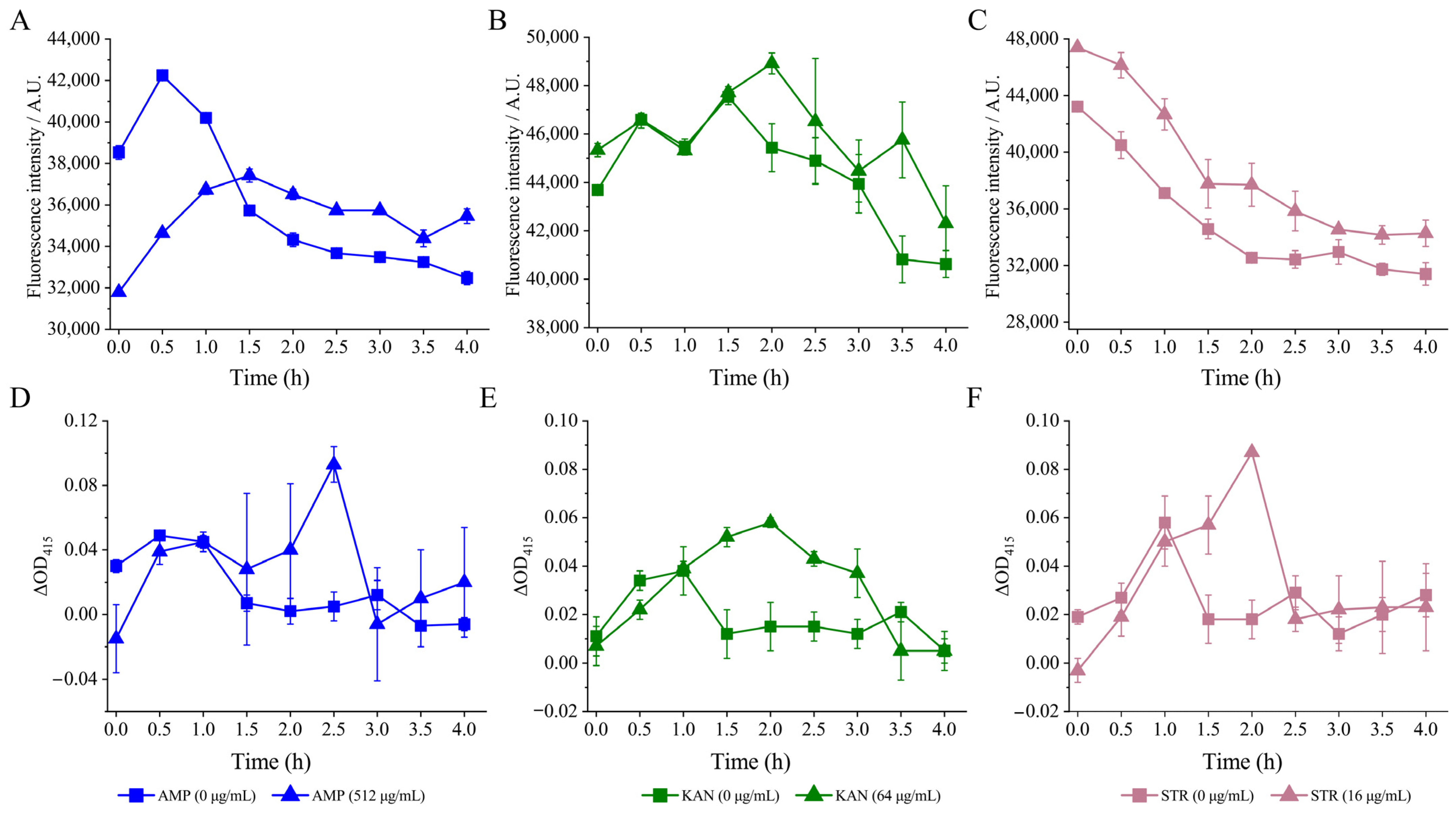
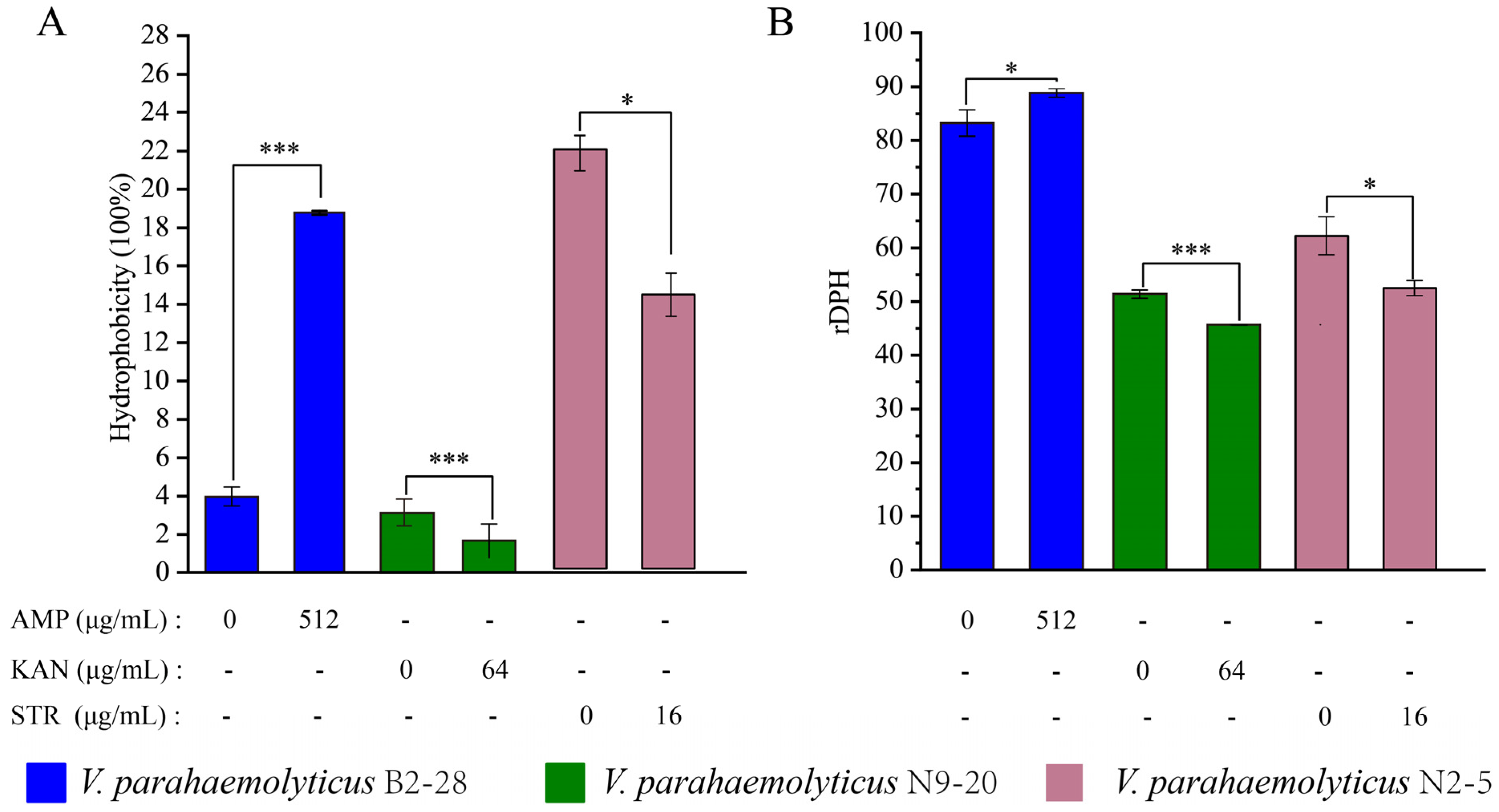
3.4.1. The Cell Membrane Permeability and Fluid and Cell Surface Hydrology of the V. parahaemolyticus Isolates under the Sub-LCs of Antibiotics
External Cell Membrane Permeability
Internal Cell Membrane Permeability
Cell Surface Hydrology and Cell Membrane Fluid
3.4.2. The Cell Structure Change of the Three V. parahaemolyticus Isolates under the Sub-LCs of Antibiotics
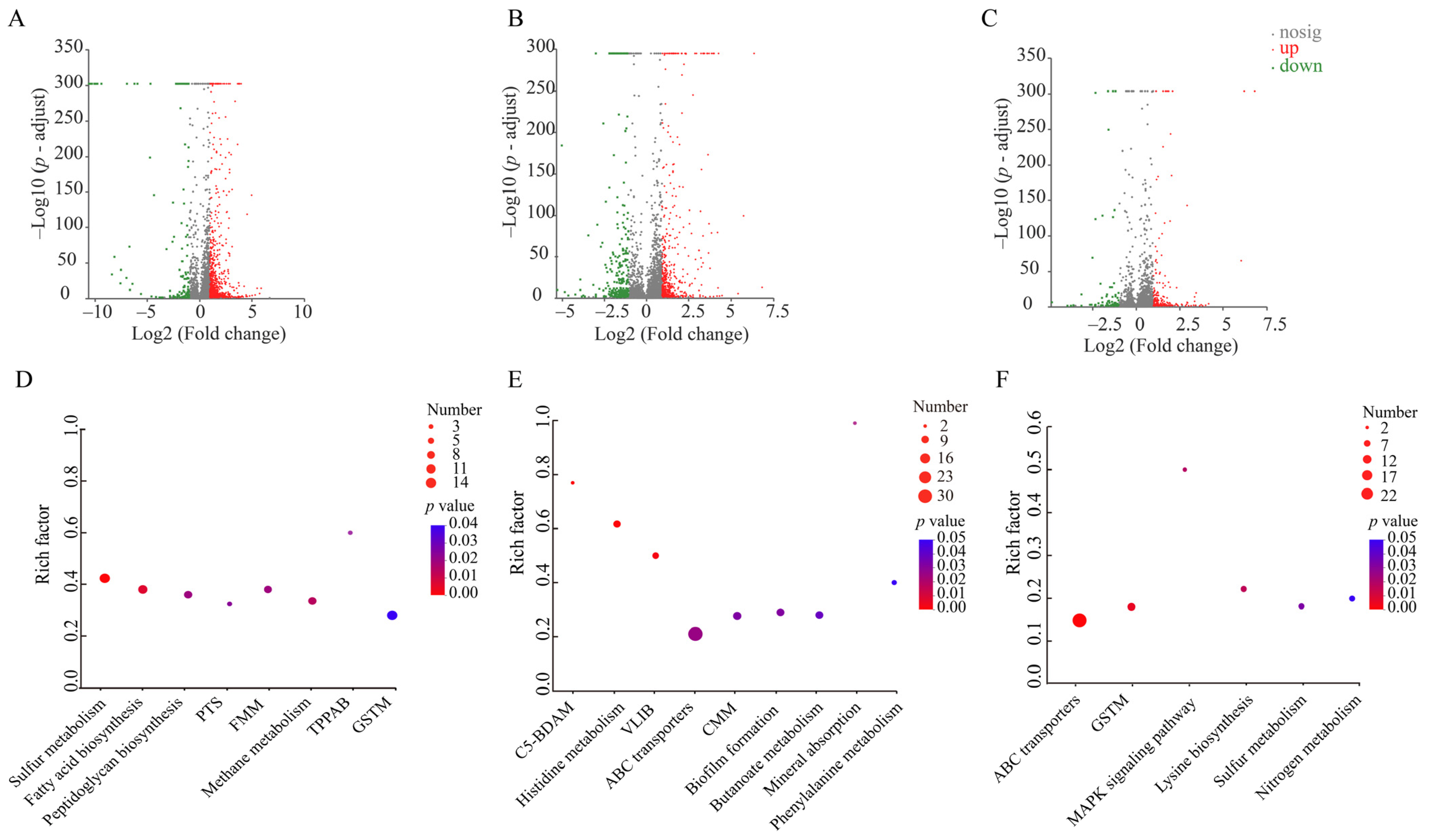
3.4.3. The Differential Transcriptomes of the Three V. parahaemolyticus Isolates Induced by the Sub-LCs of Antibiotics
The Major Changed Metabolic Pathways in V. parahaemolyticus B2-28 under AMP Stress
The Major Changed Metabolic Pathways in V. parahaemolyticus N9-20 under KAN Stress
The Major Changed Metabolic Pathways in V. parahaemolyticus N2-5 under STR Stress
3.4.4. The Molecular Basis Underlying the Antibiotic Tolerance of the V. parahaemolyticus Isolates at the Sub-LCs of the Antibiotics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, L.; Wang, J.; Chen, J.; Zhang, R.; Zhang, H.; Qi, X.; He, Y. Epidemiological characteristics of Vibrio parahaemolyticus outbreaks, Zhejiang, China, 2010–2022. Front. Microbiol. 2023, 14, 1171350. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Yan, H.; Cui, Z.; Li, H.; Zhou, W.; Liu, Z.; Zhang, H.; Manoli, T.; Mo, H.; Hu, L. Ultrasound-assisted blue light killing Vibrio parahaemolyticus to improve salmon preservation. Ultrason. Sonochemistry 2023, 95, 106389. [Google Scholar] [CrossRef] [PubMed]
- Fujino, T.; Okuno, Y.; Nakada, D.; Aoyama, A.; Fukai, K.; Mukai, T.; Ueho, T. On the bacteriological examination of shirasu-food poisoning. Med. J. Osaka Univ. 1953, 4, 299–304. [Google Scholar]
- Saito, S.; Iwade, Y.; Tokuoka, E.; Nishio, T.; Otomo, Y.; Araki, E.; Konuma, H.; Nakagawa, H.; Tanaka, H.; Sugiyama, K. Epidemiological evidence of lesser role of thermostable direct hemolysin (TDH)–related hemolysin (TRH) than TDH on Vibrio parahaemolyticus pathogenicity. Foodborne Pathog. Dis. 2015, 12, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Saetang, J.; Sukkapat, P.; Palamae, S.; Singh, P.; Senathipathi, D.N.; Buatong, J.; Benjakul, S. Multiplex PCR-lateral flow dipstick method for detection of thermostable direct hemolysin (TDH) producing V. parahaemolyticus. Biosensors 2023, 13, 698. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Yu, Q.; Tang, X.; Zhao, J.; He, X. Prevalence, antibiotic susceptibility and characterization of Vibrio parahaemolyticus isolates in China. FEMS Microbiol. Lett. 2020, 367, fnaa136. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.T.T.; Hoang, T.T.H.; Fleischmann, S.; Pham, H.N.; Lai, T.L.H.; Cam, T.T.H.; Truong, L.O.; Le, V.P.; Alter, T. Quantification and antimicrobial resistance of Vibrio parahaemolyticus in retail seafood in Hanoi, Vietnam. J. Food Prot. 2022, 85, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Lopatek, M.; Wieczorek, K.; Osek, J. Antimicrobial resistance, virulence factors, and genetic profiles of Vibrio parahaemolyticus from seafood. Appl. Environ. Microbiol. 2018, 84, e00537-18. [Google Scholar] [CrossRef] [PubMed]
- Stratev, D.; Fasulkova, R.; Krumova-Valcheva, G. Incidence, virulence genes and antimicrobial resistance of Vibrio parahaemolyticus isolated from seafood. Microb. Pathog. 2023, 177, 106050. [Google Scholar] [CrossRef]
- Dutta, D.; Kaushik, A.; Kumar, D.; Bag, S. Foodborne pathogenic Vibrios: Antimicrobial resistance. Front. Microbiol. 2021, 12, 638331. [Google Scholar] [CrossRef]
- Tan, X.; Qiao, J.; Wang, J.; Li, H.; Wang, X. Characterization of ampicillin-resistant genes in Vibrio parahaemolyticus. Microb. Pathog. 2022, 168, 105573. [Google Scholar] [CrossRef]
- Li, Z.; Junaid, M.; Chen, G.; Wang, J. Interactions and associated resistance development mechanisms between microplastics, antibiotics and heavy metals in the aquaculture environment. Rev. Aquac. 2022, 14, 1028–1045. [Google Scholar] [CrossRef]
- Jeong, H.-W.; Kim, J.-a.; Jeon, S.-J.; Choi, S.-S.; Kim, M.-K.; Yi, H.-J.; Cho, S.-J.; Kim, I.-Y.; Chon, J.-W.; Kim, D.-H. Prevalence, antibiotic-resistance, and virulence characteristics of Vibrio parahaemolyticus in restaurant fish tanks in Seoul, South Korea. Foodborne Pathog. Dis. 2020, 17, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Chen, H.; Li, N.; Liang, W. The application of the CRISPR-Cas system in antibiotic resistance. Infect. Drug Resist. 2022, 15, 4155–4168. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.E.; Boone, B.E. Next-generation sequencing strategies. Cold Spring Harb. Perspect. Med. 2019, 9, a025791. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.J.; Weimer, B.C.; Timme, R.; Lüdeke, C.H.M.; Pettengill, J.B.; Bandoy, D.D.; Weis, A.M.; Kaufman, J.; Huang, B.C.; Payne, J.; et al. Phylogenetic and biogeographic patterns of Vibrio parahaemolyticus strains from North America inferred from whole-genome sequence data. Appl. Environ. Microbiol. 2021, 87, e01403-20. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Duque, A.; Gonzalez-Muñoz, A.; Arboleda-Valencia, J.; Vivas-Aguas, L.J.; Córdoba-Meza, T.; Rodriguez-Rey, G.T.; Díaz-Guevara, P.; Martinez-Urtaza, J.; Wiesner-Reyes, M. Comparative genomics of clinical and environmental Isolates of Vibrio spp. of Colombia: Implications of traits associated with virulence and resistance. Pathogens 2021, 10, 1605. [Google Scholar] [CrossRef] [PubMed]
- Prithvisagar, K.S.; Krishna Kumar, B.; Kodama, T.; Rai, P.; Iida, T.; Karunasagar, I.; Karunasagar, I. Whole genome analysis unveils genetic diversity and potential virulence determinants in Vibrio parahaemolyticus associated with disease outbreak among cultured Litopenaeus vannamei (Pacific white shrimp) in India. Virulence 2021, 12, 1936–1949. [Google Scholar] [CrossRef]
- Yang, Q.; Dong, X.; Xie, G.; Fu, S.; Zou, P.; Sun, J.; Wang, Y.; Huang, J. Comparative genomic analysis unravels the transmission pattern and intra-species divergence of acute hepatopancreatic necrosis disease (AHPND)-causing Vibrio parahaemolyticus strains. Mol. Genet. Genom. 2019, 294, 1007–1022. [Google Scholar] [CrossRef]
- Yu, L.H.; Teh, C.S.J.; Yap, K.P.; Ung, E.H.; Thong, K.L. Comparative genomic provides insight into the virulence and genetic diversity of Vibrio parahaemolyticus associated with shrimp acute hepatopancreatic necrosis disease. Infect. Genet. Evol. 2020, 83, 104347. [Google Scholar] [CrossRef]
- Su, C.; Chen, L. Virulence, resistance, and genetic diversity of Vibrio parahaemolyticus recovered from commonly consumed aquatic products in Shanghai, China. Mar. Pollut. Bull. 2020, 160, 111554. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Yang, L.; Shao, Z.; Xie, L.; Chen, L. Identification of salt tolerance-related genes of Lactobacillus plantarum D31 and T9 strains by genomic analysis. Ann. Microbiol. 2020, 70, 10. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. Erratum: SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2015, 4, 30. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. In Gene Prediction; Kollmar, M., Ed.; Methods in Molecular Biology; Humana: New York, NY, USA, 2019; Volume 1962, pp. 1–14. [Google Scholar] [CrossRef]
- Jensen, L.J.; Julien, P.; Kuhn, M.; von Mering, C.; Muller, J.; Doerks, T.; Bork, P. eggNOG: Automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2008, 36, D250–D254. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef] [PubMed]
- Fouts, D.E. Phage_Finder: Automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 2006, 34, 5839–5851. [Google Scholar] [CrossRef] [PubMed]
- Néron, B.; Littner, E.; Haudiquet, M.; Perrin, A.; Cury, J.; Rocha, E.P.C. IntegronFinder 2.0: Identification and analysis of integrons across bacteria, with a focus on antibiotic resistance in Klebsiella. Microorganisms 2022, 10, 700. [Google Scholar] [CrossRef]
- Xie, Z.; Tang, H. ISEScan: Automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics 2017, 33, 3340–3347. [Google Scholar] [CrossRef]
- Yu, P.; Yang, L.; Wang, J.; Su, C.; Qin, S.; Zeng, C.; Chen, L. Genomic and transcriptomic analysis reveal multiple strategies for the cadmium tolerance in Vibrio parahaemolyticus N10-18 isolated from aquatic animal Ostrea gigas Thunberg. Foods 2022, 11, 3777. [Google Scholar] [CrossRef]
- González-Escalona, N.; Martinez-Urtaza, J.; Romero, J.; Espejo, R.T.; Jaykus, L.A.; DePaola, A. Determination of molecular phylogenetics of Vibrio parahaemolyticus strains by multilocus sequence typing. J. Bacteriol. 2008, 190, 2831–2840. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Piñeiro, C.; Abuín, J.M.; Pichel, J.C. Very Fast Tree: Speeding up the estimation of phylogenies for large alignments through parallelization and vectorization strategies. Bioinformatics 2020, 36, 4658–4659. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Peng, X.; Xie, L.; Chen, L. Survival and genome diversity of Vibrio parahaemolyticus isolated from edible aquatic animals. Diversity 2022, 14, 350. [Google Scholar] [CrossRef]
- Buck, J. The plate count in aquatic microbiology. Nativ. Aquat. Bact. Enumer. Act. Ecol. 1979, 1, 19–28. [Google Scholar]
- Fu, J.; Wang, Y.; Sun, M.; Xu, Y.; Chen, L. Antibacterial activity and components of the methanol-phase extract from rhizomes of pharmacophagous plant Alpinia officinarum Hance. Molecules 2022, 27, 4308. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, Y.; Yu, P.; Ren, S.; Zhu, Z.; Jin, Y.; Yan, J.; Peng, X.; Chen, L. Prophage-related gene VpaChn25_0724 contributes to cell membrane integrity and growth of Vibrio parahaemolyticus CHN25. Front. Cell. Infect. Microbiol. 2020, 10, 595709. [Google Scholar] [CrossRef] [PubMed]
- Bian, S.; Zeng, W.; Li, Q.; Li, Y.; Wong, N.K.; Jiang, M.; Zuo, L.; Hu, Q.; Li, L. Genetic structure, function, and evolution of capsule biosynthesis loci in Vibrio parahaemolyticus. Front. Microbiol. 2020, 11, 546150. [Google Scholar] [CrossRef]
- Pang, Y.; Guo, X.; Tian, X.; Liu, F.; Wang, L.; Wu, J.; Zhang, S.; Li, S.; Liu, B. Developing a novel molecular serotyping system based on capsular polysaccharide synthesis gene clusters of Vibrio parahaemolyticus. Int. J. Food Microbiol. 2019, 309, 108332. [Google Scholar] [CrossRef]
- Ciufo, S.; Kannan, S.; Sharma, S.; Badretdin, A.; Clark, K.; Turner, S.; Brover, S.; Schoch, C.L.; Kimchi, A.; DiCuccio, M. Using average nucleotide identity to improve taxonomic assignments in prokaryotic genomes at the NCBI. Int. J. Syst. Evol. Microbiol. 2018, 68, 2386–2392. [Google Scholar] [CrossRef]
- Ahsan, M.U.; Liu, Q.; Perdomo, J.E.; Fang, L.; Wang, K. A survey of algorithms for the detection of genomic structural variants from long-read sequencing data. Nat Methods 2023, 20, 1143–1158. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Wang, Q.; Wang, R.; Zhang, Y.; Lan, R.; He, F.; Yang, Q. Horizontal transfer of antibiotic resistance genes within the bacterial communities in aquacultural environment. Sci. Total Environ. 2022, 820, 153286. [Google Scholar] [CrossRef] [PubMed]
- Muchaamba, F.; Wambui, J.; Stephan, R.; Tasara, T. Cold shock proteins promote nisin tolerance in Listeria monocytogenes through modulation of cell envelope modification responses. Front. Microbiol. 2021, 12, 811939. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Liu, L.; Zhang, Y.; Yang, H.; Yan, Y.; Ding, X.; Han, Y.; Zhou, D.; Yang, R. BfvR, an AraC-family regulator, controls biofilm formation and pH6 antigen production in opposite ways in Yersinia pestis biovar Microtus. Front. Cell. Infect. Microbiol. 2018, 8, 347. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, Z.; Fei, X.; Tian, Y.; Zhou, G.; Hu, Y.; Wang, S.; Shi, H. The role of TolA, TolB, and TolR in cell morphology, OMVs production, and virulence of Salmonella Choleraesuis. AMB Express 2022, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.J.; Fang, Q.J.; Huang, H.Q.; Gong, C.G.; Hu, Y.H. HutZ is required for biofilm formation and contributes to the pathogenicity of Edwardsiella piscicida. Veter Res. 2019, 50, 76. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Deng, J.; Ren, J.; Liang, L.; Li, J.; Niu, S.; Wu, X.; Zhao, Y.; Gao, S.; Yan, F.; et al. RAP44 phage integrase-guided 50K genomic island integration in Riemerella anatipestifer. Front. Veter Sci. 2022, 9, 961354. [Google Scholar] [CrossRef] [PubMed]
- Chamblee, J.S.; Ramsey, J.; Chen, Y.; Maddox, L.T.; Ross, C.; To, K.H.; Cahill, J.L.; Young, R. Endolysin regulation in phage Mu lysis. mBio 2022, 13, e0081322. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, C.; Su, S.; Panmanee, W.; Lau, G.W.; Browne, T.; Cox, K.; Paul, A.T.; Ko, S.H.; Mortensen, J.E.; Lam, J.S.; et al. A Putative ABC transporter permease is necessary for resistance to acidified nitrite and EDTA in Pseudomonas aeruginosa under aerobic and anaerobic planktonic and biofilm conditions. Front. Microbiol. 2016, 7, 291. [Google Scholar] [CrossRef]
- Baltazar, M.; Bourgeois-Nicolaos, N.; Larroudé, M.; Couet, W.; Uwajeneza, S.; Doucet-Populaire, F.; Ploy, M.C.; Da Re, S. Activation of class 1 integron integrase is promoted in the intestinal environment. PLoS Genet. 2022, 18, e1010177. [Google Scholar] [CrossRef]
- An, X.L.; Chen, Q.L.; Zhu, D.; Zhu, Y.G.; Gillings, M.R.; Su, J.Q. Impact of wastewater treatment on the prevalence of integrons and the genetic diversity of integron gene cassettes. Appl. Environ. Microbiol. 2018, 84, e02766-17. [Google Scholar] [CrossRef] [PubMed]
- Mazel, D.; Dychinco, B.; Webb, V.A.; Davies, J. A distinctive class of integron in the Vibrio cholerae genome. Science 1998, 280, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Sarris, P.F.; Zoumadakis, C.; Panopoulos, N.J.; Scoulica, E.V. Distribution of the putative type VI secretion system core genes in Klebsiella spp. Infect. Genet. Evol. 2011, 11, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Damare, S. Cellular response of Brevibacterium casei #NIOSBA88 to arsenic and chromium—A proteomic approach. Braz. J. Microbiol. 2020, 51, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Abdel-Motaal, H.; Chen, J.; Chen, H.; Xu, T.; Meng, L.; Zhang, Z.; Meng, F.; Jiang, J. Characterization of a functionally unknown arginine-aspartate-aspartate family protein from Halobacillus andaensis and functional analysis of its conserved arginine/aspartate residues. Front. Microbiol. 2018, 9, 807. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Wang, L.; Song, N.; Yang, L.; Chen, J.; Yan, M.; Chen, H.; Zhang, R.; Li, J.; Abdel-Motaal, H.; et al. A UPF0118 family protein with uncharacterized function from the moderate halophile Halobacillus andaensis represents a novel class of Na(+)(Li(+))/H(+) antiporter. Sci. Rep. 2017, 7, 45936. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K.; Christensen, S.K.; Løbner-Olesen, A. Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 2005, 3, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Aghera, N.K.; Prabha, J.; Tandon, H.; Chattopadhyay, G.; Vishwanath, S.; Srinivasan, N.; Varadarajan, R. Mechanism of CcdA-Mediated rejuvenation of DNA gyrase. Structure 2020, 28, 562–572.e4. [Google Scholar] [CrossRef] [PubMed]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile genetic elements associated with antimicrobial resistance. Clin Microbiol Rev 2018, 31, e00088-17. [Google Scholar] [CrossRef]
- Alarcón Elvira, F.; Pardío-Sedas, V.T.; Martínez Herrera, D.; Quintana Castro, R.; Oliart Ros, R.M.; López Hernández, K.; Flores Primo, A.; Ramírez Elvira, K. Comparative survival and the cold-induced gene expression of pathogenic and nonpathogenic Vibrio Parahaemolyticus from tropical eastern oysters during cold storage. Int. J. Environ. Res. Public Health 2020, 17, 1836. [Google Scholar] [CrossRef]
- Bandyopadhyay, P.; Steinman, H.M. Legionella pneumophila catalase-peroxidases: Cloning of the katB gene and studies of KatB function. J. Bacteriol. 1998, 180, 5369–5374. [Google Scholar] [CrossRef] [PubMed]
- Eisemann, T.; Langelier, M.F.; Pascal, J.M. Structural and functional analysis of parameters governing tankyrase-1 interaction with telomeric repeat-binding factor 1 and GDP-mannose 4,6-dehydratase. J. Biol. Chem. 2019, 294, 14574–14590. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.Y.; Yan, L.; Yang, C.; Wu, Y.R.; Qin, J.L.; Hao, T.Y.; Yang, D.J.; Guo, Y.C.; Pei, X.Y.; Zhao, T.Y.; et al. Population genomics study of Vibrio alginolyticus. Hereditas 2021, 43, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.K.; Wang, C.J.; Chew, Y.; Wang, P.C.; Yin, H.S.; Kao, M.C. Functional characterization of Helicobacter pylori 26695 sedoheptulose 7-phosphate isomerase encoded by hp0857 and its association with lipopolysaccharide biosynthesis and adhesion. Biochem. Biophys. Res. Commun. 2016, 477, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Krachler, A.M.; Ham, H.; Orth, K. Outer membrane adhesion factor multivalent adhesion molecule 7 initiates host cell binding during infection by gram-negative pathogens. Proc. Natl. Acad. Sci. USA 2011, 108, 11614–11619. [Google Scholar] [CrossRef]
- Cesur, M.F.; Siraj, B.; Uddin, R.; Durmuş, S.; Çakır, T. Network-based metabolism-centered screening of potential drug targets in Klebsiella pneumoniae at genome scale. Front. Cell. Infect. Microbiol. 2019, 9, 447. [Google Scholar] [CrossRef] [PubMed]
- Meparambu Prabhakaran, D.; Ramamurthy, T.; Thomas, S. Genetic and virulence characterisation of Vibrio parahaemolyticus isolated from Indian coast. BMC Microbiol. 2020, 20, 62. [Google Scholar] [CrossRef] [PubMed]
- Che, R.X.; Xing, X.X.; Liu, X.; Qu, Q.W.; Chen, M.; Yu, F.; Ma, J.X.; Chen, X.R.; Zhou, Y.H.; God’Spower, B.O.; et al. Analysis of multidrug resistance in Streptococcus suis ATCC 700794 under tylosin stress. Virulence 2019, 10, 58–67. [Google Scholar] [CrossRef]
- Kary, S.C.; Yoneda, J.R.K.; Olshefsky, S.C.; Stewart, L.A.; West, S.B.; Cameron, A.D.S. The global regulatory cyclic AMP receptor protein (CRP) controls multifactorial fluoroquinolone susceptibility in Salmonella enterica serovar Typhimurium. Antimicrob. Agents Chemother. 2017, 61, e01666-17. [Google Scholar] [CrossRef]
- Wang, B.W.; Zhu, J.H.; Javid, B. Clinically relevant mutations in mycobacterial LepA cause rifampicin-specific phenotypic resistance. Sci. Rep. 2020, 10, 8402. [Google Scholar] [CrossRef]
- Ortiz-Padilla, M.; Portillo-Calderón, I.; Maldonado, N.; Rodríguez-Martínez, J.; de Gregorio-Iaria, B.; Merino-Bohórquez, V.; Rodríguez-Baño, J.; Pascual, Á.; Docobo-Pérez, F. Role of inorganic phosphate concentrations in in vitro activity of fosfomycin. Clin. Microbiol. Infect. 2022, 28, 302.e1–302.e4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Mao, D.; Gao, H.; Zheng, L.; Chen, Z.; Gao, Y.; Duan, Y.; Guo, J.; Luo, Y.; Ren, H. Colonization of gut microbiota by plasmid-carrying bacteria is facilitated by evolutionary adaptation to antibiotic treatment. Isme J. 2021, 16, 1284–1293. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Bao, Z.; Feng, H.; Chen, L.; Li, Q. Nitric oxide enhances resistance of Pleurotus eryngii to cadmium stress by alleviating oxidative damage and regulating of short-chain dehydrogenase/reductase family. Environ. Sci. Pollut. Res. Int. 2022, 29, 53036–53049. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Huang, Y.; Liu, P.; Yan, L.; Zhou, Y.; Yang, C.; Wu, Y.; Qin, J.; Guo, Y.; Pei, X.; et al. Population genomics of the food-borne pathogen Vibrio fluvialis reveals lineage associated pathogenicity-related genetic elements. Microb. Genom. 2022, 8, 000769. [Google Scholar] [CrossRef] [PubMed]
- Decré, D.; Arlet, G.; Bergogne-Bérézin, E.; Philippon, A. Identification of a carbenicillin-hydrolyzing beta-lactamase in Alcaligenes denitrificans subsp. xylosoxydans. Antimicrob. Agents Chemother. 1995, 39, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Sahin, O.; Barton, Y.W.; Zhang, Q. Key role of Mfd in the development of fluoroquinolone resistance in Campylobacter jejuni. PLoS Pathog. 2008, 4, e1000083. [Google Scholar] [CrossRef] [PubMed]
- Yakout, M.A.; Ali, G.H. A novel parC mutation potentiating fluoroquinolone resistance in Klebsiella pneumoniae and Escherichia coli clinical isolates. J. Infect. Dev. Ctries. 2022, 16, 314–319. [Google Scholar] [CrossRef] [PubMed]
- El-Razik, K.A.A.; Arafa, A.A.; Hedia, R.H.; Ibrahim, E.S. Tetracycline resistance phenotypes and genotypes of coagulase-negative Staphylococcal isolates from bubaline mastitis in Egypt. Veter World 2017, 10, 702–710. [Google Scholar] [CrossRef]
- Jabeen, S.; Yap, H.Y.; Abdullah, F.F.J.; Zakaria, Z.; Isa, N.M.; Tan, Y.C.; Joo, Y.S.; Satharasinghe, D.A.; Omar, A.R. Complete genome sequence analysis and characterization of selected iron regulation genes of Pasteurella multocida serotype A Strain PMTB2.1. Genes 2019, 10, 81. [Google Scholar] [CrossRef]
- Pal, C.; Bengtsson-Palme, J.; Rensing, C.; Kristiansson, E.; Larsson, D.G. BacMet: Antibacterial biocide and metal resistance genes database. Nucleic Acids Res. 2014, 42, D737–D743. [Google Scholar] [CrossRef]
- Bartsch, A.; Llabrés, S.; Pein, F.; Kattner, C.; Schön, M.; Diehn, M.; Tanabe, M.; Munk, A.; Zachariae, U.; Steinem, C. High-resolution experimental and computational electrophysiology reveals weak β-lactam binding events in the porin PorB. Sci. Rep. 2019, 9, 1264. [Google Scholar] [CrossRef]
- Bessa, L.J.; Ferreira, M.; Gameiro, P. Evaluation of membrane fluidity of multidrug-resistant isolates of Escherichia coli and Staphylococcus aureus in presence and absence of antibiotics. J. Photochem. Photobiol. B 2018, 181, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, L.; Liu, P.; Jin, Y.; Qin, S.; Chen, L. Identification of antibacterial components in the methanol-phase extract from edible herbaceous plant Rumex madaio Makino and their antibacterial action Modes. Molecules 2022, 27, 660. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, M.; Vischer, N.O.E.; Strahl, H.; Hamoen, L.W. Assessing membrane fluidity and visualizing fluid membrane domains in bacteria using fluorescent membrane dyes. Bio Protoc. 2018, 8, e3063. [Google Scholar] [CrossRef] [PubMed]
- Pinto, R.; Tang, Q.X.; Britton, W.J.; Leyh, T.S.; Triccas, J.A. The Mycobacterium tuberculosis cysD and cysNC genes form a stress-induced operon that encodes a tri-functional sulfate-activating complex. Microbiology 2004, 150, 1681–1686. [Google Scholar] [CrossRef]
- Espeland, L.O.; Georgiou, C.; Klein, R.; Bhukya, H.; Haug, B.E.; Underhaug, J.; Mainkar, P.S.; Brenk, R. An experimental toolbox for structure-based hit discovery for P. aeruginosa FabF, a promising target for antibiotics. ChemMedChem 2021, 16, 2715–2726. [Google Scholar] [CrossRef] [PubMed]
- Alves, J.; Westling, L.; Peters, E.C.; Harris, J.L.; Trauger, J.W. Cloning, expression, and enzymatic activity of Acinetobacter baumannii and Klebsiella pneumoniae acetyl-coenzyme A carboxylases. Anal. Biochem. 2011, 417, 103–111. [Google Scholar] [CrossRef]
- Lobritz, M.A.; Andrews, I.W.; Braff, D.; Porter, C.B.M.; Gutierrez, A.; Furuta, Y.; Cortes, L.B.G.; Ferrante, T.; Bening, S.C.; Wong, F.; et al. Increased energy demand from anabolic-catabolic processes drives β-lactam antibiotic lethality. Cell Chem. Biol. 2022, 29, 276–286.e4. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.A.; Kobus, S.; Czech, L.; Hoeppner, A.; Zarzycki, J.; Erb, T.J.; Lauterbach, L.; Dickschat, J.S.; Bremer, E.; Smits, S.H.J. The architecture of the diaminobutyrate acetyltransferase active site provides mechanistic insight into the biosynthesis of the chemical chaperone ectoine. J. Biol. Chem. 2020, 295, 2822–2838. [Google Scholar] [CrossRef]
- Akhova, A.; Nesterova, L.; Shumkov, M.; Tkachenko, A. Cadaverine biosynthesis contributes to decreased Escherichia coli susceptibility to antibiotics. Res. Microbiol. 2021, 172, 103881. [Google Scholar] [CrossRef]
- Pearce, S.R.; Mimmack, M.L.; Gallagher, M.P.; Gileadi, U.; Hyde, S.C.; Higgins, C.F. Membrane topology of the integral membrane components, OppB and OppC, of the oligopeptide permease of Salmonella typhimurium. Mol. Microbiol. 1992, 6, 47–57. [Google Scholar] [CrossRef]
- Fukami-Kobayashi, K.; Tateno, Y.; Nishikawa, K. Domain dislocation: A change of core structure in periplasmic binding proteins in their evolutionary history. J. Mol. Biol. 1999, 286, 279–290. [Google Scholar] [CrossRef]
- Kröger, P.; Shanmugaratnam, S.; Ferruz, N.; Schweimer, K.; Höcker, B. A comprehensive binding study illustrates ligand recognition in the periplasmic binding protein PotF. Structure 2021, 29, 433–443.e4. [Google Scholar] [CrossRef]
- Gregory, G.J.; Dutta, A.; Parashar, V.; Boyd, E.F. Investigations of dimethylglycine, glycine betaine, and ectoine uptake by a betaine-carnitine-choline transporter family transporter with diverse substrate specificity in Vibrio Species. J. Bacteriol. 2020, 202, e00314-20. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, C.P.; Encinas, M.V.; Araya, M.A.; Pérez, J.M.; Tantaleán, J.C.; Fuentes, D.E.; Calderón, I.L.; Pichuantes, S.E.; Vásquez, C.C. Biochemical characterization of a thermostable cysteine synthase from Geobacillus stearothermophilus V. Biochimie 2004, 86, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Rosa, B.; Marchetti, M.; Paredi, G.; Amenitsch, H.; Franko, N.; Benoni, R.; Giabbai, B.; De Marino, M.G.; Mozzarelli, A.; Ronda, L.; et al. Combination of SAXS and protein painting discloses the three-dimensional organization of the bacterial cysteine synthase complex, a potential target for enhancers of antibiotic action. Int. J. Mol. Sci. 2019, 20, 5219. [Google Scholar] [CrossRef]
- Getz, L.J.; Thomas, N.A. The Transcriptional regulator HlyU positively regulates expression of exsA, leading to Type III secretion system 1 activation in Vibrio parahaemolyticus. J. Bacteriol. 2018, 200, e00653-17. [Google Scholar] [CrossRef]
- Isogai, S.; Takagi, H. Enhancement of lysine biosynthesis confers high-temperature stress tolerance to Escherichia coli cells. Appl. Microbiol. Biotechnol. 2021, 105, 6899–6908. [Google Scholar] [CrossRef] [PubMed]
- Beceiro, A.; Tomás, M.; Bou, G. Antimicrobial resistance and virulence: A successful or deleterious association in the bacterial world? Clin. Microbiol. Rev. 2013, 26, 185–230. [Google Scholar] [CrossRef]
- Carter, A.P.; Clemons, W.M.; Brodersen, D.E.; Morgan-Warren, R.J.; Wimberly, B.T.; Ramakrishnan, V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature 2000, 407, 340–348. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, X.; Wang, G.; Yao, J.; Wei, J.; Liu, Z.; Lin, X.; Liu, Y. Quantitative proteomics reveals the antibiotics adaptation mechanism of Aeromonas hydrophila under kanamycin stress. J. Proteom. 2022, 264, 104621. [Google Scholar] [CrossRef] [PubMed]
- Reygaert, W.C. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Almuhaideb, E.; Chintapenta, L.K.; Abbott, A.; Parveen, S.; Ozbay, G. Assessment of Vibrio parahaemolyticus levels in oysters (Crassostrea virginica) and seawater in Delaware Bay in relation to environmental conditions and the prevalence of molecular markers to identify pathogenic Vibrio parahaemolyticus strains. PLoS ONE 2020, 15, e0242229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Deng, Y.; Feng, J.; Guo, Z.; Chen, H.; Wang, B.; Hu, J.; Lin, Z.; Su, Y. Functional characterization of VscCD, an important component of the type III secretion system of Vibrio harveyi. Microb. Pathog. 2021, 157, 104965. [Google Scholar] [CrossRef] [PubMed]
- Abushattal, S.; Vences, A.; Osorio, C.R. A virulence gene typing scheme for Photobacterium damselae subsp. piscicida, the causative agent of fish photobacteriosis, reveals a high prevalence of plasmid-encoded virulence factors and of type III secretion system genes. Aquaculture 2020, 521, 735057. [Google Scholar] [CrossRef]
- Akeda, Y.; Okayama, K.; Kimura, T.; Dryselius, R.; Kodama, T.; Oishi, K.; Iida, T.; Honda, T. Identification and characterization of a type III secretion-associated chaperone in the type III secretion system 1 of Vibrio parahaemolyticus. FEMS Microbiol Lett 2009, 296, 18–25. [Google Scholar] [CrossRef]
- Tam, V.C.; Suzuki, M.; Coughlin, M.; Saslowsky, D.; Biswas, K.; Lencer, W.I.; Faruque, S.M.; Mekalanos, J.J. Functional analysis of VopF activity required for colonization in Vibrio cholerae. mBio 2010, 1, e00289-10. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, R.; Qi, X.; Zhou, B.; Wang, J.; Chen, Y.; Zhang, H. Epidemiology of foodborne disease outbreaks caused by Vibrio parahaemolyticus during 2010–2014 in Zhejiang Province, China. Food Control 2017, 77, 110–115. [Google Scholar] [CrossRef]


| Genome Feature | V. parahaemolyticus Isolate | ||
|---|---|---|---|
| B2-28 | N2-5 | N9-20 | |
| Genome size (bp) | 5,381,824 | 5,368,856 | 5,009,026 |
| G + C (%) | 45.14 | 45.28 | 45.40 |
| DNA scaffold | 324 | 71 | 42 |
| Total predicted gene | 5692 | 5564 | 4796 |
| Protein-coding gene | 5610 | 5191 | 4709 |
| RNA gene (rRNA and tRNA) | 137 | 127 | 146 |
| Genes assigned to COG | 4626 | 4092 | 3886 |
| Genes with unknown function | 984 | 1099 | 823 |
| GI | 4 | 14 | 6 |
| Prophage | 1 | 1 | 0 |
| In | 27 | 7 | 4 |
| IS | 2 | 2 | 1 |
| V. parahaemolyticus Strain | MIC (μg/mL) | Sub-LC (μg/mL) | Fatality Rate (%) | |
|---|---|---|---|---|
| B2-28 | AMP | 100,000 | 512 | 10.38 |
| 256 | 11.38 | |||
| STR | 128 | 32 | 15.73 | |
| 16 | 10.95 | |||
| N9-20 | AMP | 50,000 | 512 | 11.93 |
| 256 | 12.75 | |||
| KAN | 256 | 64 | 10.02 | |
| 32 | 8.27 | |||
| STR | 128 | 64 | 25.78 | |
| 32 | 11.06 | |||
| N2-5 | AMP | 50,000 | 2048 | 16.17 |
| 1024 | 5.56 | |||
| KAN | 128 | 64 | 13.01 | |
| 32 | 13.78 | |||
| STR | 128 | 32 | 18.64 | |
| 16 | 10.45 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Yu, P.; Wang, J.; Zhao, T.; Zhao, Y.; Pan, Y.; Chen, L. Genomic and Transcriptomic Analyses Reveal Multiple Strategies for Vibrio parahaemolyticus to Tolerate Sub-Lethal Concentrations of Three Antibiotics. Foods 2024, 13, 1674. https://doi.org/10.3390/foods13111674
Yang L, Yu P, Wang J, Zhao T, Zhao Y, Pan Y, Chen L. Genomic and Transcriptomic Analyses Reveal Multiple Strategies for Vibrio parahaemolyticus to Tolerate Sub-Lethal Concentrations of Three Antibiotics. Foods. 2024; 13(11):1674. https://doi.org/10.3390/foods13111674
Chicago/Turabian StyleYang, Lianzhi, Pan Yu, Juanjuan Wang, Taixia Zhao, Yong Zhao, Yingjie Pan, and Lanming Chen. 2024. "Genomic and Transcriptomic Analyses Reveal Multiple Strategies for Vibrio parahaemolyticus to Tolerate Sub-Lethal Concentrations of Three Antibiotics" Foods 13, no. 11: 1674. https://doi.org/10.3390/foods13111674
APA StyleYang, L., Yu, P., Wang, J., Zhao, T., Zhao, Y., Pan, Y., & Chen, L. (2024). Genomic and Transcriptomic Analyses Reveal Multiple Strategies for Vibrio parahaemolyticus to Tolerate Sub-Lethal Concentrations of Three Antibiotics. Foods, 13(11), 1674. https://doi.org/10.3390/foods13111674