
SVR Chemometrics to Quantify β-Lactoglobulin and α-Lactalbumin in Milk Using MIR

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials, Samples, and Standards
2.2. Reagents for the Kjeldahl Method
2.3. Mid-Infrared Spectroscopy (MIR)
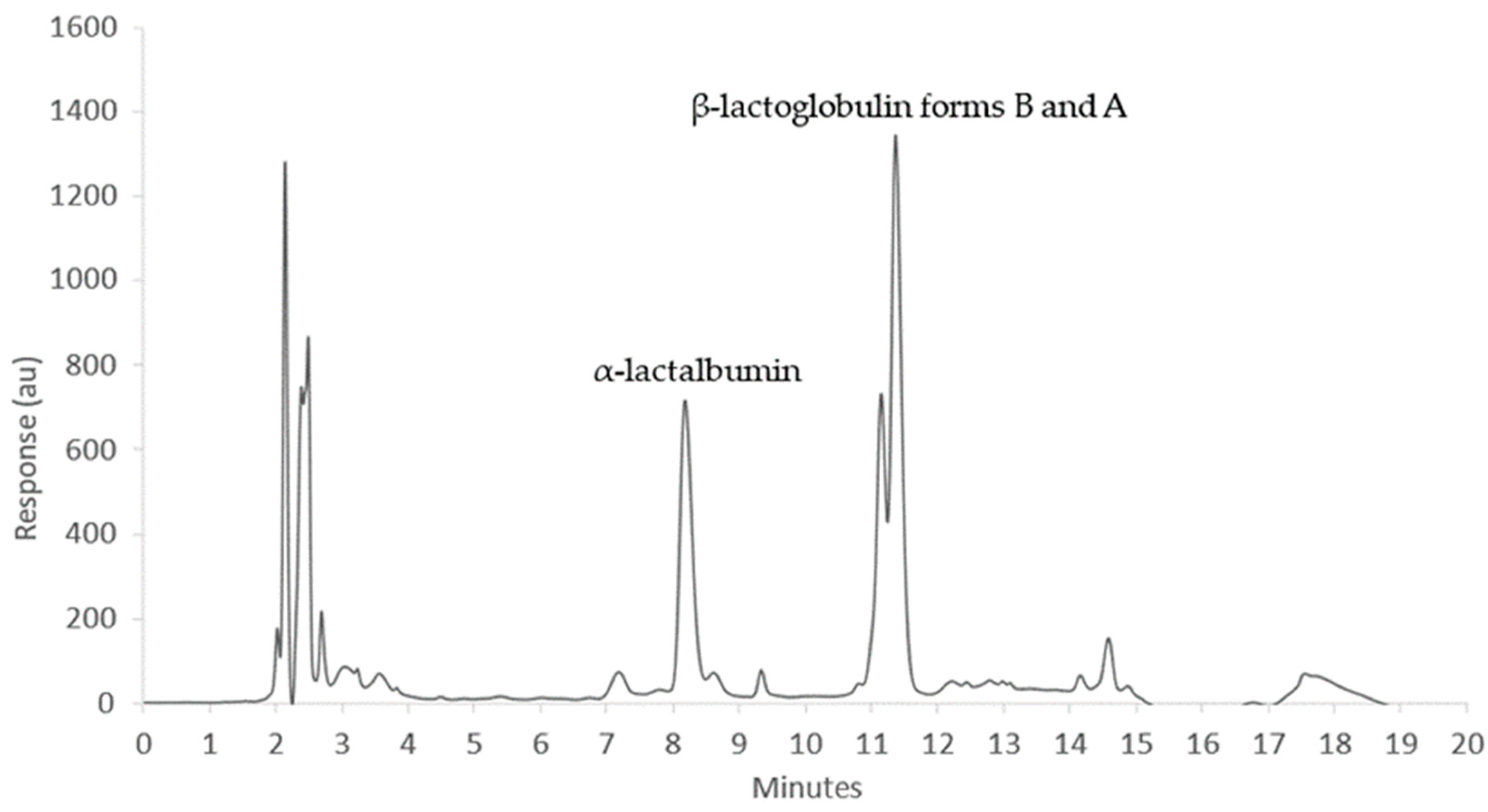
2.4. High Performance Liquid Chromatography (HPLC)
2.4.1. Sample Handling
2.4.2. Chromatography
2.4.3. Calibrations
2.4.4. Extraction Efficiency
- is the protein in spiked sample;
- is the protein in unspiked sample;
- is the concentration of spike.
2.5. Kjeldahl
- Vs and Vb (mL): Titrant acid used for test portion and blank;
- M: Molarity of the acid solution;
- W(g): Test portion weight.
2.6. Chemometrics Analysis
2.6.1. Data Description
2.6.2. Outlier Detection
- is the ith vector in the PLS residual matrix ;
- is the MIR spectra;
- is the PLS loadings matrix;
- is the PLS scores matrix and is its ith vector;
- is the number of PLS components used;
- is the standard deviation of jth PLS component.
2.6.3. Data Partitioning
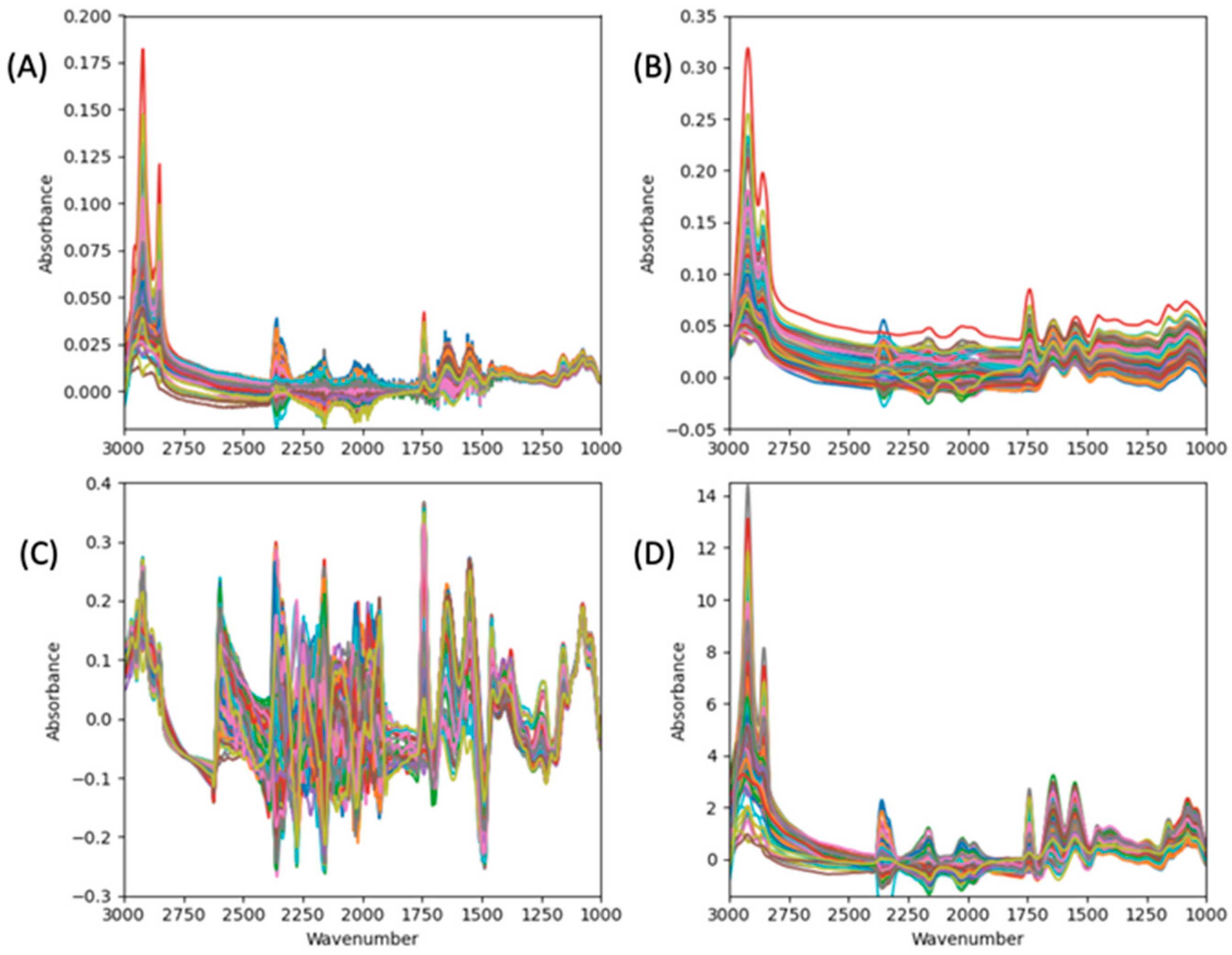
2.6.4. Spectral Preprocessing
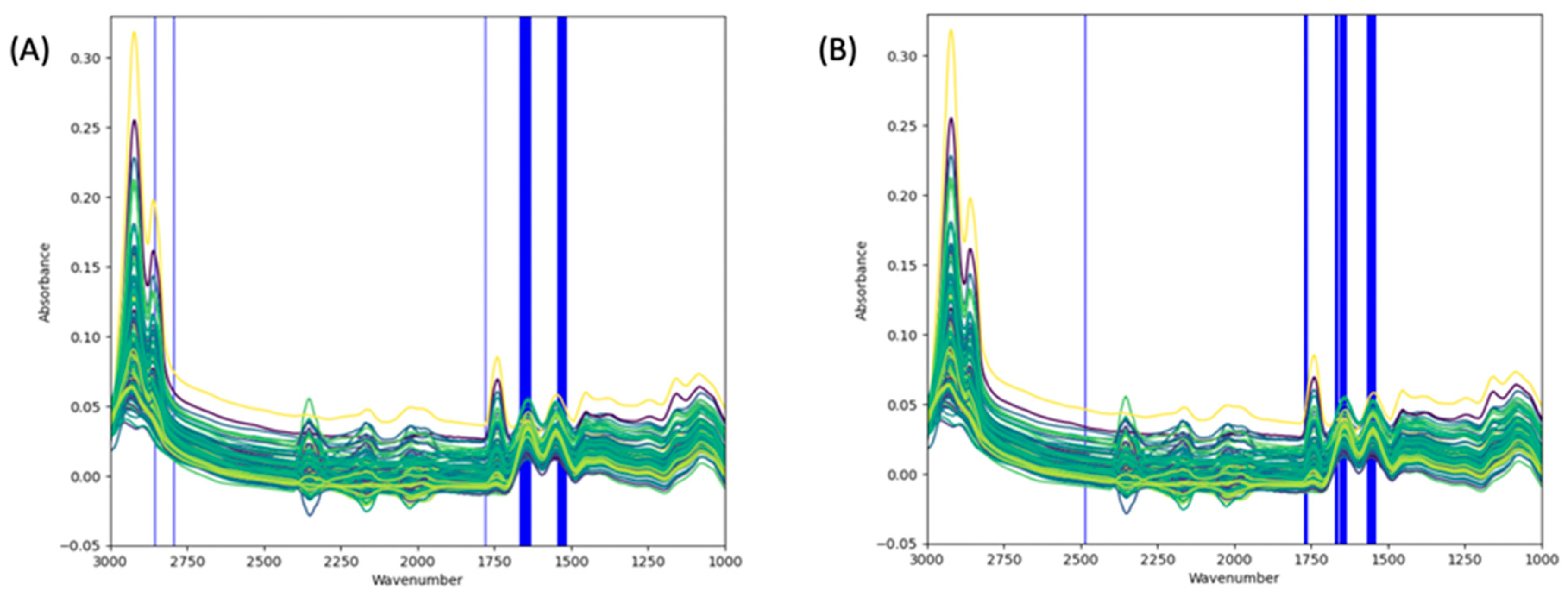
2.6.5. Wavenumber Selection
2.6.6. Regression Analysis
Partial Least Square (PLS)
- is the concentration values of and ;
- is the PLS scores matrix with respect to Y;
- is the residual matrix with respect to Y;
- is the PLS regression coefficients.
Support Vector Regression (SVR)
- where
- is the one of linear, polynomial, or RBF kernels;
- is the predicted value;
- is the target output;
- is the regularization parameter;
- and are tolerance limits.
Ridge Regression
- is the MIR spectra;
- is the ridge regression coefficient vector and is the intercept;
- is the regularization parameter or penalty term, and . Setting turn Equation (8) to which is the linear regression cost function;
- is the predicted concentration value usually denoted by ;
- is the actual concentration value.
3. Results
3.1. Descriptive Analysis of Protein Content in the Dataset and Spectra Preprocessing
3.2. Spectra Interpretation and Regions of Interest
3.3. Chemometric Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smithers, G.W. Whey and Whey Proteins—From ‘Gutter-to-Gold. ’ Int. Dairy J. 2008, 18, 695–704. [Google Scholar] [CrossRef]
- Tsermoula, P.; Khakimov, B.; Nielsen, J.H.; Engelsen, S.B. WHEY—The Waste-Stream That Became More Valuable than the Food Product. Trends Food Sci. Technol. 2021, 118, 230–241. [Google Scholar] [CrossRef]
- McGrath, B.A.; Fox, P.F.; McSweeney, P.L.H.; Kelly, A.L. Composition and Properties of Bovine Colostrum: A Review. Dairy Sci. Technol. 2016, 96, 133–158. [Google Scholar] [CrossRef]
- Zhang, W.; Lu, J.; Chen, B.; Gao, P.; Song, B.; Zhang, S.; Pang, X.; Hettinga, K.; Lyu, J. Comparison of Whey Proteome and Glycoproteome in Bovine Colostrum and Mature Milk. J. Agric. Food Chem. 2023, 71, 10863–10876. [Google Scholar] [CrossRef]
- Ng-Kwai-Hang, K.F.; Hayes, J.F.; Moxley, J.E.; Monardes, H.G. Variation in Milk Protein Concentrations Associated with Genetic Polymorphism and Environmental Factors. J. Dairy Sci. 1987, 70, 563–570. [Google Scholar] [CrossRef]
- Regester, G.O.; Smithers, G.W. Seasonal Changes in the β-Lactoglobulin, α-Lactalbumin, Glycomacropeptide, and Casein Content of Whey Protein Concentrate. J. Dairy Sci. 1991, 74, 796–802. [Google Scholar] [CrossRef]
- Li, S.; Ye, A.; Singh, H. Seasonal Variations in Composition, Properties, and Heat-Induced Changes in Bovine Milk in a Seasonal Calving System. J. Dairy Sci. 2019, 102, 7747–7759. [Google Scholar] [CrossRef]
- Hogarth, C.J.; Fitzpatrick, J.L.; Nolan, A.M.; Young, F.J.; Pitt, A.; Eckersall, P.D. Differential Protein Composition of Bovine Whey: A Comparison of Whey from Healthy Animals and from Those with Clinical Mastitis. Proteomics 2004, 4, 2094–2100. [Google Scholar] [CrossRef]
- Litwińczuk, Z.; Król, J.; Brodziak, A.; Barłowska, J. Changes of Protein Content and Its Fractions in Bovine Milk from Different Breeds Subject to Somatic Cell Count. J. Dairy Sci. 2011, 94, 684–691. [Google Scholar] [CrossRef]
- Minj, S.; Anand, S. Whey Proteins and Its Derivatives: Bioactivity, Functionality, and Current Applications. Dairy 2020, 1, 233–258. [Google Scholar] [CrossRef]
- Khalesi, M.; FitzGerald, R.J. Investigation of the Flowability, Thermal Stability and Emulsification Properties of Two Milk Protein Concentrates Having Different Levels of Native Whey Proteins. Food Res. Int. 2021, 147, 110576. [Google Scholar] [CrossRef]
- Canellada, F.; Laca, A.; Laca, A.; Díaz, M. Environmental Impact of Cheese Production: A Case Study of a Small-Scale Factory in Southern Europe and Global Overview of Carbon Footprint. Sci. Total Environ. 2018, 635, 167–177. [Google Scholar] [CrossRef]
- Hinrichs, J. Incorporation of Whey Proteins in Cheese. Int. Dairy J. 2001, 11, 495–503. [Google Scholar] [CrossRef]
- Delikanli, B.; Ozcan, T. Effects of Various Whey Proteins on the Physicochemical and Textural Properties of Set Type Nonfat Yoghurt. Int. J. Dairy Technol. 2014, 67, 495–503. [Google Scholar] [CrossRef]
- Lanigan, J.; Singhal, A. Early Nutrition and Long-Term Health: A Practical Approach: Symposium on ‘Early Nutrition and Later Disease: Current Concepts, Research and Implications. ’ Proc. Nutr. Soc. 2009, 68, 422–429. [Google Scholar] [CrossRef]
- Almeida, C.C.; Mendonça Pereira, B.F.; Leandro, K.C.; Costa, M.P.; Spisso, B.F.; Conte-Junior, C.A. Bioactive Compounds in Infant Formula and Their Effects on Infant Nutrition and Health: A Systematic Literature Review. Int. J. Food Sci. 2021, 2021, 8850080. [Google Scholar] [CrossRef]
- Wagner, J.; Biliaderis, C.G.; Moschakis, T. Whey Proteins: Musings on Denaturation, Aggregate Formation and Gelation. Crit. Rev. Food Sci. Nutr. 2020, 60, 3793–3806. [Google Scholar] [CrossRef]
- Blanpain-Avet, P.; André, C.; Khaldi, M.; Bouvier, L.; Petit, J.; Six, T.; Jeantet, R.; Croguennec, T.; Delaplace, G. Predicting the Distribution of Whey Protein Fouling in a Plate Heat Exchanger Using the Kinetic Parameters of the Thermal Denaturation Reaction of β-Lactoglobulin and the Bulk Temperature Profiles. J. Dairy Sci. 2016, 99, 9611–9630. [Google Scholar] [CrossRef]
- Deeth, H.; Bansal, N. Chapter 1—Whey Proteins: An Overview. In Whey Proteins; Deeth, H.C., Bansal, N., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 1–50. ISBN 978-0-12-812124-5. [Google Scholar]
- Li, Z.; Liu, Z.; Mu, H.; Liu, Y.; Zhang, Y.; Wang, Q.; Quintero, L.E.E.; Li, X.; Chen, S.; Gong, Y.; et al. The Stability and Spicy Taste Masking Effect of Capsaicin Loaded α-Lactalbumin Micelles Formulated in Defatted Cheese. Food Funct. 2022, 13, 12258–12267. [Google Scholar] [CrossRef]
- Liu, B.; Thum, C.; Wang, Q.; Feng, C.; Li, T.; Damiani Victorelli, F.; Li, X.; Chang, R.; Chen, S.; Gong, Y.; et al. The Fortification of Encapsulated Soy Isoflavones and Texture Modification of Soy Milk by α-Lactalbumin Nanotubes. Food Chem. 2023, 419, 135979. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, W.; Li, Z.; Liu, B.; Hu, Y.; Chen, S.; Vries, R.d.; Yuan, Y.; Quintero, L.E.E.; Hou, G.; et al. The Stability and Bioavailability of Curcumin Loaded α-Lactalbumin Nanocarriers Formulated in Functional Dairy Drink. Food Hydrocoll. 2022, 131, 107807. [Google Scholar] [CrossRef]
- Liu, B.; Liu, B.; Wang, R.; Li, Y. α-Lactalbumin Self-Assembled Nanoparticles with Various Morphologies, Stiffnesses, and Sizes as Pickering Stabilizers for Oil-in-Water Emulsions and Delivery of Curcumin. J. Agric. Food Chem. 2021, 69, 2485–2492. [Google Scholar] [CrossRef]
- Burke, N.; Zacharski, K.A.; Southern, M.; Hogan, P.; Ryan, M.P.; Adley, C.C.; Burke, N.; Zacharski, K.A.; Southern, M.; Hogan, P.; et al. The Dairy Industry: Process, Monitoring, Standards, and Quality. In Descriptive Food Science; IntechOpen: London, UK, 2018; ISBN 978-1-78984-595-2. [Google Scholar]
- Rodriguez-Otero, J.L.; Hermida, M.; Centeno, J. Analysis of Dairy Products by Near-Infrared Spectroscopy: A Review. J. Agric. Food Chem. 1997, 45, 2815–2819. [Google Scholar] [CrossRef]
- Barbano, D.M.; Lynch, J.M.; Fleming, J.R. Direct and Indirect Determination of True Protein Content of Milk by Kjeldahl Analysis: Collaborative Study. J. Assoc. Off. Anal. Chem. 1991, 74, 281–288. [Google Scholar] [CrossRef]
- Feldsine, P.; Abeyta, C.; Andrews, W.H. AOAC International Methods Committee Guidelines for Validation of Qualitative and Quantitative Food Microbiological Official Methods of Analysis. J. AOAC Int. 2002, 85, 1187–1200. [Google Scholar] [CrossRef]
- Hicks, T.D.; Kuns, C.M.; Raman, C.; Bates, Z.T.; Nagarajan, S. Simplified Method for the Determination of Total Kjeldahl Nitrogen in Wastewater. Environments 2022, 9, 55. [Google Scholar] [CrossRef]
- Lynch, J.M.; Barbano, D.M. Kjeldahl Nitrogen Analysis as a Reference Method for Protein Determination in Dairy Products. J. AOAC Int. 1999, 82, 1389–1398. [Google Scholar] [CrossRef]
- Rhee, K.C. Determination of Total Nitrogen. Curr. Protoc. Food Anal. Chem. 2001, 00, B1.2.1–B1.2.9. [Google Scholar] [CrossRef]
- Barbano, D.M.; Clark, J.L.; Dunham, C.E.; Flemin, R.J. Kjeldahl Method for Determination of Total Nitrogen Content of Milk: Collaborative Study. J. Assoc. Off. Anal. Chem. 1990, 73, 849–859. [Google Scholar] [CrossRef]
- Sáez-Plaza, P.; Michałowski, T.; Navas, M.J.; Asuero, A.G.; Wybraniec, S. An Overview of the Kjeldahl Method of Nitrogen Determination. Part I. Early History, Chemistry of the Procedure, and Titrimetric Finish. Crit. Rev. Anal. Chem. 2013, 43, 178–223. [Google Scholar] [CrossRef]
- De Marchi, M.; Penasa, M.; Zidi, A.; Manuelian, C.L. Invited Review: Use of Infrared Technologies for the Assessment of Dairy Products—Applications and Perspectives. J. Dairy Sci. 2018, 101, 10589–10604. [Google Scholar] [CrossRef]
- Mendes, E.; Duarte, N. Mid-Infrared Spectroscopy as a Valuable Tool to Tackle Food Analysis: A Literature Review on Coffee, Dairies, Honey, Olive Oil and Wine. Foods 2021, 10, 477. [Google Scholar] [CrossRef]
- Federal University of Juiz De Fora; Anjos, V. Near And Mid Infrared Spectroscopy To Assess Milk Products Quality: A Review Of Recent Applications. J. Dairy Res. Technol. 2020, 3, 1–10. [Google Scholar] [CrossRef]
- Saxton, R.; McDougal, O.M. Whey Protein Powder Analysis by Mid-Infrared Spectroscopy. Foods 2021, 10, 1033. [Google Scholar] [CrossRef]
- Zappi, A.; Marassi, V.; Giordani, S.; Kassouf, N.; Roda, B.; Zattoni, A.; Reschiglian, P.; Melucci, D. Extracting Information and Enhancing the Quality of Separation Data: A Review on Chemometrics-Assisted Analysis of Volatile, Soluble and Colloidal Samples. Chemosensors 2023, 11, 45. [Google Scholar] [CrossRef]
- Amsaraj, R.; Ambade, N.D.; Mutturi, S. Variable Selection Coupled to PLS2, ANN and SVM for Simultaneous Detection of Multiple Adulterants in Milk Using Spectral Data. Int. Dairy J. 2021, 123, 105172. [Google Scholar] [CrossRef]
- Andrade, J.; Pereira, C.G.; Almeida, J.C.d., Jr.; Viana, C.C.R.; Neves, L.N.d.O.; Silva, P.H.F.d.; Bell, M.J.V.; Anjos, V.d.C.d. FTIR-ATR Determination of Protein Content to Evaluate Whey Protein Concentrate Adulteration. LWT 2019, 99, 166–172. [Google Scholar] [CrossRef]
- Mota, L.F.M.; Pegolo, S.; Baba, T.; Peñagaricano, F.; Morota, G.; Bittante, G.; Cecchinato, A. Evaluating the Performance of Machine Learning Methods and Variable Selection Methods for Predicting Difficult-to-Measure Traits in Holstein Dairy Cattle Using Milk Infrared Spectral Data. J. Dairy Sci. 2021, 104, 8107–8121. [Google Scholar] [CrossRef]
- Neto, H.A.; Tavares, W.L.F.; Ribeiro, D.C.S.Z.; Alves, R.C.O.; Fonseca, L.M.; Campos, S.V.A. On the Utilization of Deep and Ensemble Learning to Detect Milk Adulteration. BioData Min. 2019, 12, 13. [Google Scholar] [CrossRef]
- Zhu, X.; Guo, W.; Kang, F.; Kong, F.; Zhu, Q. Determination of Protein Content of Raw Fresh Cow’s Milk Using Dielectric Spectroscopy Combined with Chemometric Methods. Food Bioprocess Technol. 2016, 9, 2092–2102. [Google Scholar] [CrossRef]
- Soyeurt, H.; Grelet, C.; McParland, S.; Calmels, M.; Coffey, M.; Tedde, A.; Delhez, P.; Dehareng, F.; Gengler, N. A Comparison of 4 Different Machine Learning Algorithms to Predict Lactoferrin Content in Bovine Milk from Mid-Infrared Spectra. J. Dairy Sci. 2020, 103, 11585–11596. [Google Scholar] [CrossRef]
- Bonfatti, V.; Di Martino, G.; Carnier, P. Effectiveness of Mid-Infrared Spectroscopy for the Prediction of Detailed Protein Composition and Contents of Protein Genetic Variants of Individual Milk of Simmental Cows. J. Dairy Sci. 2011, 94, 5776–5785. [Google Scholar] [CrossRef]
- Tonolini, M.; Sørensen, K.M.; Skou, P.B.; Ray, C.; Engelsen, S.B. Prediction of α-Lactalbumin and β-Lactoglobulin Composition of Aqueous Whey Solutions Using Fourier Transform Mid-Infrared Spectroscopy and Near-Infrared Spectroscopy. Appl. Spectrosc. 2021, 75, 718–727. [Google Scholar] [CrossRef]
- Kaylegian, K.E.; Houghton, G.E.; Lynch, J.M.; Fleming, J.R.; Barbano, D.M. Calibration of Infrared Milk Analyzers: Modified Milk Versus Producer Milk1. J. Dairy Sci. 2006, 89, 2817–2832. [Google Scholar] [CrossRef]
- Wojciechowski, K.L.; Barbano, D.M. Prediction of Fatty Acid Chain Length and Unsaturation of Milk Fat by Mid-Infrared Milk Analysis1. J. Dairy Sci. 2016, 99, 8561–8570. [Google Scholar] [CrossRef]
- Portnoy, M.; Coon, C.; Barbano, D.M. Infrared Milk Analyzers: Milk Urea Nitrogen Calibration. J. Dairy Sci. 2021, 104, 7426–7437. [Google Scholar] [CrossRef]
- Lynch, J.M.; Barbano, D.M.; Fleming, J.R.; Barbano Laboratory; California Department of Food and Agriculture; Dairy One; Dairy Quality Control Institute, Inc.; Land O’Lakes; State of Wisconsin Department of Agriculture; U.S. Department of Agriculture (USDA) Atlanta Milk Market Administrator Laboratory; et al. Determination of the Total Nitrogen Content of Hard, Semihard, and Processed Cheese by the Kjeldahl Method: Collaborative Study. J. AOAC Int. 2002, 85, 445–455. [Google Scholar] [CrossRef]
- Santos, P.M.; Pereira-Filho, E.R.; Rodriguez-Saona, L.E. Rapid Detection and Quantification of Milk Adulteration Using Infrared Microspectroscopy and Chemometrics Analysis. Food Chem. 2013, 138, 19–24. [Google Scholar] [CrossRef]
- Duraipandian, S.; Bergholt, M.S.; Zheng, W.; Ho, K.Y.; Teh, M.; Yeoh, K.G.; So, J.B.Y.; Shabbir, A.; Huang, Z. Real-Time Raman Spectroscopy for in Vivo, Online Gastric Cancer Diagnosis during Clinical Endoscopic Examination. J. Biomed. Opt. 2012, 17, 081418. [Google Scholar] [CrossRef]
- Mujica, L.; Rodellar, J.; Fernández, A.; Güemes, A. Q-Statistic and T2-Statistic PCA-Based Measures for Damage Assessment in Structures. Struct. Health Monit. 2011, 10, 539–553. [Google Scholar] [CrossRef]
- Thennadil, S.N.; Dewar, M.; Herdsman, C.; Nordon, A.; Becker, E. Automated Weighted Outlier Detection Technique for Multivariate Data. Control Eng. Pract. 2018, 70, 40–49. [Google Scholar] [CrossRef]
- Lörchner, C.; Horn, M.; Berger, F.; Fauhl-Hassek, C.; Glomb, M.A.; Esslinger, S. Quality Control of Spectroscopic Data in Non-Targeted Analysis—Development of a Multivariate Control Chart. Food Control 2022, 133, 108601. [Google Scholar] [CrossRef]
- Mishra, P.; Passos, D. A Synergistic Use of Chemometrics and Deep Learning Improved the Predictive Performance of Near-Infrared Spectroscopy Models for Dry Matter Prediction in Mango Fruit. Chemom. Intell. Lab. Syst. 2021, 212, 104287. [Google Scholar] [CrossRef]
- Yang, K.; An, C.; Zhu, J.; Guo, W.; Lu, C.; Zhu, X. Comparison of Near-Infrared and Dielectric Spectra for Quantitative Identification of Bovine Colostrum Adulterated with Mature Milk. J. Dairy Sci. 2022, 105, 8638–8649. [Google Scholar] [CrossRef]
- Galvão, R.K.H.; Araujo, M.C.U.; José, G.E.; Pontes, M.J.C.; Silva, E.C.; Saldanha, T.C.B. A Method for Calibration and Validation Subset Partitioning. Talanta 2005, 67, 736–740. [Google Scholar] [CrossRef]
- Ferreira, R.d.A.; Teixeira, G.; Peternelli, L.A. Kennard-Stone Method Outperforms the Random Sampling in the Selection of Calibration Samples in SNPs and NIR Data. Ciênc. Rural 2021, 52, e20201072. [Google Scholar] [CrossRef]
- Li, X.; Kong, W.; Shi, W.; Shen, Q. A Combination of Chemometrics Methods and GC–MS for the Classification of Edible Vegetable Oils. Chemom. Intell. Lab. Syst. 2016, 155, 145–150. [Google Scholar] [CrossRef]
- Mishra, P.; Biancolillo, A.; Roger, J.M.; Marini, F.; Rutledge, D.N. New Data Preprocessing Trends Based on Ensemble of Multiple Preprocessing Techniques. TrAC Trends Anal. Chem. 2020, 132, 116045. [Google Scholar] [CrossRef]
- Torniainen, J.; Afara, I.O.; Prakash, M.; Sarin, J.K.; Stenroth, L.; Töyräs, J. Open-Source Python Module for Automated Preprocessing of near Infrared Spectroscopic Data. Anal. Chim. Acta 2020, 1108, 1–9. [Google Scholar] [CrossRef]
- Nørgaard, L.; Saudland, A.; Wagner, J.; Nielsen, J.P.; Munck, L.; Engelsen, S.B. Interval Partial Least-Squares Regression (iPLS): A Comparative Chemometric Study with an Example from Near-Infrared Spectroscopy. Appl. Spectrosc. 2000, 54, 413–419. [Google Scholar] [CrossRef]
- Leardi, R. Application of Genetic Algorithm–PLS for Feature Selection in Spectral Data Sets. J. Chemom. 2000, 14, 643–655. [Google Scholar] [CrossRef]
- Leardi, R.; Boggia, R.; Terrile, M. Genetic Algorithms as a Strategy for Feature Selection. J. Chemom. 1992, 6, 267–281. [Google Scholar] [CrossRef]
- Ostertag, F.; Schmidt, C.M.; Berensmeier, S.; Hinrichs, J. Development and Validation of an RP-HPLC DAD Method for the Simultaneous Quantification of Minor and Major Whey Proteins. Food Chem. 2021, 342, 128176. [Google Scholar] [CrossRef]
- Elgar, D.F.; Norris, C.S.; Ayers, J.S.; Pritchard, M.; Otter, D.E.; Palmano, K.P. Simultaneous Separation and Quantitation of the Major Bovine Whey Proteins Including Proteose Peptone and Caseinomacropeptide by Reversed-Phase High-Performance Liquid Chromatography on Polystyrene–Divinylbenzene. J. Chromatogr. A 2000, 878, 183–196. [Google Scholar] [CrossRef]
- Akiba, T.; Sano, S.; Yanase, T.; Ohta, T.; Koyama, M. Optuna: A Next-Generation Hyperparameter Optimization Framework. In Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Anchorage, AK, USA, 4–8 August 2019; Association for Computing Machinery: New York, NY, USA, 2019; pp. 2623–2631. [Google Scholar]
- Niero, G.; Penasa, M.; Gottardo, P.; Cassandro, M.; De Marchi, M. Short Communication: Selecting the Most Informative Mid-Infrared Spectra Wavenumbers to Improve the Accuracy of Prediction Models for Detailed Milk Protein Content. J. Dairy Sci. 2016, 99, 1853–1858. [Google Scholar] [CrossRef]
- Dabrowska, A.; David, M.; Freitag, S.; Andrews, A.M.; Strasser, G.; Hinkov, B.; Schwaighofer, A.; Lendl, B. Broadband Laser-Based Mid-Infrared Spectroscopy Employing a Quantum Cascade Detector for Milk Protein Analysis. Sens. Actuators B Chem. 2022, 350, 130873. [Google Scholar] [CrossRef]
- Ayvaz, H.; Sierra-Cadavid, A.; Aykas, D.P.; Mulqueeney, B.; Sullivan, S.; Rodriguez-Saona, L.E. Monitoring Multicomponent Quality Traits in Tomato Juice Using Portable Mid-Infrared (MIR) Spectroscopy and Multivariate Analysis. Food Control 2016, 66, 79–86. [Google Scholar] [CrossRef]
- Müller-Maatsch, J.; van Ruth, S.M. Handheld Devices for Food Authentication and Their Applications: A Review. Foods 2021, 10, 2901. [Google Scholar] [CrossRef]
- Crocombe, R.A. Portable Spectroscopy. Appl. Spectrosc. 2018, 72, 1701–1751. [Google Scholar] [CrossRef]
- Kappacher, C.; Trübenbacher, B.; Losso, K.; Rainer, M.; Bonn, G.K.; Huck, C.W. Portable vs. Benchtop NIR-Sensor Technology for Classification and Quality Evaluation of Black Truffle. Molecules 2022, 27, 589. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Mean | SD | Min. | Max. |
|---|---|---|---|---|
| True Protein (%) | 3.1506 | 0.6776 | 2.0034 | 4.2631 |
| Casein (%) | 2.5336 | 0.5529 | 1.6120 | 3.4028 |
| Whey (%) | 0.6170 | 0.1426 | 0.2763 | 0.9075 |
| β-LG (mg/mL) | 3.3500 | 0.7600 | 2.2200 | 4.6000 |
| α-LA (mg/mL) | 1.6000 | 0.2900 | 1.0800 | 2.0800 |
| Preprocessing | β-LG | α-LA | n_Comps |
|---|---|---|---|
| Baseline + normalize+ | |||
| SavGol(filter_win = 115, | 93% | 94% | 16 |
| poly_order = 1, deriv_order = 0) | |||
| normalize + SavGol(filter_win = 99, | 93% | 93% | 20 |
| poly_order = 3, deriv_order = 0) | |||
| SavGol(filter_win = 115, | 93% | 93% | 20 |
| poly_order = 2, deriv_order = 0) | |||
| LSNV + normalize + | |||
| SavGol(filter_win = 99, | 77% | 80% | 8 |
| poly_order = 0, deriv_order = 2) | |||
| SNV + SavGol(filter_win = 77, | 66% | 64% | 8 |
| poly_order = 3, deriv_order = 0) | |||
| EMSC + SavGol(filter_win = 191, | 28% | 31% | 5 |
| poly_order = 1, deriv_order = 1) |
| Model | Parameter | Search Space | -LG_Opt | -LA_Opt |
|---|---|---|---|---|
| C | loguniform(5 × 10−³, 1 × 10³) | 792.3681 | 96.3447 | |
| epsilon | uniform (0.01, 0.9) | 0.0311 | 0.01069 | |
| SVR | kernel | [‘linear’, ‘rbf’, ‘poly’] | linear | linear |
| degree | [1,2,3,4] | 3 | 1 | |
| gamma | Loguniform (1 × 10−5, 1 × 105) | 0.0126 | 284.4739 | |
| alpha | Loguniform (1 × 10−5, 10) | 0.00078 | 0.00095 | |
| [‘auto’, ‘svd’, ‘cholesky’, | ||||
| Ridge | solver | ‘lsqr’, ‘sparse_cg’, ‘sag’ | lsqr | sparse_cg |
| , ‘saga’] | ||||
| fit_intercept | [True, False] | TRUE | TRUE | |
| LR | fit_intercept | [True, False] | TRUE | TRUE |
| copy_X | [True, False] | FALSE | FALSE | |
| PLS | n_comps | range (1,20) | 14 | 14 |
| KS(P) | RS(P) | LOROCV | LOSOCV | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CM | Protein | RMSE | RMSE | RMSE | RMSE | |||||
| Raw | PLS | β−LG | 93.00% | 0.21 | 89.70% | 0.23 | 92.10% | 0.15 | 90.60% | 0.22 |
| α−LA | 93.80% | 0.08 | 86.80% | 0.1 | 90.70% | 0.06 | 89.40% | 0.09 | ||
| SVR | β−LG | 92.70% | 0.21 | 85.90% | 0.28 | 88.90% | 0.18 | 87.40% | 0.26 | |
| α−LA | 95.30% | 0.07 | 86.80% | 0.1 | 90.50% | 0.06 | 89.30% | 0.09 | ||
| Ridge | β−LG | 92.40% | 0.22 | 87.50% | 0.26 | 89.50% | 0.18 | 88.00% | 0.25 | |
| α−LA | 94.20% | 0.07 | 86.80% | 0.1 | 89.80% | 0.06 | 88.60% | 0.1 | ||
| LR | β−LG | 88.70% | 0.27 | 88.90% | 0.25 | −9.7 × 1018 | 1.3 × 108 | −8.7× 1018 | 2.1× 108 | |
| α−LA | 89.50% | 0.1 | 88.80% | 0.1 | −6.7 × 1018 | 6.30 × 108 | −1.30 × 1019 | 1.0 × 109 | ||
| OP+GA | PLS | β−LG | 92.30% | 0.21 | 90.00% | 0.23 | 92.60% | 0.15 | 91.70% | 0.21 |
| α−LA | 93.40% | 0.08 | 89.00% | 0.09 | 92.20% | 0.05 | 91.10% | 0.08 | ||
| SVR | β−LG | 94.70% | 0.18 | 90.50% | 0.23 | 92.60% | 0.15 | 91.80% | 0.21 | |
| α−LA | 96.50% | 0.06 | 89.20% | 0.09 | 92.70% | 0.05 | 91.90% | 0.08 | ||
| Ridge | β−LG | 93.50% | 0.2 | 90.40% | 0.23 | 92.60% | 0.15 | 91.60% | 0.21 | |
| α−LA | 95.80% | 0.06 | 88.80% | 0.1 | 92.30% | 0.05 | 91.50% | 0.08 | ||
| LR | β−LG | 81.20% | 0.34 | 85.40% | 0.28 | 89.10% | 0.18 | 88.10% | 0.25 | |
| α−LA | 90.00% | 0.1 | 86.40% | 0.1 | 91.20% | 0.06 | 90.00% | 0.09 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babatunde, H.A.; Collins, J.; Lukman, R.; Saxton, R.; Andersen, T.; McDougal, O.M. SVR Chemometrics to Quantify β-Lactoglobulin and α-Lactalbumin in Milk Using MIR. Foods 2024, 13, 166. https://doi.org/10.3390/foods13010166
Babatunde HA, Collins J, Lukman R, Saxton R, Andersen T, McDougal OM. SVR Chemometrics to Quantify β-Lactoglobulin and α-Lactalbumin in Milk Using MIR. Foods. 2024; 13(1):166. https://doi.org/10.3390/foods13010166
Chicago/Turabian StyleBabatunde, Habeeb Abolaji, Joseph Collins, Rianat Lukman, Rose Saxton, Timothy Andersen, and Owen M. McDougal. 2024. "SVR Chemometrics to Quantify β-Lactoglobulin and α-Lactalbumin in Milk Using MIR" Foods 13, no. 1: 166. https://doi.org/10.3390/foods13010166
APA StyleBabatunde, H. A., Collins, J., Lukman, R., Saxton, R., Andersen, T., & McDougal, O. M. (2024). SVR Chemometrics to Quantify β-Lactoglobulin and α-Lactalbumin in Milk Using MIR. Foods, 13(1), 166. https://doi.org/10.3390/foods13010166