
Simultaneous Rapid Determination of Seven Alternaria Toxins in Tuberous Crops during Storage Using QuEChERS Coupled with Ultrahigh-Performance Liquid Chromatography-Tandem Mass Spectrometry
 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
2.2. Samples
2.3. Toxin Extraction from Tubers Using the QuEChERS Procedure
2.4. Toxin Determination Using UPLC–MS/MS
2.5. Evaluation of Recovery and Matrix Effect
2.6. Method Validation
3. Results and Discussion
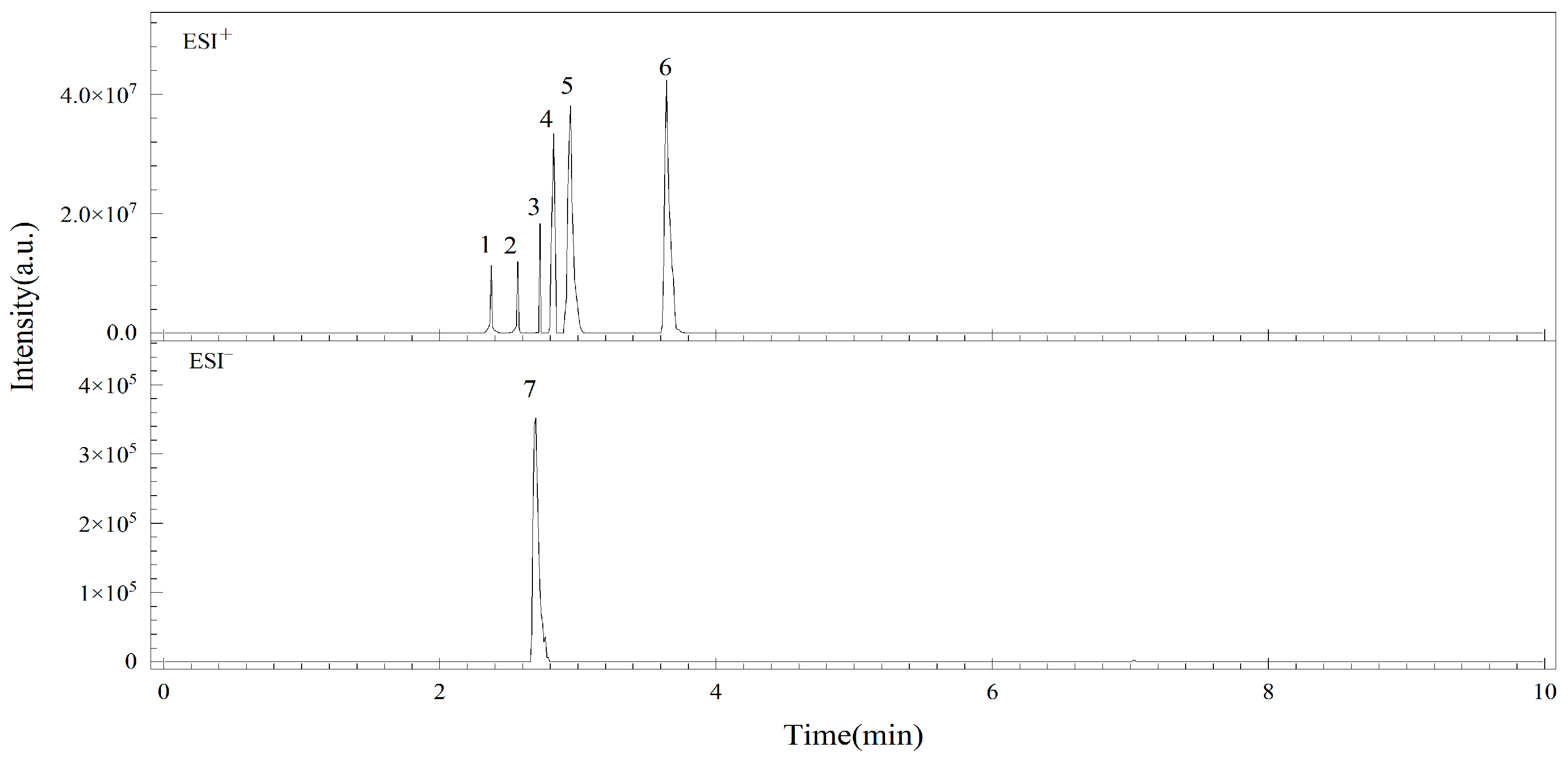
3.1. Optimization of MS/MS Conditions
3.2. Optimization of UPLC Conditions
3.2.1. Selection of UPLC Columns
3.2.2. Mobile Phase Selection
3.3. Optimization of Sample Pretreatment
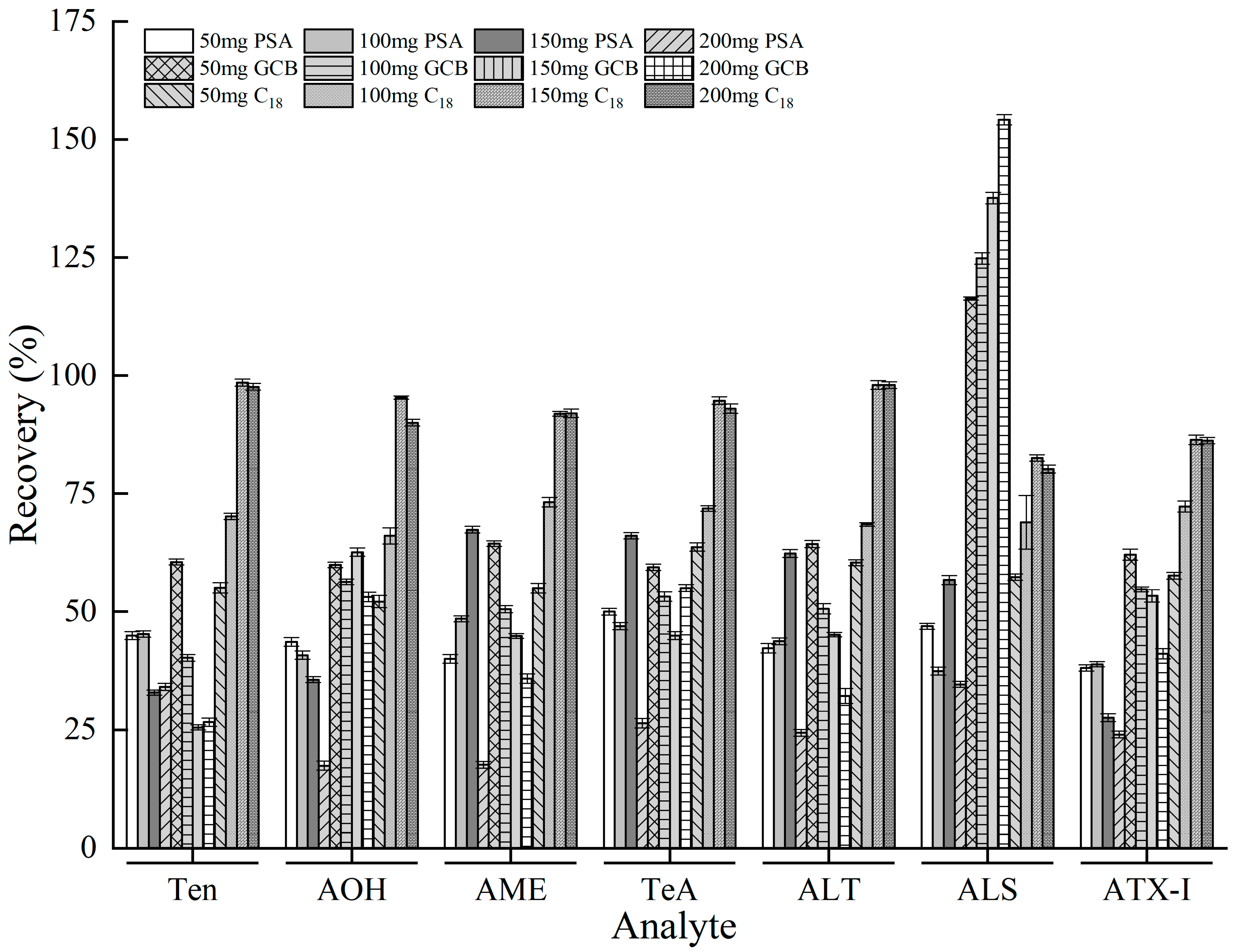
3.4. Optimization of QuEChERS Clean-Up
3.5. Method Validation
3.5.1. Calibration and Method Validation
3.5.2. Matrix Effect
3.6. Measurement of Real Samples during Storage
3.7. Comparison among Analytical Methods for Alternaria Toxins
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Xie, C.H. Potato Industry: Status and Development. J. Chin. Huazhong Agric. Univ. (Soc. Sci. Ed.) 2012, 17, 1–4. [Google Scholar]
- Li, D.; Wu, C.; Yin, H.Q. Current Status and Countermeasures of Potato Industry in Enshi Autonomous Prefecture. J. Chin. Chin. Potato J. 2010, 1, 3–5. [Google Scholar]
- Tymon, L.S.; Peever, T.L.; Johnson, D.A. Identification and Enumeration of Small-Spored Alternaria Species Associated with Potato in the U.S. Northwest. Plant Dis. 2015, 3, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Xue, L.W.; Ding, P.; Jiang, J.H. Simultaneous Determination of Three Common Mycotoxins in Sweet Potato by Pressure Capillary Electrochromatography Combined with SPE. In Proceedings of the Sixth National Congress of the Chinese Society of Biological Engineering and the Ninth Annual Academic Conference, Shanghai, China, 6 November 2015. [Google Scholar]
- Patriarca, A.; Vaamonde, G.; Pinto, V.F. References. In Encyclopedia of Food Microbiology, 2nd ed.; Batt, C.A., Tortorello, M.L., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 19, pp. 54–60. ISBN 978-0-12-384733-1. [Google Scholar]
- Asta, C.D.; Cirlini, M.; Falavigna, C. Chapter Three-Mycotoxins from Alternaria: Toxicological Implications. Adv. Mol. Toxicol. 2014, 8, 107–121. [Google Scholar]
- Arcella, D.; Eskola, M.; Gómez-Ruiz, J.A. Dietary exposure assessment to Alternaria toxins in the European population, EFSA report. EFSA J. 2016, 14, 4654–4686. [Google Scholar]
- Budde-Rodriguez, S.; Pasche, J.S.; Mallik, I.; Gudmestad, N.C. Sensitivity of Alternaria spp. from potato to pyrimethanil, cyprodinil, and fludioxonil. Crop. Prot. 2022, 152, 105855–105863. [Google Scholar] [CrossRef]
- Fernández-Cruz, M.L.; Mansilla, M.L.; Tadeo, J.L. Mycotoxins in fruits and their processed products: Analysis, occurrence and health implications. J. Adv. Res. 2010, 1, 113–122. [Google Scholar] [CrossRef]
- Sedaghati, E.; Hokmabadi, H. References. In Encyclopedia of Food and Health; Motarjemi, Y., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 3, pp. 331–339. ISBN 978-0-12-378613-5. [Google Scholar]
- Tralamazza, S.M.; Piacentini, K.C.; Iwase, C.; Rocha, L.O. Toxigenic Alternaria species: Impact in cereals worldwide. Curr. Opin. Food. Sci. 2018, 23, 57–63. [Google Scholar] [CrossRef]
- Aichinger, G.; Favero, G.D.; Warth, B.; Marko, D. Alternaria toxins—Still emerging? Compr. Rev. Food Sci. F 2021, 20, 4390–4406. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Q.; Mao, X.; Sun, Q.H.; Wei, Z.X.; Li, J.; You, Y.L.; Zhao, J.Q.; Jiang, G.B.; Wu, Y.N.; Wang, L.P.; et al. Alternaria Mycotoxins: An Overview of Toxicity, Metabolism, and Analysis in Food. J. Agric. Food Chem. 2021, 69, 7817–7830. [Google Scholar] [CrossRef]
- Moretti, A.; Sarrocco, S. References. In Encyclopedia of Food and Health; Motarjemi, Y., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 12, pp. 162–168. ISBN 978-0-12-378613-5. [Google Scholar]
- Walravens, J.; Mikula, H.; Rychlik, M.; Asam, S.; Saeger, S.D. Validated UPLC-MS/MS Methods to Quantitate Free and Conjugated Alternaria Toxins in Commercially Available Tomato Products, Fruit and Vegetable Juices in Belgium. J. Agric. Food Chem. 2016, 64, 5101–5109. [Google Scholar] [CrossRef] [PubMed]
- Gotthardt, M.; Asam, S.; Gunkel, K.; Moghaddam, A.F.; Rychlik, M. Quantitation of Six Alternaria Toxins in Infant Foods Applying Stable Isotope Labeled Standards. Front. Microbiol. 2019, 10, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Jiang, N.; Xian, H.; Wei, D.Z.; Feng, X.Y. A single-step solid phase extraction for the simultaneous determination of 8 mycotoxins in fruits by ultra-high performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2016, 1429, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Ryu, D. Worldwide Occurrence of Mycotoxins in Cereals and Cereal Derived Food Products: Public Health Perspectives of Their Co-Occurrence. J. Agric. Food Chem. 2017, 65, 7034–7051. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Schaab, M.R.; Southwood, G.; Tor, E.R.; Aston, L.S.; Song, W.; Eitzer, B.; Majumdar, S.; Lapainis, T.; Ma, H. A Collaborative Study: Determination of Mycotoxins in Corn, Peanut Butter, and Wheat Flour Using Stable Isotope Dilution Assay (SIDA) and Liquid Chromatography–Tandem Mass Spectrometry (LC-MS/MS). J. Agric. Food Chem. 2017, 65, 7138–7152. [Google Scholar] [CrossRef] [PubMed]
- Lattanzio, V.M.T.; Solfrizzo, M.; Visconti, A.; Angelo, V. Simultaneous determination of aflatoxins, ochratoxin A andFusarium toxins in maize by liquid chromatography/tandem mass spectrometry after multitoxin immunoaffinity cleanup. Rapid Commun. Mass Spectrom. 2007, 21, 3253–3261. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.B.; Dong, L.; Liu, J.; Lu, L.; Zhao, M.M.; Ding, H.; Wang, X.H.; Hu, D.J.; Zhou, Y.X. Research progress of QuEChERS in the detection of mycotoxins in food. J. Chin. Food Sci. 2014, 35, 286–291. [Google Scholar]
- Zhang, Y.; Lan, F.; Zhang, F.; Shen, J.C.; Chu, X.G. Determination of 41 hormones in cereal feeds by liquid chromatography-tandem mass spectrometry. J. Chin. Chem. 2011, 29, 523–524. [Google Scholar] [CrossRef]
- Logrieco, A.; Moretti, A.; Solfrizzo, M. Alternaria toxins and plant diseases: An overview of origin, occurrence and risks. World Mycotoxin J. 2009, 2, 129–140. [Google Scholar] [CrossRef]
- Michele, S.; Lucia, G.; Rita, B.; Martina, C.; Angela, P.; Ivan, P. Multimycotoxin Analysis by LC-MS/MS in Cereal Food and Feed: Comparison of Different Approaches for Extraction, Purification, and Calibration. J. AOAC Int. 2018, 101, 647–657. [Google Scholar]
- Woo, S.Y.; Ryu, S.Y.; Tian, F.; Lee, S.Y.; Park, S.B.; Chun, H.S. Simultaneous Determination of Twenty Mycotoxins in the Korean Soybean Paste Doenjang by LC-MS/MS with Immunoaffinity Cleanup. Toxins 2019, 11, 594. [Google Scholar] [CrossRef] [PubMed]
- Rubert, J.; Dzuman, Z.; Vaclavikova, M.; Zachariasova, M.; Soler, C.; Hajslova, J. Analysis of mycotoxins in barley using ultra high liquid chromatography high resolution mass spectrometry: Comparison of efficiency and efficacy of different extraction procedures. Talanta 2012, 99, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.B.; Yang, J.H.; Niu, X.K.; Tangni, E.K.; Zhao, Z.H.; Han, Z.A. Reliable and accurate UHPLC-MS/MS method for screening of Aspergillus, Penicillium and Alternaria mycotoxins in orange, grape and apple juices. Ana. Methods 2021, 13, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Vogelgesang, J.; Hädrich, J. Limits of detection, identification anddetermination: A statistical approach for practitioners. Accredit. Qual. Assur. 1998, 3, 242–255. [Google Scholar] [CrossRef]
- Song, H.T.; Li, C.Y.; Wan, Y.Y.; Ding, X.S.; Tan, X.Y.; Dai, G.L.; Liu, S.J.; Ju, W.Z. Screen astragalosides from Huangqi injections by LC-TOF-MS-basedmass defect filtering approach. China J. Chin. Mater. Med. 2017, 42, 686–695. [Google Scholar]
- Berardis, S.; Paola, E.L.; Montevecchi, G.; Garbini, D.; Masino, F.; Antonelli, A.; Melucci, D. Determination of four Alternaria alternata mycotoxins by QuEChERS approach coupled with liquid chromatography-tandem mass spectrometry in tomato-based and fruit-based products. Food Res. Int. 2018, 106, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Wong, J.W.; Zhang, K.; Hayward, D.G.; Lee, N.S.; Trucksess, M.W. Multi-mycotoxin Analysis of Finished Grain and Nut Products Using High-Performance Liquid Chromatography–Triple-Quadrupole Mass Spectrometry. J. Agric. Food Chem. 2013, 61, 4771–4782. [Google Scholar] [CrossRef]
- González-Jartín, J.M.; Alfonso, A.; Rodríguez, I.; Sainz, M.J.; Vieytes, M.R.; Botana, L.M. A QuEChERS based extraction procedure coupled to UPLC-MS/MS detection for mycotoxins analysis in beer. Food Chem. 2019, 275, 703–710. [Google Scholar] [CrossRef]
- Dong, H.; Xian, Y.; Xiao, K.; Wu, Y.; Zhu, L.; He, J. Development and comparison of single-step solid phase extraction and QuEChERS clean-up for the analysis of 7 mycotoxins in fruits and vegetables during storage by UHPLC-MS/MS. Food Chem. 2018, 274, 471–479. [Google Scholar] [CrossRef]
- Karageorgou, E.; Myridakis, A.; Stephanou, E.G.; Samanidou, V. Multiresidue LC-MS/MS analysis of cephalosporins and quinolones in milk following ultrasound-assisted matrix solid-phase dispersive extraction combined with the quick, easy, cheap, effective, rugged, and safe methodology. J. Sep. Sci. 2013, 36, 2020–2027. [Google Scholar] [CrossRef]
- Wilkowska, A.; Biziuk, M. Determination of pesticide residues in food matrices using the QuEChERS methodology. Food Chem. 2011, 125, 803–812. [Google Scholar] [CrossRef]
- Zhang, C.; Deng, Y.; Zheng, J.; Zhang, Y.; Liao, C.; Su, L.; Zhou, Y.; Gong, D.; Chen, L. The application of the QuEChERS methodology in the determination of antibiotics in food: A review. TRAC-Trend Anal. Chem. 2019, 118, 517–537. [Google Scholar] [CrossRef]
- Zhou, J.; Xu, J.J.; Cai, Z.X.; Huang, B.F.; Jin, M.C.; Ren, Y.P. Simultaneous determination of five Alternaria toxins in cereals using QuEChERS-based methodology. J. Chromatogr. B 2017, 1068, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Han, S.; Lu, M.L.; Wang, P.Y.; Han, J.; Wang, J.H. Modified QuEChERS method combined with ultra-high performance liquid chromatography tandem mass spectrometry for the simultaneous determination of 26 mycotoxins in sesame butter. J. Chromatogr. B 2014, 970, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Chen, L.; Xian, Y.; Hou, X.; Liang, M.; Dong, H.; Chen, J. Quantitative analysis of fourteen heterocyclic aromatic amines in bakery products by a modified QuEChERS method coupled to ultra-high performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS). Food Chem. 2019, 298, 125048–125065. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Gao, G.T.; Ha, Y.M.; Fan, J.L.; Chen, Y.T. Research Progress of Alternaria Mycotoxins in Cereals. J. Chin. Acta Agric. Nucl. Sin. 2017, 31, 334–341. [Google Scholar]
- Monbaliu, S.; Poucke, C.V.; Detavernier, C. Occurrence of mycotoxins in feed as analyzed by a multi-mycotoxin LC-MS/MS Method. J. Agric. Food Chem. 2010, 58, 66–71. [Google Scholar] [CrossRef]
- Reinholds, I.; Bogdanova, E.; Pugajeva, I.; Alksne, L.; Bartkevics, V. Determination of fungi and multi-class mycotoxins in camelia sinensis and herbal teas and dietary exposure assessment. Toxins 2020, 12, 555. [Google Scholar] [CrossRef]
- Lan, F.; Wang, X.Y.; Yao, J.; Jiang, W.; Xu, J.J.; Sun, L.; Wang, Z.X. Determination of Typical Alternaria Toxins in Edible Vegetable Oil by Ultra-high Liquid Performance Chromatography-Tandem Mass Spectrometry. J. Chin. Food Sci. 2022, 43, 338–343. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | Retention Time/min | Ionization Mode | Parent Ion (m/z) | Daughter Ion (m/z) | Cone Voltage/V | Collision Energy/eV |
|---|---|---|---|---|---|---|
| Ten | 2.85 | ESI+ | 415.4 | 132.1 * 115.1 | 25 | 13.18 |
| AME | 3.55 | ESI+ | 259.2 | 258.2 * 128.1 | 25 | 30.25 |
| AOH | 2.73 | ESI+ | 273.2 | 185.1 * 213.2 | 25 | 40.25 |
| TeA | 2.62 | ESI+ | 198.2 | 125.1 * 153.1 | 25 | 15.12 |
| ALT | 2.37 | ESI+ | 293.2 | 257.2 * 275.4 | 25 | 12.8 |
| ALS | 2.56 | ESI+ | 291.2 | 227.2 * 255.2 | 25 | 30.18 |
| ATX-I | 2.68 | ESI− | 353.1 | 333.3 * 315.25 | 25 | 8.10 |
| Analyte | ME | Linear Range(μg/L) | Linear Equation | R2 | LOD (μg/kg) | LOQ (μg/kg) | Low Spike Levels | Medium Spike Levels | High Spike Levels | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean Recovery (%) (n = 6) | RSDr (%) (n = 6) | RSDwR (%) (n = 24) | Mean Recovery (%) (n = 6) | RSDr (%) (n = 6) | RSDwR (%) (n = 24) | Mean Recovery (%) (n = 6) | RSDr (%) (n = 6) | RSDwR (%) (n = 24) | |||||||
| TeA | 115.02 | 2.5–500 | y = 8513.11x + 992.03 | 0.9978 | 0.27 | 0.89 | 96.21 | 3.56 | 4.21 | 97.03 | 3.67 | 4.30 | 102.37 | 3.77 | 4.56 |
| AOH | 113.90 | y = 7580.95x + 579.78 | 0.9996 | 0.25 | 0.83 | 95.66 | 4.21 | 4.52 | 94.69 | 3.96 | 4.69 | 97.21 | 4.06 | 4.82 | |
| AME | 133.20 | y = 16842.20x + 3351.28 | 0.9990 | 0.70 | 2.31 | 92.01 | 4.35 | 4.52 | 93.25 | 4.69 | 4.79 | 95.21 | 4.58 | 4.36 | |
| ALS | 92.71 | y = 481x + 226.58 | 0.9973 | 0.32 | 1.06 | 93.61 | 3.52 | 4.02 | 94.50 | 3.69 | 4.50 | 96.00 | 4.01 | 4.15 | |
| ALT | 84.50 | y = 976.24x + 102.31 | 0.9987 | 0.52 | 1.72 | 104.00 | 5.18 | 6.31 | 83.24 | 5.69 | 6.29 | 96.21 | 5.54 | 6.58 | |
| ATX-I | 123.51 | y = 5015.64x + 568.28 | 0.9997 | 0.61 | 2.01 | 86.65 | 6.23 | 6.52 | 92.51 | 6.17 | 6.58 | 83.20 | 5.98 | 6.39 | |
| Ten | 117.72 | y = 8277.02x + 253.09 | 0.9939 | 0.50 | 1.65 | 93.21 | 6.55 | 7.26 | 90.51 | 6.49 | 7.19 | 89.03 | 6.50 | 3.69 | |
| Analytes | Taro (μg/kg, %) | Potato (μg/kg, %) | Sweet Potato (μg/kg, %) | Yam (μg/kg, %) | Cassava (μg/kg, %) |
|---|---|---|---|---|---|
| 5.14, 16.77 1 | - | 5.21, 33.33 | 5.08, 33.33 | - | |
| Ten | 5.46, 33.33 | - | 5.38, 33.33 | 5.54, 33.33 | - |
| 5.61, 33.33 | - | 6.98, 66.67 | 7.46, 66.67 | - | |
| - | 1.02, 16.77 | 2.01, 33.33 | - | - | |
| AME | - | 2.07, 16.77 | 2.65, 33.33 | - | - |
| - | 2.22, 33.33 | 3.25, 66.67 | - | - | |
| - | 3.01, 16.77 | 2.74, 33.33 | - | 2.79, 33.33 | |
| AOH | - | 3.18, 33.33 | 2.75, 33.33 | - | 4.23, 33.33 |
| - | 2.22, 33.33 | 4.73, 33.33 | - | 4.01, 66.67 | |
| 2.13, 16.77 | 3.07, 33.33 | 1.04, 33.33 | - | - | |
| TeA | 2.21, 16.77 | 3.33, 33.33 | 1.61, 33.33 | - | - |
| 2.59, 16.77 | 3.87, 33.33 | 2.82, 33.33 | - | - | |
| 9.47, 33.33 | 9.83, 33.33 | 9.53, 16.77 | 9.49, 33.33 | 9.79, 33.33 | |
| ALS | 5.13, 66.67 | 10.97, 33.33 | 9.61, 16.77 | 9.50, 33.33 | 9.88, 66.67 |
| 14.51, 66.67 | 11.01, 33.33 | 9.81, 33.33 | 9.57, 83.33 | 10.55, 83.33 | |
| - | 0.05, 16.77 | - | - | - | |
| ATX-I | - | 0.15, 16.77 | - | - | - |
| - | 0.12, 16.77 | - | - | - |
| Analyte | Sample | Method | Detection Technique | LOD (μg/kg) | LOQ (μg/kg) | Recovery (%) | Ref. |
|---|---|---|---|---|---|---|---|
| ALT, AOH, AME | Sweet Potato | SPE | Pressure Capillary Electrochromatography | 0.10–0.9 | 0.33–2.97 | 81.3–103.3 | [4] |
| ALT, AOH, Ten, AME, TeA | Fruits | SPE | UPLC–MS/MS. | 0.33 | 1.09 | 87.4–100.5 | [17] |
| ALT, AOH, TEN, AME, TeA | Cereals | DLLME | UPLC–MS/MS | 0.61–48.20 | 2.01–120.10 | 72.7–109.1 | [37] |
| ALT, AOH, Ten, AME | Feed | SPE | UPLC–MS/MS | 0.15–3.0 | 0.50–10.00 | 97.0–104.8 | [41] |
| AOH, ALT | Barley | QuEChERS | UPLC–MS/MS | 0.13–0.30 | 0.45–1.01 | 85.7–89.4 | [42] |
| TeA, AME, AOH, ALT, Ten | Edible Vegetable Oil | QuEChERS | UPLC–MS/MS | 0.06–0.12 | 2.00–20.00 | 80.5–105.2 | [43] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xing, J.; Wu, X.; Xu, X.; Cheng, H.; Shen, J.; Zheng, R.; Mao, L.; Luo, X.; Mu, Y.; Liu, Y. Simultaneous Rapid Determination of Seven Alternaria Toxins in Tuberous Crops during Storage Using QuEChERS Coupled with Ultrahigh-Performance Liquid Chromatography-Tandem Mass Spectrometry. Foods 2023, 12, 862. https://doi.org/10.3390/foods12040862
Xing J, Wu X, Xu X, Cheng H, Shen J, Zheng R, Mao L, Luo X, Mu Y, Liu Y. Simultaneous Rapid Determination of Seven Alternaria Toxins in Tuberous Crops during Storage Using QuEChERS Coupled with Ultrahigh-Performance Liquid Chromatography-Tandem Mass Spectrometry. Foods. 2023; 12(4):862. https://doi.org/10.3390/foods12040862
Chicago/Turabian StyleXing, Jiali, Xi Wu, Xiaorong Xu, Hai Cheng, Jian Shen, Ruihang Zheng, Lingyan Mao, Xiaohu Luo, Yinghua Mu, and Yu Liu. 2023. "Simultaneous Rapid Determination of Seven Alternaria Toxins in Tuberous Crops during Storage Using QuEChERS Coupled with Ultrahigh-Performance Liquid Chromatography-Tandem Mass Spectrometry" Foods 12, no. 4: 862. https://doi.org/10.3390/foods12040862
APA StyleXing, J., Wu, X., Xu, X., Cheng, H., Shen, J., Zheng, R., Mao, L., Luo, X., Mu, Y., & Liu, Y. (2023). Simultaneous Rapid Determination of Seven Alternaria Toxins in Tuberous Crops during Storage Using QuEChERS Coupled with Ultrahigh-Performance Liquid Chromatography-Tandem Mass Spectrometry. Foods, 12(4), 862. https://doi.org/10.3390/foods12040862