
An Accurate, Rapid and Cost-Effective Method for T-nos Detection Based on CRISPR/Cas12a



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Optimisation of Conventional PCR Methods
2.3. The Cas12a Bulk Assay
2.4. Statistics and Reproducibility
2.5. Real-Time PCR Studies to Detect NOS
2.6. Construction and Purification of Reference Plasmid DNA
3. Results
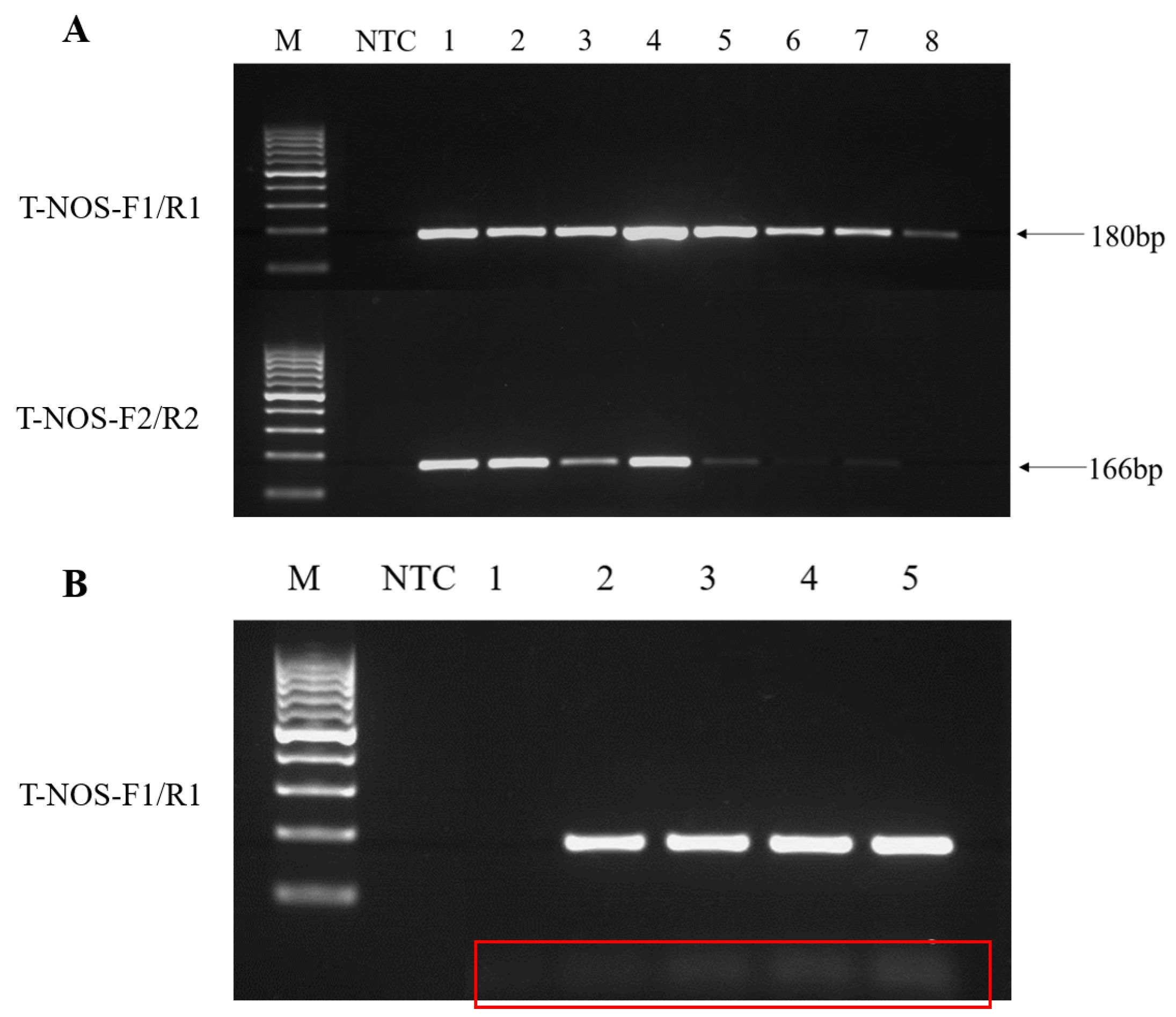
3.1. Optimisation of the PCR System and Amplification Conditions
3.2. Optimisation of the In-Tube CRISPR Detection System
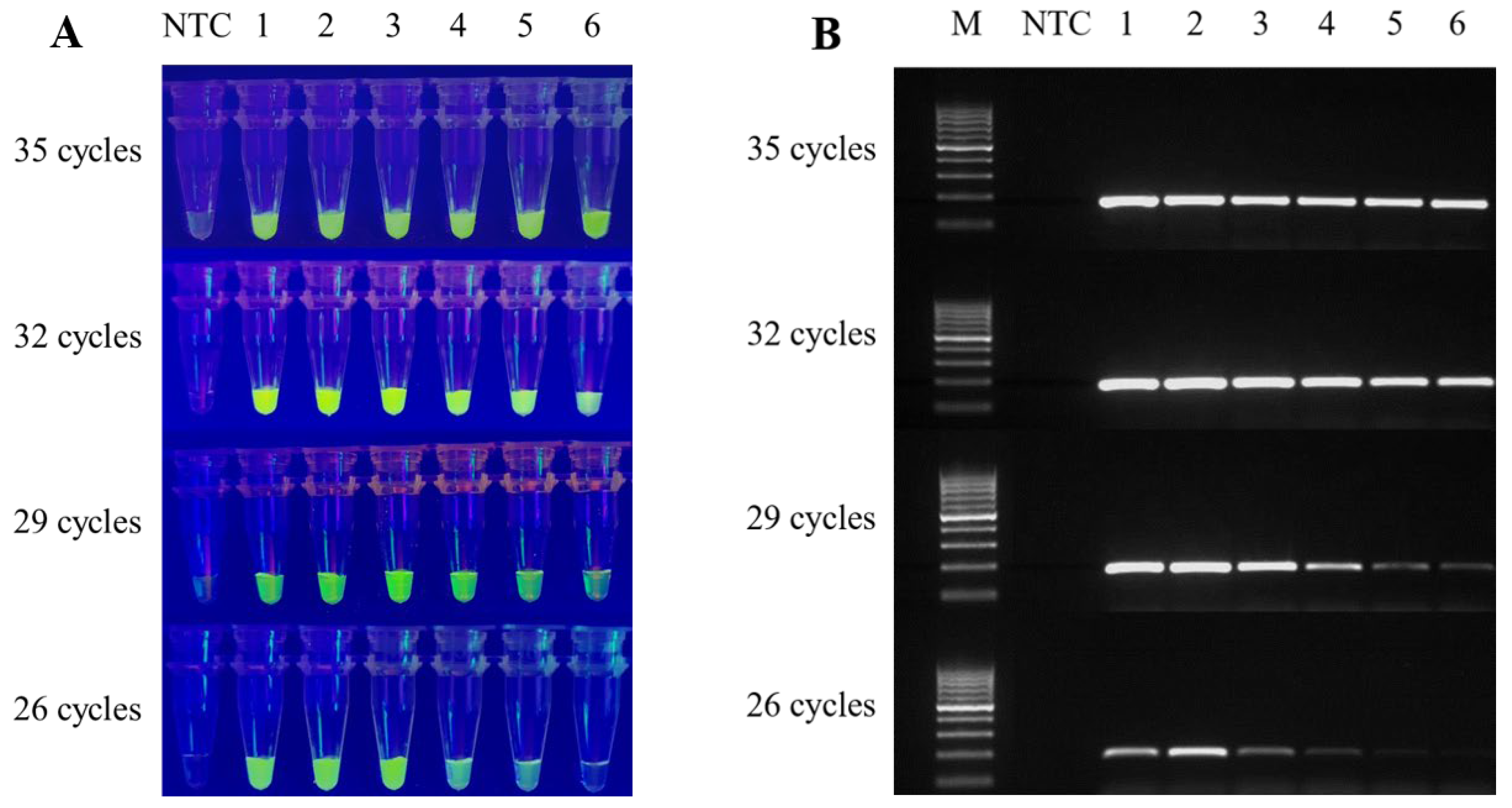
3.3. Combining the CRISPR Assay with Conventional PCR, and Optimising the Reaction Time
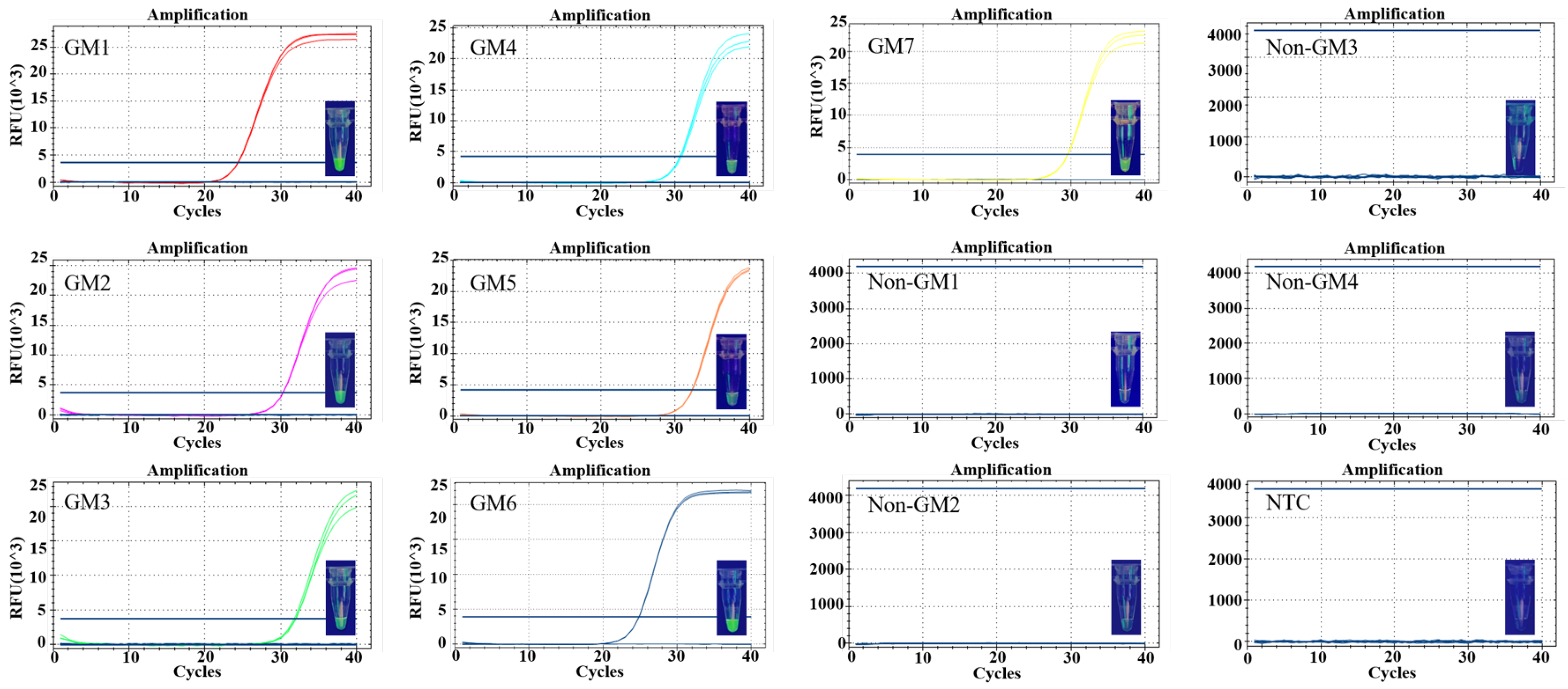
3.4. Specificity and Sensitivity of Detection
3.5. Detection of Actual Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Querci, M.; Van den Bulcke, M.; Zel, J.; Van den Eede, G.; Broll, H. New Approaches in GMO Detection. Anal. Bioanal. Chem. 2010, 396, 1991–2002. [Google Scholar] [CrossRef]
- Fraiture, M.A.; Herman, P.; Taverniers, I.; Loose, M.D.; Deforce, D. Current and New Approaches in GMO Detection: Challenges and Solutions. BioMed Res. Int. 2015, 2015, 392872. [Google Scholar] [CrossRef]
- Moghissi, A.A.; Jaeger, L.; Shafei, D.A.; Bloom, L.L. Regulatory Science Requirements of Labeling of Genetically Modified Food. Crit. Rev. Biotechnol. 2018, 38, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Kou, J.P.; Tang, Q.L.; Zhang, X.F. Agricultural GMO Safety Administration in China. J. Integr. Agric. 2015, 14, 2157–2165. [Google Scholar] [CrossRef]
- Buh Gašparič, M.; Tengs, T.; Paz, J.L.; Holst-Jensen, A.; Pla, M.; Esteve, T.V.; Zel, J.; Gruden, K. Comparison of Nine Different Real-Time Pcr Chemistries for Qualitative and Quantitative Applications in Gmo Detection. Anal. Bioanal. Chem. 2010, 396, 2023–2029. [Google Scholar] [CrossRef] [PubMed]
- Kaba, J.; Rola, M.; Materniak, M.; Kuźmak, J.; Nowicki, M. Isolation and Characterization of Caprine Arthritis Encephalitis Virus in Goats from Poland. Pol. J. Vet. Sci. 2009, 12, 183–188. [Google Scholar]
- Ma, L.; Zeng, F.; Huang, B.; Zhu, Y.; Wu, M.; Xu, F.; Xiao, L.; Huang, R.; Ma, J.; Cong, F.; et al. Point-of-Care Diagnostic Assay for Rapid Detection of Porcine Deltacoronavirus Using the Recombinase Polymerase Amplification Method. Transbound. Emerg. Dis. 2019, 66, 1324–1331. [Google Scholar] [CrossRef]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA Detection Using Recombination Proteins. PLoS Biol. 2006, 4, e204. [Google Scholar] [CrossRef]
- Chen, X.; Wang, X.; Jin, N.; Zhou, Y.; Huang, S.; Miao, Q.; Zhu, Q.; Xu, J. Endpoint Visual Detection of Three Genetically Modified Rice Events by Loop-Mediated Isothermal Amplification. Int. J. Mol. Sci. 2012, 13, 14421–14433. [Google Scholar] [CrossRef]
- Wang, X.F.; Fu, Z.F.; Chen, X.Y.; Peng, C.; Xu, X.L.; Wei, W.; Li, F.W.; Xu, J.F. Use of a Novel Metal Indicator to Judge Loop-Mediated Isothermal Amplification for Detecting the 35s Promoter. Anal. Bioanal. Chem. 2017, 409, 881–889. [Google Scholar] [CrossRef]
- Craw, P.; Balachandran, W. Isothermal Nucleic Acid Amplification Technologies for Point-of-Care Diagnostics: A Critical Review. Lab Chip 2012, 12, 2469–2486. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, F.; Li, Q.; Wang, L.; Fan, C. Isothermal Amplification of Nucleic Acids. Chem. Rev. 2015, 115, 12491–12545. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Hoshika, S.; Hutter, D.; Bradley, K.M.; Benner, S.A. Recombinase-Based Isothermal Amplification of Nucleic Acids with Self-Avoiding Molecular Recognition Systems (Samrs). Chembiochem 2014, 15, 2268–2274. [Google Scholar] [CrossRef] [PubMed]
- Godde, J.S.; Bickerton, A. The Repetitive DNA Elements Called Crisprs and Their Associated Genes: Evidence of Horizontal Transfer among Prokaryotes. J. Mol. Evol. 2006, 62, 718–729. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic Acid Detection with Crispr-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Tian, X.; Doudna, J.A. Crispr-Cas12a Target Binding Unleashes Single-Stranded Dnase Activity. Cold Spring Harb. Lab. 2017, 360, 436–439. [Google Scholar] [CrossRef]
- Harrington, L.B.; Burstein, D.; Chen, J.S.; Páez-Espino, D.; Ma, E.; Witte, I.P.; Cofsky, J.C.; Kyrpides, N.C.; Banfield, J.F.; Doudna, J.A. Programmed DNA Destruction by Miniature Crispr-Cas14 Enzymes. Science 2018, 362, 839–842. [Google Scholar] [CrossRef]
- Li, S.Y.; Cheng, Q.X.; Liu, J.K.; Nie, X.Q.; Zhao, G.P.; Wang, J. Crispr-Cas12a Has Both Cis- and Trans-Cleavage Activities on Single-Stranded DNA. Cell Res. 2018, 28, 491–493. [Google Scholar] [CrossRef]
- Yue, H.; Shu, B.; Tian, T.; Xiong, E.H.; Huang, M.Q.; Zhu, D.B.; Sun, J.; Liu, Q.; Wang, S.C.; Li, Y.R.; et al. Droplet Cas12a Assay Enables DNA Quantification from Unamplified Samples at the Single-Molecule Level. Nano Lett. 2021, 21, 4643–4653. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Shu, B.W.; Jiang, Y.Z.; Ye, M.M.; Liu, L.; Guo, Z.H.; Han, Z.P.; Wang, Z.; Zhou, X.M. An Ultralocalized Cas13a Assay Enables Universal and Nucleic Acid Amplification-Free Single-Molecule RNA Diagnostics. ACS Nano 2021, 15, 1167–1178. [Google Scholar] [CrossRef]
- Liu, H.; Wang, J.; Li, P.; Bai, L.; Jia, J.; Pan, A.; Long, X.; Cui, W.; Tang, X. Rapid Detection of P–35s and T-Nos in Genetically Modified Organisms by Recombinase Polymerase Amplification Combined with a Lateral Flow Strip. Food Control 2020, 107, 106775. [Google Scholar] [CrossRef]
- Yang, L.; Shen, H.; Pan, A.; Chen, J.; Huang, C.; Zhang, D. Screening and Construct-Specific Detection Methods of Transgenic Huafan No 1 Tomato by Conventional and Real-Time Pcr. J. Sci. Food Agric. 2005, 85, 2159–2166. [Google Scholar] [CrossRef]
- Losey, J.E.; Rayor, L.S.; Carter, M.E. Transgenic Pollen Harms Monarch Larvae. Nature 1999, 399, 214. [Google Scholar] [CrossRef]
- Finamore, A.; Roselli, M.; Britti, S.; Monastra, G.; Ambra, R.; Turrini, A.; Mengheri, E. Intestinal and Peripheral Immune Response to Mon810 Maize Ingestion in Weaning and Old Mice. J. Agric. Food Chem. 2008, 56, 11533–11539. [Google Scholar] [CrossRef] [PubMed]
- Amin, L.; Mahadi, Z.; Samian, A.L.; Ibrahim, R. Risk Perception Towards Food Safety Issues: Gm Foods Versus Non-Gm Foods. J. Food Agric. Environ. 2013, 11, 28–35. [Google Scholar]
- Kleter, G.A.; Kuiper, H.A. Genetically Modified (Gm) Food Safety: Pesticide Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Hoboken, NJ, USA, 2007. [Google Scholar]
- Wu, Z.Q.; Fu, X.B. Trangenesis: Principle, technologies and application. Res. Prog. Biotechnol. Eur. 2019, 9, 1–11. [Google Scholar]
- Reiting, R.; Broll, H.; Waiblinger, H.-U.; Grohmann, L. Collaborative Study of a T-Nos Real-Time Pcr Method for Screening of Genetically Modified Organisms in Food Products. J. Verbrauch. Lebensm. 2007, 2, 116–121. [Google Scholar] [CrossRef]
- Waiblinger, H.-U.; Ernst, B.; Anderson, A.; Pietsch, K. Validation and Collaborative Study of a P35s and T-Nos Duplex Real-Time Pcr Screening Method to Detect Genetically Modified Organisms in Food Products. Eur. Food Res. Technol. 2008, 226, 1221–1228. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, Y.; Li, W.; Zhang, L.; Li, Y.; Li, X.; Li, J.; Zhu, L.; Wu, G. Development of a General Method for Detection and Quantification of the P35s Promoter Based on Assessment of Existing Methods. Sci. Rep. 2014, 4, 7358. [Google Scholar] [CrossRef] [PubMed]
- Asiello, A.J.; Baeumner, A.J. Miniaturized Isothermal Nucleic Acid Amplification, a Review. Lab Chip 2011, 11, 1420–1430. [Google Scholar] [CrossRef] [PubMed]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N. Loop-Mediated Isothermal Amplification of DNA. Nucleic Acids Res. 2000, 28, e63. [Google Scholar] [CrossRef] [PubMed]
- Randhawa, G.J.; Singh, M.; Morisset, D.; Payal, S.; Jana, Z. Loop-Mediated Isothermal Amplification: Rapid Visual and Real-Time Methods for Detection of Genetically Modified Crops. J. Agric. Food Chem. 2013, 61, 11338–11346. [Google Scholar] [CrossRef] [PubMed]
- Daher, R.K.; Stewart, G.; Boissinot, M.; Bergeron, M.G. Recombinase Polymerase Amplification for Diagnostic Applications. Clin. Chem. J. Am. Assoc. Clin. Chem. 2016, 62, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liu, B.; Tang, D.; Niessner, R.; Chen, G.; Knopp, D. DNA-Based Hybridization Chain Reaction for Amplified Bioelectronic Signal and Ultrasensitive Detection of Proteins. Anal. Chem. 2012, 84, 5392–5399. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Macdonald, J.; von Stetten, F. Review: A Comprehensive Summary of a Decade Development of the Recombinase Polymerase Amplification. Analyst 2018, 144, 31–67. [Google Scholar] [CrossRef] [PubMed]
- Salisu, I.B.; Shahid, A.A.; Yaqoob, A.; Ali, Q.; Bajwa, K.S.; Rao, A.Q.; Husnain, T. Molecular Approaches for High Throughput Detection and Quantification of Genetically Modified Crops: A Review. Front. Plant Sci. 2017, 8, 1670. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, F.; Qian, C.; Wu, C.; Ye, Z.; Wang, L.; Qian, W.; Ping, J.; Wu, J.; Ying, Y. Counting DNA Molecules with Visual Segment-Based Readouts in Minutes. Chem. Commun. 2018, 54, 1105–1108. [Google Scholar] [CrossRef] [PubMed]
- Broeders, S.; Huber, I.; Grohmann, L.; Berben, G.; Taverniers, I.; Mazzara, M.; Roosens, N.H.C.; Morisset, D. Guidelines for Validation of Qualitative Real-Time Pcr Methods. Trends Food Sci. Technol. 2014, 37, 115–126. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Peng, C.; Ding, L.; Su, Z.; Chen, X.; Wang, X.; Sun, M.; Xu, J. An Accurate, Rapid and Cost-Effective Method for T-nos Detection Based on CRISPR/Cas12a. Foods 2023, 12, 615. https://doi.org/10.3390/foods12030615
Wang Y, Peng C, Ding L, Su Z, Chen X, Wang X, Sun M, Xu J. An Accurate, Rapid and Cost-Effective Method for T-nos Detection Based on CRISPR/Cas12a. Foods. 2023; 12(3):615. https://doi.org/10.3390/foods12030615
Chicago/Turabian StyleWang, Yuling, Cheng Peng, Lin Ding, Zhixun Su, Xiaoyun Chen, Xiaofu Wang, Meihao Sun, and Junfeng Xu. 2023. "An Accurate, Rapid and Cost-Effective Method for T-nos Detection Based on CRISPR/Cas12a" Foods 12, no. 3: 615. https://doi.org/10.3390/foods12030615
APA StyleWang, Y., Peng, C., Ding, L., Su, Z., Chen, X., Wang, X., Sun, M., & Xu, J. (2023). An Accurate, Rapid and Cost-Effective Method for T-nos Detection Based on CRISPR/Cas12a. Foods, 12(3), 615. https://doi.org/10.3390/foods12030615