
Determination of Mycotoxins in Dried Fruits Using LC-MS/MS—A Sample Homogeneity, Troubleshooting and Confirmation of Identity Study


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cryogenic Milling
2.3. Sample Preparation
2.4. Particle Size Analysis
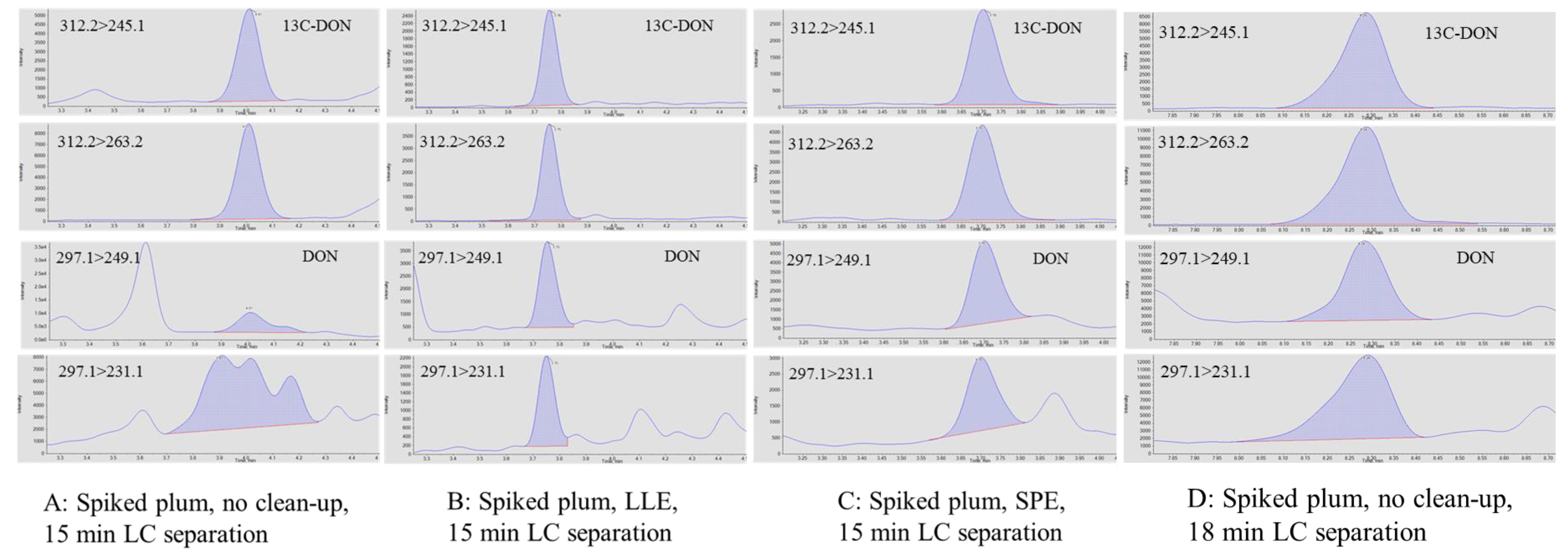
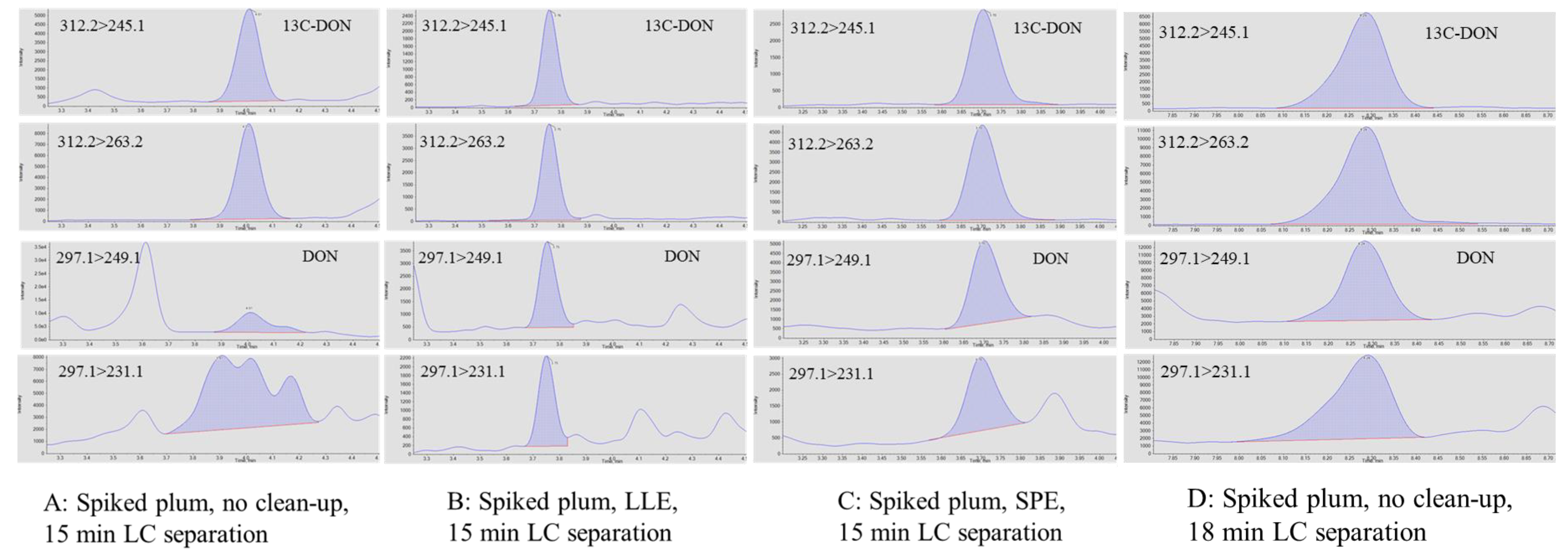
2.5. Troubleshooting Procedures for the Determination of DON in Plums
- Liquid-liquid extraction (LLE): Sample extracts (0.5 mL, 50% ACN) were mixed with dichloromethane (DCM) (1.0 mL) in a 15 mL tube, which was vigorously vortexed for 1 min and left at room temperature for 2 min until phase separation was observed. Half a milliliter of the bottom layer (DCM phase) was pipetted into a 1.5 mL LC vial for LC-MS analysis.
- Solid phase extraction (SPE): SPE was conducted using a vacuum manifold with a collection rack inserted inside. Florisil SPE cartridges (1.0 mL, 100 mg, Restek, College Station, PA, USA) were conditioned sequentially with 0.5 mL of isopropyl alcohol (IPA)/DCM (v/v, 50/50) and hexane. After an aliquot (0.5 mL) of the sample extracts was loaded, the SPE cartridges were washed using 5% DCM in hexane (0.5 mL) and DON was eluted and collected using 1.0 mL DCM in the 15 mL disposable centrifuge tubes on the collection rack. Elution rate was controlled at approximately 2 drops/s.
- LC separation: The elution gradient of the original LC method was evaluated to improve the separation of target analytes from matrix interreferences. The column, mobile phases, flow rate, injection volume, and column temperature of the method remained the same. In the original LC method, 10 mM ammonium formate/0.1% formic acid/water (A) and 10 mM ammonium formate/0.1% formic acid/methanol (B) were used as mobile phases. Gradient elution started at 5% B, ramped to 40% B in 2 min and then to 100% B by 10 min via linear gradient mode, held for 2.5 min, and changed to 5% B at 12 min, followed by 3 min of column conditioning. The modified gradient elution started at 5% B and held for 4 min, ramped to 100% B by 12 min via linear gradient mode, held for 2.5 min, and changed to 5% B at 15 min, followed by 3 min of column conditioning. Same flow rate, 0.3 mL/min was used for both gradients.
2.6. LC-MS Information Dependent Analysis (IDA) and Enhanced Product Ion (EPI) Analysis
3. Results and Discussion
3.1. Homogeneity Test
3.2. Recovery Studies
3.3. Determination of DON Using Liquid-Liquid Extraction and LC Separation
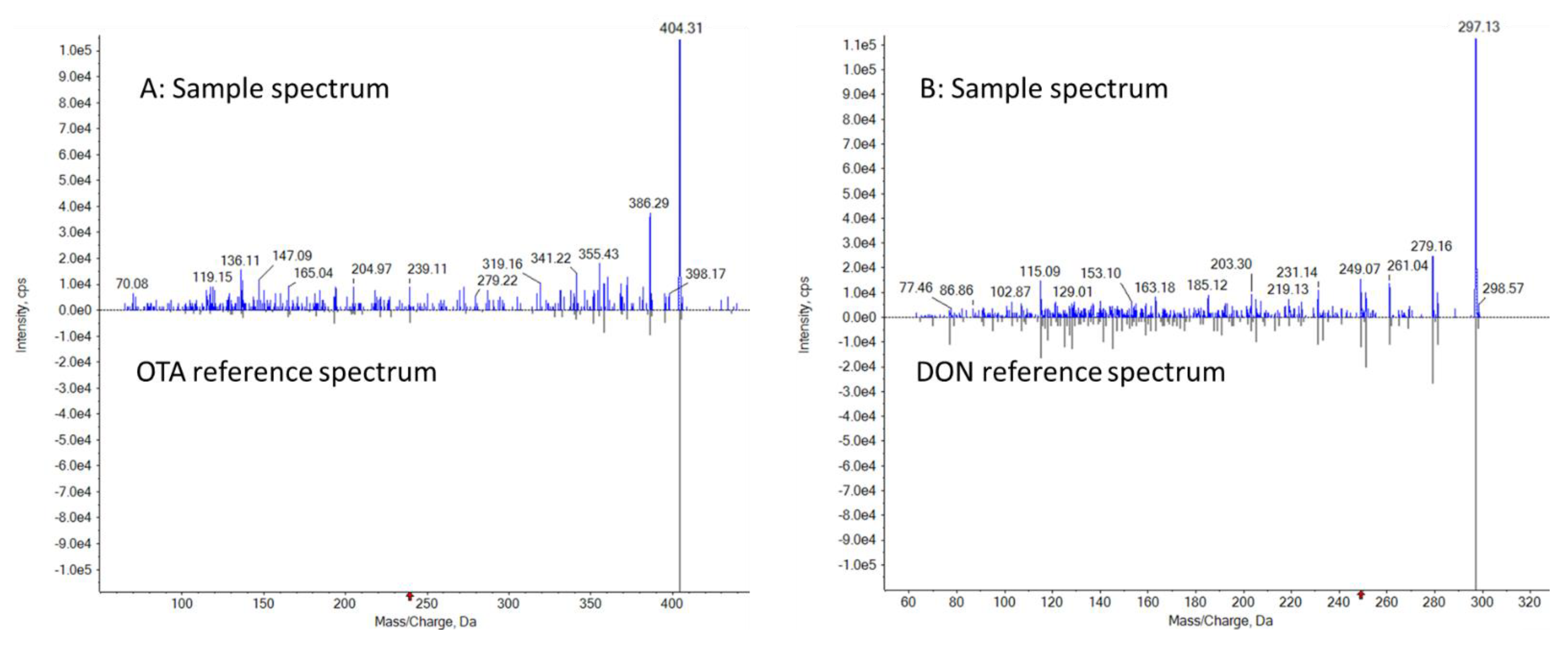
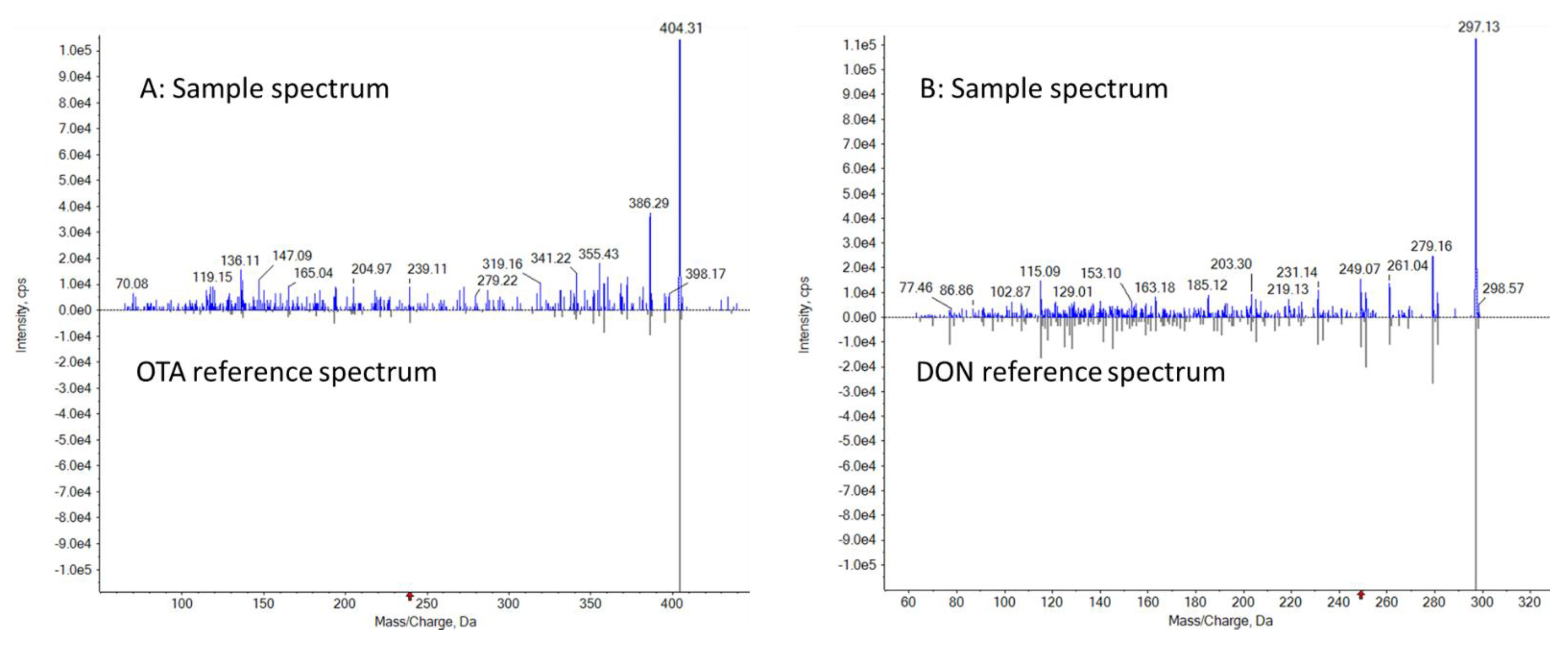
3.4. Analysis of Incurred Samples and Confirmation Using LC-MS-EPI Library
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Keast, D.R.; O’Neil, C.E.; Jones, J.M. Dried fruit consumption is associated with improved diet quality and reduced obesity in US adults: National Health and Nutrition Examination Survey, 1999–2004. Nutr. Res. 2011, 316, 460–467. [Google Scholar] [CrossRef] [PubMed]
- USDA. Commodity Specification for Dried Fruit. USA; 2019. Available online: https://www.ams.usda.gov/sites/default/files/media/CommoditySpecificationDriedFruitAugust%202019.pdf (accessed on 17 February 2022).
- USDA. Raisins: World Markets and Trade. USA; 2018. Available online: https://apps.fas.usda.gov/psdonline/circulars/raisins.pdf (accessed on 17 February 2022).
- Trucksess, M.W.; Scott, P.M. Mycotoxins in botanicals and dried fruits: A review. Food Addit. Contam. 2008, 25, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Cruz, M.; Mansilla, M.L.; Tadeo, J.L. Mycotoxins in fruits and their processed products: Analysis, occurrence and health implications. J. Adv. Res. 2010, 1, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Ozer, H.; Ozay, G. Mycotoxin risks and toxigenic fungi in date, prune and dried apricot among Mediterranean crops. Phytopathol. Mediterr. 2012, 51, 148–157. [Google Scholar]
- Drusch, S.; Ragab, W. Mycotoxins in fruits, fruit juices, and dried fruits. J. Food Prot. 2003, 66, 1514–1527. [Google Scholar] [CrossRef]
- EC. Commission Regulation (EC) No. 1881/2006 of 19 December 2006 Setting Maximum Levels for Certain Contaminants in Foodstuffs. 2006. Available online: https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=celex%3A32006R1881 (accessed on 17 February 2022).
- US FDA. Molecular Biology and Natural Toxins. In Compliance Program Guidance Manual; US FDA: Silver Spring, MD, USA, 2021. [Google Scholar]
- EU. The Rapid Alert System for Food and Feed Annual Report; EU: Maastricht, The Netherlands, 2016; Available online: https://ec.europa.eu/food/safety/rasff-food-and-feed-safety-alerts/reports-and-publications_en (accessed on 17 February 2022).
- EU. The Rapid Alert System for Food and Feed Annual Report; EU: Maastricht, The Netherlands, 2017; Available online: https://ec.europa.eu/food/safety/rasff-food-and-feed-safety-alerts/reports-and-publications_en (accessed on 17 February 2022).
- EU. The Rapid Alert System for Food and Feed Annual Report; EU: Maastricht, The Netherlands, 2018; Available online: https://ec.europa.eu/food/safety/rasff-food-and-feed-safety-alerts/reports-and-publications_en (accessed on 17 February 2022).
- EU. The Rapid Alert System for Food and Feed Annual Report; EU: Maastricht, The Netherlands, 2019; Available online: https://ec.europa.eu/food/safety/rasff-food-and-feed-safety-alerts/reports-and-publications_en (accessed on 17 February 2022).
- EU. The Rapid Alert System for Food and Feed Annual Report; EU: Maastricht, The Netherlands, 2020; Available online: https://ec.europa.eu/food/safety/rasff-food-and-feed-safety-alerts/reports-and-publications_en (accessed on 17 February 2022).
- Gilbert, J. Overview of mycotoxin methods, present status and future needs. Nat. Toxins 1999, 7, 347–352. [Google Scholar] [CrossRef]
- Sforza, S.; Dall’asta, C.; Marchelli, R. Recent advances in mycotoxin determination in food and feed by hyphenated chromatographic techniques/mass spectrometry. Mass Spectrom. Rev. 2006, 25, 54–76. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, A.L.; Caruso, G.; Cavaliere, C.; Foglia, P.; Samperi, R.; Laganà, A. Multiclass mycotoxin analysis in food, environmental and biological matrices with chromatography/mass spectrometry. Mass Spectrom. Rev. 2012, 31, 466–503. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhang, Z.; Hu, X.; Zhang, Q. Advanced hyphenated chromatographic-mass spectrometry in mycotoxin determination: Current status and prospects. Mass Spectrom. Rev. 2013, 32, 420–452. [Google Scholar] [CrossRef]
- Sulyok, M.; Krska, R.; Schuhmacher, R. Application of a liquid chromatography-tandem mass spectrometric method to multi-mycotoxin determination in raw cereals and evaluation of matrix effects. Food Addit. Contam. 2007, 24, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Rychlik, M.; Asam, S. Stable isotope dilution assays in mycotoxin analysis. Anal. Bioanal. Chem. 2008, 390, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Trufelli, H.P.; Famiglini, P.G.; Cappiello, A. An overview of matrix effects in liquid chromatography-mass spectrometry. Mass Spectrom. Rev. 2011, 30, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Schaab, M.R.; Southwood, G.; Tor, E.R.; Aston, L.S.; Song, W.; Eitzer, B.; Majumdar, S.; Lapainis, T.; Mai, H.; et al. A Collaborative Study: Determination of Mycotoxins in Corn, Peanut Butter, and Wheat Flour Using Stable Isotope Dilution Assay (SIDA) and Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS). J. Agric. Food Chem. 2017, 65, 7138–7152. [Google Scholar] [CrossRef] [PubMed]
- Reference Materials—Guidance for Characterization and Assessment of Homogeneity and Stability; ISO GUIDE 35:2017; International Organisation for Standardisation (ISO): Geneva, Switzerland, 2017.
- Zhang, K.; Wong, J.W.; Yang, P.; Hayward, D.G.; Sakuma, T.; Zou, Y.; Schreiber, A.; Borton, C.; Nguyen, T.V.; Kaushik, B.; et al. Protocol for an electrospray ionization tandem mass spectral product ion library: Development and application for identification of 240 pesticides in foods. Anal. Chem. 2012, 84, 5677–5684. [Google Scholar] [CrossRef] [PubMed]
- Particle Size Analysis—Laser Diffraction Methods, Part 1: General Principles International Organisation for Standardisation (ISO); ISO13320-1; ISO: Geneva, Switzerland, 1999.
- Deanne, F.M.E.R. Particle-Size Analysis: A Comparison of Various Methods II. Part. Part. Syst. Charact. 1997, 14, 278–282. [Google Scholar]
- Hackley, V.; Lum, L.; Gintautas, V.; Ferraris, C. Particle Size Analysis by Laser Diffraction Spectrometry: Application to Cementitious Powders; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2004. [Google Scholar]
- AOAC Official Method 977.16 Sampling for Aflatoxins Preparation for Sample Procedure First Action; AOAC: Rockville, MD, USA, 1977.
- Diana Di Mavungu, J.S.; Monbaliu, J.S.; Scippo, M.L.; Maghuin-Rogister, G.; Schneider, Y.G.; Larondelle, Y.; Callebaut, A.; Robbens, J.; Van Peteghem, C.; De Saeger, S. LC-MS/MS multi-analyte method for mycotoxin determination in food supplements. Food Addit. Contam. 2009, 26, 885–895. [Google Scholar] [CrossRef] [Green Version]
- Vaclavikova, M.; MacMahon, S.; Zhang, K.; Begley, T.H. Application of single immunoaffinity clean-up for simultaneous determination of regulated mycotoxins in cereals and nuts. Talanta 2013, 117, 345–351. [Google Scholar] [CrossRef]
- Zhang, K.; Wong, J.W.; Hayward, D.G.; Vaclavikova, M.; Liao, C.D.; Trucksess, M.W. Determination of mycotoxins in milk-based products and infant formula using stable isotope dilution assay and liquid chromatography tandem mass spectrometry. J. Agric. Food Chem. 2013, 61, 6265–6273. [Google Scholar] [CrossRef]
- Sulyok, M.; Stadler, D.; Steiner, D.; Krska, R. Validation of an LC-MS/MS-based dilute-and-shoot approach for the quantification of > 500 mycotoxins and other secondary metabolites in food crops: Challenges and solutions. Anal. Bioanal. Chem. 2020, 412, 2607–2620. [Google Scholar] [CrossRef] [Green Version]
- Pitard, F.F. Pierre Gy’s Sampling Theory and Sampling Practice; CRC Press: Boca Raton, FL, USA, 1989. [Google Scholar]
- FDA. CVM GFI #118 Mass Spectrometry for Confirmation of Identity of Animal Drug Residues. 2003. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/cvm-gfi-118-mass-spectrometry-confirmation-identity-animal-drug-residues (accessed on 25 January 2022).
- US FDA. Advisory Levels for Deoxynivalenol (DON) in Finished Wheat Products for Human Consumption and Grains and Grain By-Products Used for Animal Feed; US FDA: Silver Spring, MD, USA, 2010. [Google Scholar]
- FAO/WHO. Food Standards Programme, Codex Alimentarius Commission, 38th Session Geneva, Switzerland, 6–11 July 2015, Report of the Ninth Session of the CODEX Committee on Contaminants in Foods, New Delhi, India, 16–20 March 2015. Available online: www.fao.org/input/download/report/923/REP15_CFe.pdf (accessed on 17 February 2022).
- Smallwood, I.M. Handbook of Organic Solvent Properties; John Wiley & Sons Inc.: Hoboken, NJ, USA, 1996. [Google Scholar]
- US FDA. Guidelines for the Validation of Chemical Methods for the FDA Foods Program, 3rd ed.; US FDA: Silver Spring, MD, USA, 2019. [Google Scholar]
- Schürmann, A.; Dvorak, V.; Crüzer, C.; Butcher, P.; Kaufmann, A. False-positive liquid chromatography/tandem mass spectrometric confirmation of sebuthylazine residues using the identification points system according to EU directive 2002/657/EC due to a biogenic insecticide in tarragon. Rapid Commun. Mass Spectrom. 2009, 23, 1196–1200. [Google Scholar] [CrossRef]
- Gros, M.; Petrović, M.; Barceló, D. Tracing pharmaceutical residues of different therapeutic classes in environmental waters by using liquid chromatography/quadrupole-linear ion trap mass spectrometry and automated library searching. Anal. Chem. 2009, 81, 898–912. [Google Scholar] [CrossRef] [PubMed]
- Dresen, S.; Ferreirós, N.; Gnann, H.; Zimmermann, R.; Weinmann, W. Detection and identification of 700 drugs by multi-target screening with a 3200 Q TRAP LC-MS/MS system and library searching. Anal. Bioanal. Chem. 2010, 396, 2425–2434. [Google Scholar] [PubMed]
- NIST. Basis for Interpretation of the Library Search Results, NIST/EPA/NIH Mass Spectral. Library (NIST 17) and NIST Mass Spectral Search Program (Version 2.3) User’s Guide. Available online: https://chemdata.nist.gov/dokuwiki/lib/exe/fetch.php?media=chemdata:nist17:nistms_ver23man.pdf (accessed on 25 January 2022).
- SCIEX OS for Triple Quadrupole Systems Software User Guide. Available online: https://sciex.com/content/dam/SCIEX/pdf/customer-docs/user-guide/sciex-os-tnt-user-guide-en.pdf (accessed on 25 January 2022).


{kind=link}
{kind=link}
{kind=link}
| Dried Fruits and QC | Average Particle Size (RSD%), n = 10 | ||
|---|---|---|---|
| Dv10 (µm) | Dv50 (µm) | Dv90 (µm) | |
| Cranberry | 23 (2) | 165 (2) | 556 (4) |
| Raisin | 16 (8) | 119 (8) | 426 (13) |
| Plum | 21 (2) | 126 (3) | 435 (8) |
| Fig | 27 (3) | 198 (3) | 567 (5) |
| NIST1565 (measured) | 31 (2) | 225 (1) | 550 (2) |
| NIST1565 (reference) | NA | NA | 588 (3) |
| CRM3310 (measured) | 41 (2) | 71 (1) | 116 (1) |
| CRM3310 (reference) | 39 (5) | 73 (4) | 105 (6) |
| Fortification Conc. | Matrix | Average Spike Recovery (RSD)%, n = 3 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AFB1 | AFB2 | AFG1 | AFG2 | DON | FB1 | FB2 | FB3 | OTA | T-2 | HT-2 | ZON | ||
| L-1 (1/10 ng/g) | Cranberry | 89 (4) | 105 (2) | 101 (10) | 99 (13) | ND | 88 (21) | 87 (5) | 92 (2) | 101 (13) | 97 (6) | ND | 107 (12) |
| Fig | 105 (2) | 109 (7) | 100 (4) | 113 (1) | ND | 255 (4) | 124 (1) | ND | 103 (5) | 104 (8) | 100 (19) | 127 (12) | |
| Plum | 114 (1) | 112 (5) | 104 (3) | 145 (2) | ND | 110 (6) | 122 (1) | 115 (4) | 101 (4) | 106 (9) | 103 (9) | 99 (5) | |
| Raisin | 106 (2) | 101 (2) | 94 (3) | 99 (3) | 115 (12) | 120 (7) | 121 (3) | 118 (4) | 135 (1) | 103 (4) | 94 (11) | 111 (7) | |
| L-2 (10/100 ng/g) | Cranberry | 87 (6) | 98 (10) | 104 (5) | 113 (5) | 98 (4) | 88 (2) | 92 (5) | 90 (3) | 95 (12) | 98 (5) | 89 (5) | 100 (7) |
| Fig | 105 (3) | 106 (3) | 105 (4) | 107 (1) | 125 (6) | 120 (2) | 111 (2) | 120 (1) | 104 (1) | 94 (6) | 88 (1) | 107 (2) | |
| Plum | 112 (2) | 112 (3) | 105 (1) | 124 (3) | ND | 118 (4) | 117 (2) | 114 (2) | 113 (4) | 115 (1) | 106 (5) | 108 (4) | |
| Raisin | 104 (2) | 104 (5) | 101 (3) | 102 (4) | 107 (6) | 105 (6) | 113 (2) | 110 (1) | 111 (5) | 105 (3) | 94 (9) | 99 (1) | |
| L-3 (100/1000 ng/g) | Cranberry | 77 (5) | 93 (5) | 87 (4) | 90 (6) | 94 (5) | 93 (6) | 90 (8) | 97 (3) | 89 (4) | 96 (8) | 92 (9) | 98 (6) |
| Fig | 104 (2) | 107 (4) | 104 (4) | 110 (1) | 107 (2) | 110 (2) | 110 (1) | 111 (5) | 107 (1) | 101 (6) | 92 (5) | 104 (4) | |
| Plum | 113 (1) | 114 (4) | 111 (5) | 122 (8) | 117 (5) | 121 (3) | 116 (1) | 113 (4) | 110 (1) | 114 (5) | 109 (11) | 114 (1) | |
| Raisin | 93 (3) | 107 (2) | 91 (14) | 86 (8) | 99 (5) | 105 (3) | 108 (2) | 111 (6) | 92 (13) | 102 (5) | 86 (14) | 91 (8) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, K.; Tan, S.; Xu, D. Determination of Mycotoxins in Dried Fruits Using LC-MS/MS—A Sample Homogeneity, Troubleshooting and Confirmation of Identity Study. Foods 2022, 11, 894. https://doi.org/10.3390/foods11060894
Zhang K, Tan S, Xu D. Determination of Mycotoxins in Dried Fruits Using LC-MS/MS—A Sample Homogeneity, Troubleshooting and Confirmation of Identity Study. Foods. 2022; 11(6):894. https://doi.org/10.3390/foods11060894
Chicago/Turabian StyleZhang, Kai, Steven Tan, and David Xu. 2022. "Determination of Mycotoxins in Dried Fruits Using LC-MS/MS—A Sample Homogeneity, Troubleshooting and Confirmation of Identity Study" Foods 11, no. 6: 894. https://doi.org/10.3390/foods11060894
APA StyleZhang, K., Tan, S., & Xu, D. (2022). Determination of Mycotoxins in Dried Fruits Using LC-MS/MS—A Sample Homogeneity, Troubleshooting and Confirmation of Identity Study. Foods, 11(6), 894. https://doi.org/10.3390/foods11060894