
Multiclass Comparative Analysis of Veterinary Drugs, Mycotoxins, and Pesticides in Bovine Milk by Ultrahigh-Performance Liquid Chromatography–Hybrid Quadrupole–Linear Ion Trap Mass Spectrometry



Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Apparatus and Chromatographic Conditions
2.3. Sample Preparation
2.4. Sample Collection and Analysis
3. Results and Discussion
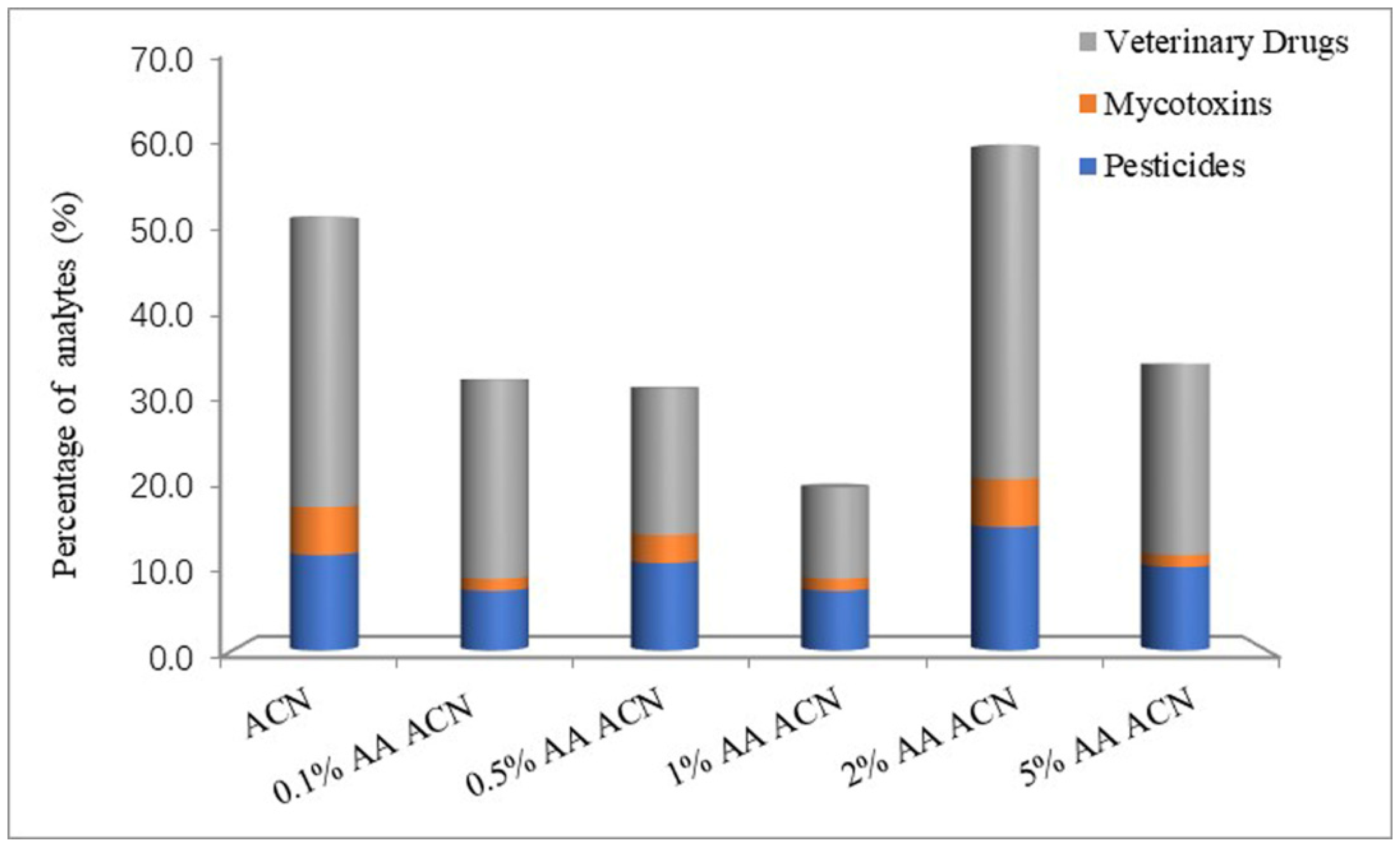
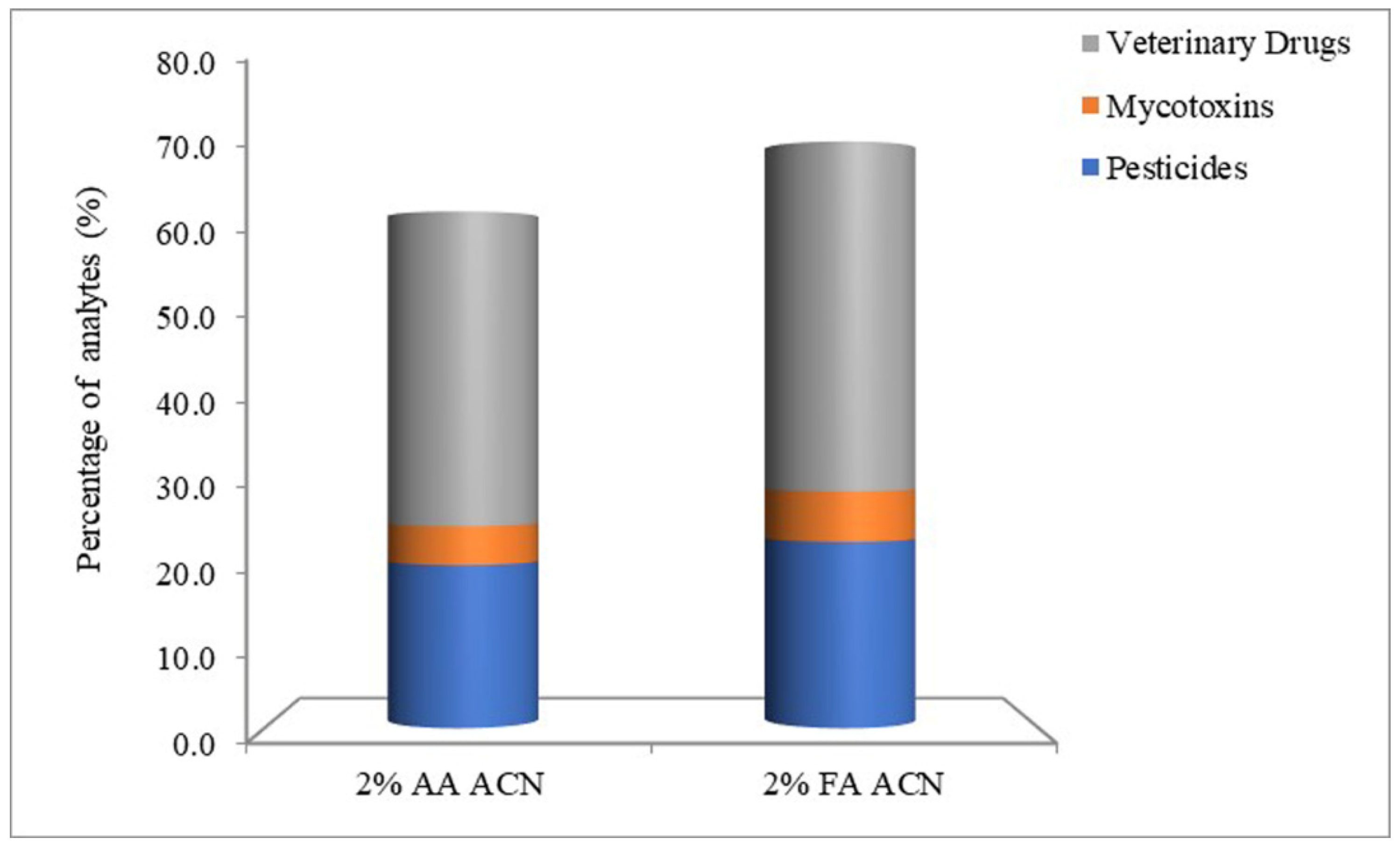
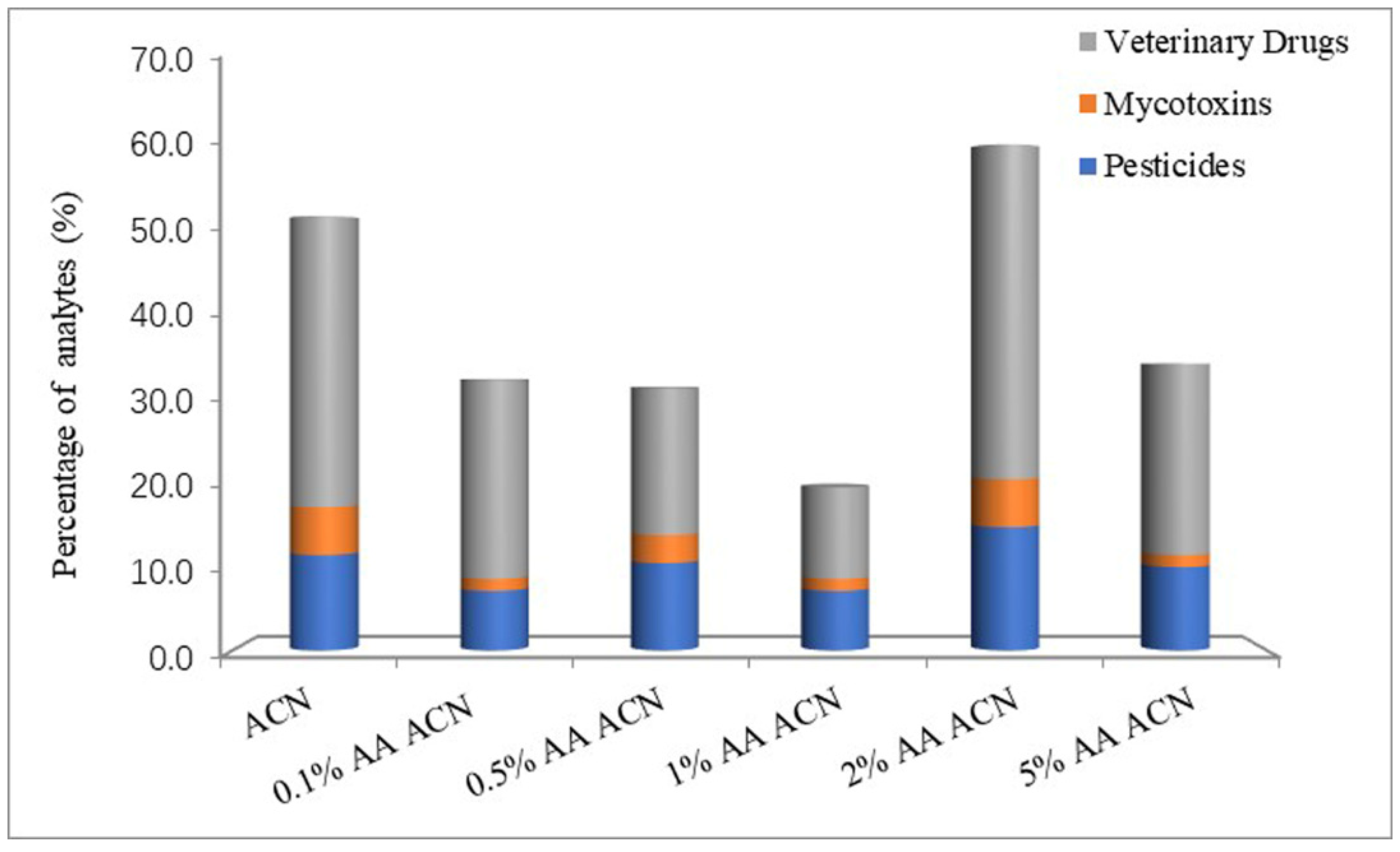
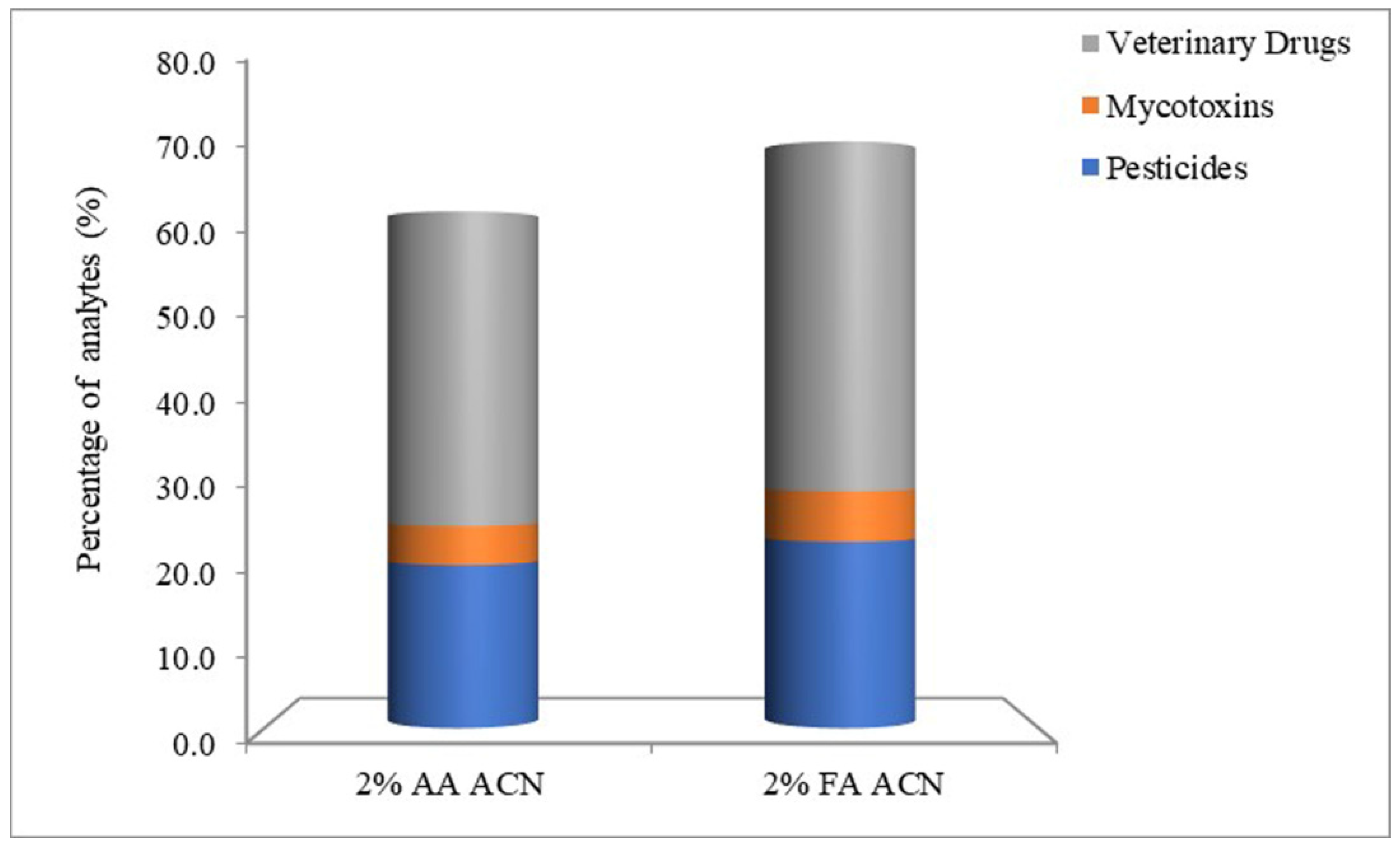
3.1. Optimization of Sample Preparation
3.2. Optimization of UHPLC-Qtrap-MS
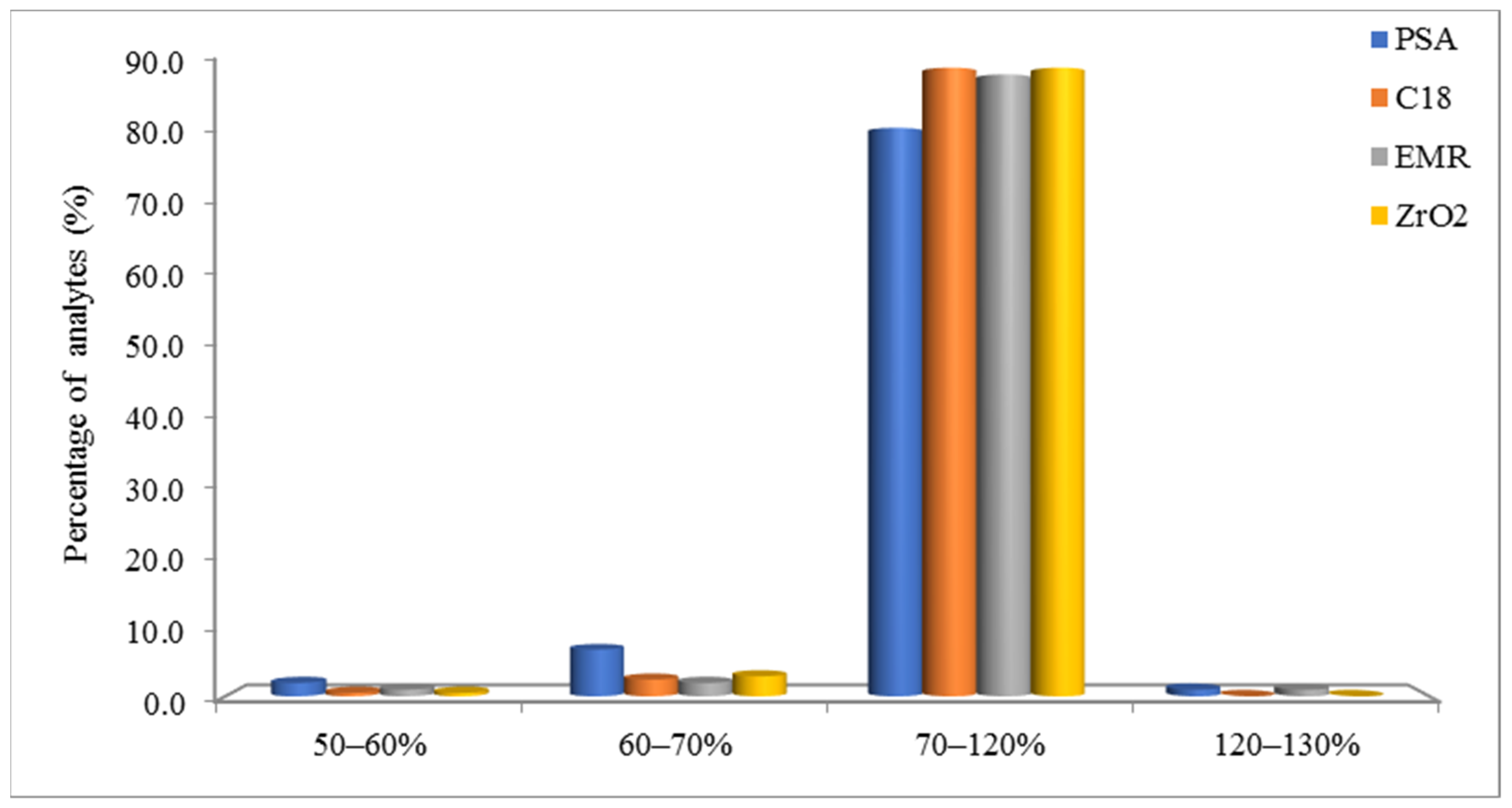
3.3. Method Validation
3.4. Application of the Developed Method to Milk Samples
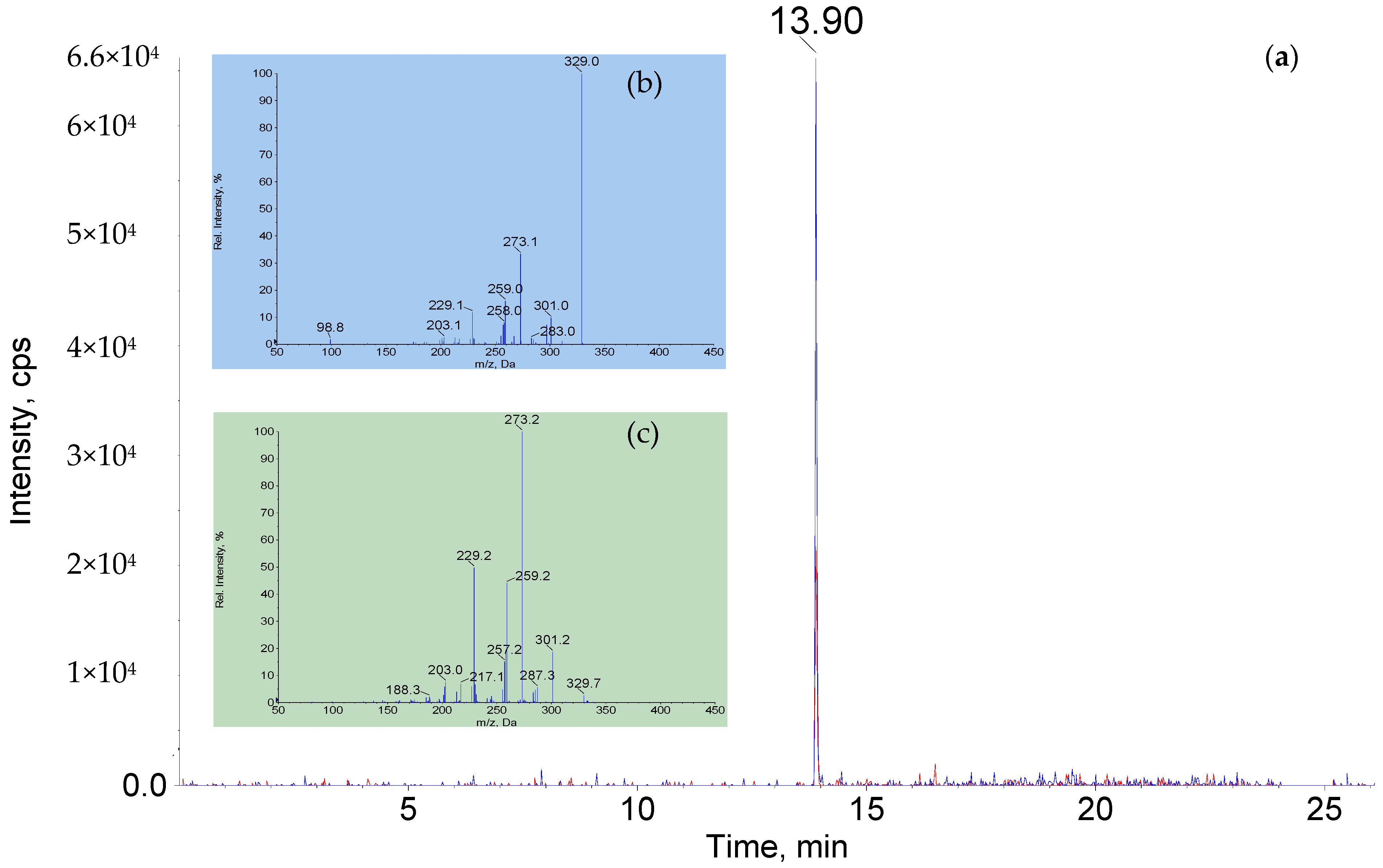
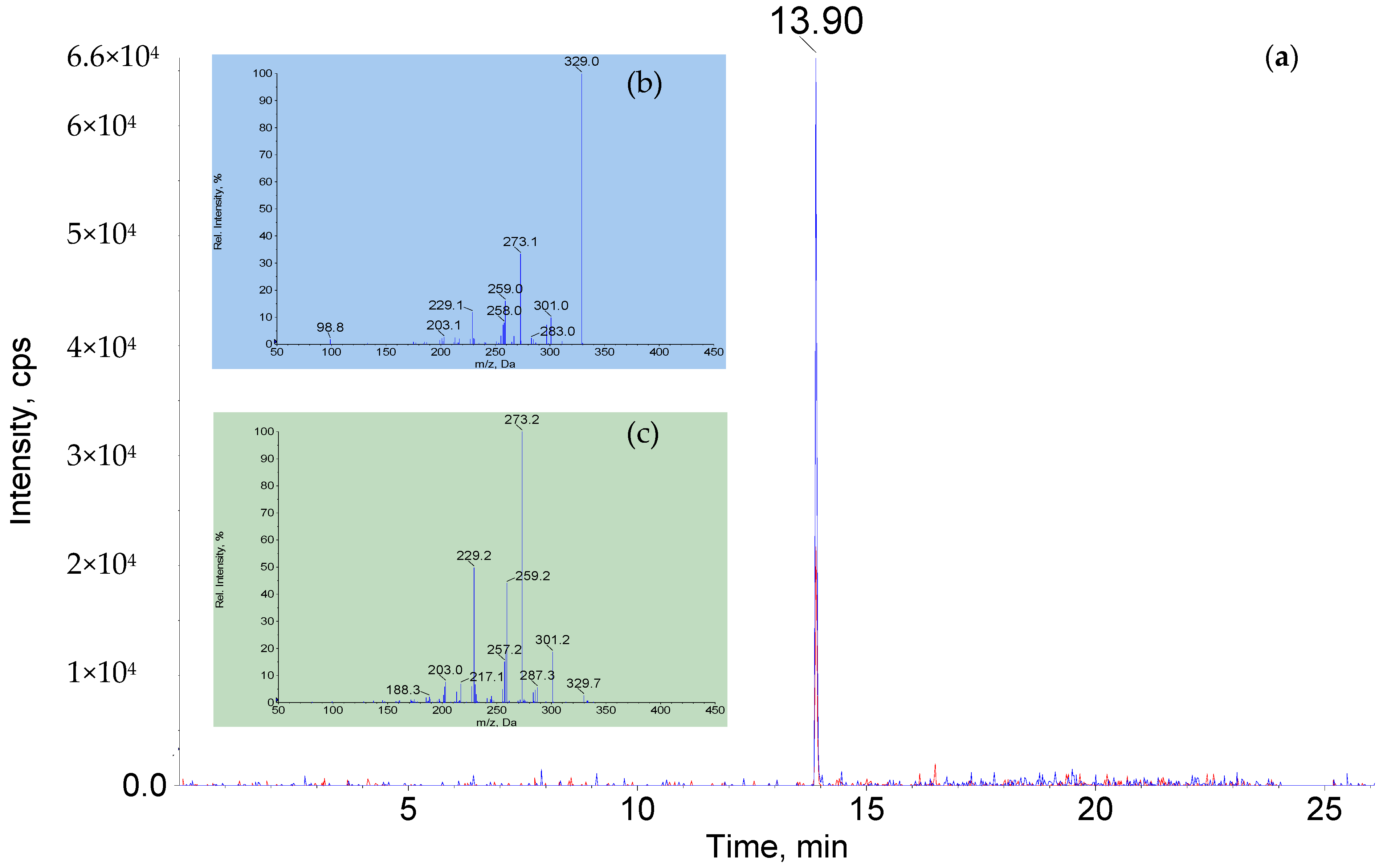
3.5. Qualitative and Quantitative Confirmation
3.6. Comparative Analysis of Contaminants in Milk
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Manav, Ö.G.; Dinç-Zor, Ş.; Alpdoğan, G. Optimization of a Modified Quechers Method by Means of Experimental Design for Multiresidue Determination of Pesticides in Milk and Dairy Products by Gc–Ms. Microchem. J. 2019, 144, 124–129. [Google Scholar] [CrossRef]
- Jadhav, M.R.; Pudale, A.; Raut, P.; Utture, S.; Shabeer, T.A.; Banerjee, K. A unified approach for high-throughput quantitative analysis of the residues of multi-class veterinary drugs and pesticides in bovine milk using LC-MS/MS and GC–MS/MS. Food Chem. 2019, 272, 292–305. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, X.; Yang, M.; Yang, L.; Han, X.; Jiang, X.; Zhao, B. High sensitive detection of penicillin G residues in milk by surface-enhanced Raman scattering. Talanta 2017, 167, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Yahaya, N.; Sanagi, M.M.; Mitome, T.; Nishiyama, N.; Ibrahim, W.A.W.; Nur, H. Dispersive Micro-Solid Phase Extraction Combined with High-Performance Liquid Chromatography for the Determination of Three Penicillins in Milk Samples. Food Anal. Methods 2015, 8, 1079–1087. [Google Scholar] [CrossRef]
- Junza, A.; Dorival-Garcia, N.; Zafra-Gomez, A.; Barron, D.; Ballesteros, O.; Barbosa, J.; Navalon, A. Multiclass Method for the Determination of Quinolones and Beta-Lactams, in Raw Cow Milk Using Dispersive Liquid-Liquid Microextraction and Ultra High Performance Liquid Chromatography-Tandem Mass Spectrometry. J. Chromatogr. A 2014, 1356, 10–22. [Google Scholar] [CrossRef]
- Chen, L.; Huang, X. Sensitive Monitoring of Fluoroquinolones in Milk and Honey Using Multiple Monolithic Fiber Solid-Phase Microextraction Coupled to Liquid Chromatography Tandem Mass Spectrometry. J. Agric. Food Chem. 2016, 64, 8684–8693. [Google Scholar] [CrossRef]
- Meng, Z.; Shi, Z.; Liang, S.; Dong, X.; Li, H.; Sun, H. Residues investigation of fluoroquinolones and sulphonamides and their metabolites in bovine milk by quantification and confirmation using ultra-performance liquid chromatography–tandem mass spectrometry. Food Chem. 2015, 174, 597–605. [Google Scholar] [CrossRef]
- Chen, Y.; Kong, D.; Liu, L.; Song, S.; Kuang, H.; Xu, C. Development of an ELISA and Immunochromatographic Assay for Tetracycline, Oxytetracycline, and Chlortetracycline Residues in Milk and Honey Based on the Class-Specific Monoclonal Antibody. Food Anal. Methods 2015, 9, 905–914. [Google Scholar] [CrossRef]
- Wang, J.; Leung, D.; Chow, W.; Chang, J.; Wong, J.W. Development and Validation of a Multiclass Method for Analysis of Veterinary Drug Residues in Milk Using Ultrahigh Performance Liquid Chromatography Electrospray Ionization Quadrupole Orbitrap Mass Spectrometry. J. Agric. Food Chem. 2015, 63, 9175–9187. [Google Scholar] [CrossRef]
- Dasenaki, M.; Bletsou, A.A.; Koulis, G.A.; Thomaidis, N. Qualitative Multiresidue Screening Method for 143 Veterinary Drugs and Pharmaceuticals in Milk and Fish Tissue Using Liquid Chromatography Quadrupole-Time-of-Flight Mass Spectrometry. J. Agric. Food Chem. 2015, 63, 4493–4508. [Google Scholar] [CrossRef]
- Flores-Flores, M.E.; González-Peñas, E. An LC–MS/MS method for multi-mycotoxin quantification in cow milk. Food Chem. 2017, 218, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Zheng, N.; Wen, F.; Guo, L.; Fu, C.; Ouyang, H.; Zhong, L.; Wang, J.; Lei, S. Multi-mycotoxins analysis in raw milk by ultra high performance liquid chromatography coupled to quadrupole orbitrap mass spectrometry. Food Control 2018, 84, 305–311. [Google Scholar] [CrossRef]
- Flores-Flores, M.E.; Lizarraga, E.; de Cerain, A.L.; González-Peñas, E. Presence of Mycotoxins in Animal Milk: A Review. Food Control 2015, 53, 163–176. [Google Scholar] [CrossRef]
- Huang, L.C.; Zheng, N.; Zheng, B.Q.; Wen, F.; Cheng, J.B.; Han, R.W.; Xu, X.M.; Li, S.L.; Wang, J.Q. Simultaneous Determination of Aflatoxin M1, Ochratoxin a, Zearalenone and Alpha-Zearalenol in Milk by Uhplc-Ms/Ms. Food Chem. 2014, 146, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Shuib, N.S.; Makahleh, A.; Salhimi, S.M.; Saad, B. Determination of aflatoxin M 1 in milk and dairy products using high performance liquid chromatography-fluorescence with post column photochemical derivatization. J. Chromatogr. A 2017, 1510, 51–56. [Google Scholar] [CrossRef]
- Dos Anjos, M.R.; Castro, I.M.; Mde, L.S.; de Lima, V.V.; de Aquino-Neto, F.R. Multiresidue Method for Simultaneous Analysis of Aflatoxin M1, Avermectins, Organophosphate Pesticides and Milbemycin in Milk by Ultra-Performance Liquid Chromatography Coupled to Tandem Mass Spectrometry. Food Addit. Contam. Part A Chem. Anal. Control Exp. Risk Assess. 2016, 33, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Rejczak, T.; Tuzimski, T. Quechers-Based Extraction with Dispersive Solid Phase Extraction Clean-up Using Psa and Zro2-Based Sorbents for Determination of Pesticides in Bovine Milk Samples by Hplc-Dad. Food Chem. 2017, 217, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Selvi, C.; Paramasivam, M.; Rajathi, D.S.; Chandrasekaran, S. Multiresidue Analysis of Organochlorine Pesticides in Milk, Egg and Meat by GC–ECD and Confirmation by GC–MS. Bull. Environ. Contam. Toxicol. 2012, 89, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Commission Decision 2010/37/EC of 22 December 2009 on Pharmacologically Active Substances and Their Classification Regarding Maximum Residue Limits in Foodstuffs of Animal Origin. Off. J. Eur. Union 2010, L15, 1–72. [Google Scholar]
- Ministry of Agriculture of the People’s Republic of China. Announcement 2002/235 Concerning the Maximum Residue Limit of Veterinary Drug of Animal Foodstuff. Available online: http://www.agri.gov.cn/blgg/t20030226_59300.htm (accessed on 17 January 2019).
- Guo, P.; Chen, Y.; Yue, C.; Yu, G. Simultaneous determination of clavulanic acid and tazobactam in bovine milk by HPLC. Food Addit. Contam. Part A Chem. Anal. Control Exp. Risk Assess. 2017, 34, 617–623. [Google Scholar] [CrossRef]
- Muscarella, M.; Magro, S.L.; Palermo, C.; Centonze, D. A confirmatory method for aflatoxin M(1) determination in milk based on immunoaffinity cleanup and high-performance liquid chromatography with fluorometric detection. Methods Mol. Biol. 2011, 739, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Shamsipur, M.; Yazdanfar, N.; Ghambarian, M. Combination of solid-phase extraction with dispersive liquid–liquid microextraction followed by GC–MS for determination of pesticide residues from water, milk, honey and fruit juice. Food Chem. 2016, 204, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Armorini, S.; Yeatts, J.E.; Mullen, K.A.E.; Mason, S.E.; Mehmeti, E.; Anderson, K.L.; Washburn, S.P.; Baynes, R.E. Development of a HS-SPME-GC-MS/MS Method for the Quantitation of Thymol and Carvacrol in Bovine Matrices and To Determine Residue Depletion in Milk and Tissues. J. Agric. Food Chem. 2016, 64, 7856–7865. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Yu, Q.-W.; He, H.-B.; Lu, Q.; Shi, Z.-G.; Feng, Y.-Q. Nickel Oxide Nanoparticle-Deposited Silica Composite Solid-Phase Extraction for Benzimidazole Residue Analysis in Milk and Eggs by Liquid Chromatography–Mass Spectrometry. J. Agric. Food Chem. 2015, 64, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Castilla-Fernández, D.; Moreno-González, D.; Beneito-Cambra, M.; Molina-Díaz, A. Critical assessment of two sample treatment methods for multiresidue determination of veterinary drugs in milk by UHPLC-MS/MS. Anal. Bioanal. Chem. 2019, 411, 1433–1442. [Google Scholar] [CrossRef]
- Turnipseed, S.B.; Lohne, J.J.; Storey, J.M.; Andersen, W.; Young, S.L.; Carr, J.R.; Madson, M.R. Challenges in Implementing a Screening Method for Veterinary Drugs in Milk Using Liquid Chromatography Quadrupole Time-of-Flight Mass Spectrometry. J. Agric. Food Chem. 2014, 62, 3660–3674. [Google Scholar] [CrossRef]
- Jank, L.; Martins, M.T.; Arsand, J.B.; Motta, T.M.C.; Feijó, T.C.; Castilhos, T.D.S.; Hoff, R.B.; Barreto, F.; Pizzolato, T.M. Liquid Chromatography–Tandem Mass Spectrometry Multiclass Method for 46 Antibiotics Residues in Milk and Meat: Development and Validation. Food Anal. Methods 2017, 10, 2152–2164. [Google Scholar] [CrossRef]
- Freitas, S.K.B.; Paim, A.P.S.; e Silva, P.T.S. Development of a LC-IT-TOF MS Procedure to Quantify Veterinary Drug Residues in Milk Employing a QuEChERS Approach. Food Anal. Methods 2014, 7, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Stolker, A.A.; Rutgers, P.; Oosterink, E.; Lasaroms, J.J.; Peters, R.J.; van Rhijn, J.A.; Nielen, M.W. Comprehensive screening and quantification of veterinary drugs in milk using UPLC-ToF-MS. Anal. Bioanal. Chem. 2008, 391, 2309–2322. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Zhao, M.; Xi, Y.; Mao, Q.; Zhou, X.; Chen, D.; Yan, P. Nontargeted Screening and Determination of Sulfonamides: A Dispersive Micro Solid-Phase Extraction Approach to the Analysis of Milk and Honey Samples Using Liquid Chromatography–High-Resolution Mass Spectrometry. J. Agric. Food Chem. 2017, 65, 1984–1991. [Google Scholar] [CrossRef] [PubMed]
- Moretti, S.; Cruciani, G.; Romanelli, S.; Rossi, R.; Saluti, G.; Galarini, R. Multiclass method for the determination of 62 antibiotics in milk. Biol. Mass Spectrom. 2016, 51, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Chiesa, L.; Di Cesare, F.; Nobile, M.; Villa, R.; Decastelli, L.; Martucci, F.; Fontana, M.; Pavlovic, R.; Arioli, F.; Panseri, S. Antibiotics and Non-Targeted Metabolite Residues Detection as a Comprehensive Approach toward Food Safety in Raw Milk. Foods 2021, 10, 544. [Google Scholar] [CrossRef] [PubMed]
- Vidal, J.L.M.; Frenich, A.G.; Aguilera-Luiz, M.M.; Romero-González, R. Development of fast screening methods for the analysis of veterinary drug residues in milk by liquid chromatography-triple quadrupole mass spectrometry. Anal. Bioanal. Chem. 2010, 397, 2777–2790. [Google Scholar] [CrossRef] [PubMed]
- Chirollo, C.; Vassallo, A.; Piaz, F.D.; Lamagna, B.; Tortora, G.; Neglia, G.; De Tommasi, N.; Severino, L. Investigation of the Persistence of Penicillin G and Dihydrostreptomycin Residues in Milk of Lactating Buffaloes (Bubalus bubalis) Using Ultra-High-Performance Liquid Chromatography and Tandem Mass Spectrometry. J. Agric. Food Chem. 2018, 66, 6388–6393. [Google Scholar] [CrossRef]
- Wittenberg, J.B.; Simon, K.A.; Wong, J.W. Targeted Multiresidue Analysis of Veterinary Drugs in Milk-Based Powders Using Liquid Chromatography–Tandem Mass Spectrometry (LC-MS/MS). J. Agric. Food Chem. 2017, 65, 7288–7293. [Google Scholar] [CrossRef]
- Jedziniak, P.; Szprengier-Juszkiewicz, T.; Pietruk, K.; Sledzińska, E.; Zmudzki, J. Determination of non-steroidal anti-inflammatory drugs and their metabolites in milk by liquid chromatography–tandem mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 2955–2963. [Google Scholar] [CrossRef]
- Whelan, M.; Kinsella, B.; Furey, A.; Moloney, M.; Cantwell, H.; Lehotay, S.J.; Danaher, M. Determination of anthelmintic drug residues in milk using ultra high performance liquid chromatography-tandem mass spectrometry with rapid polarity switching. J. Chromatogr. A 2010, 1217, 4612–4622. [Google Scholar] [CrossRef]
- Wang, X.; Li, P. Rapid screening of mycotoxins in liquid milk and milk powder by automated size-exclusion SPE-UPLC–MS/MS and quantification of matrix effects over the whole chromatographic run. Food Chem. 2015, 173, 897–904. [Google Scholar] [CrossRef]
- Campone, L.; Piccinelli, A.L.; Celano, R.; Russo, M.; Rastrelli, L. Rapid analysis of aflatoxin M1 in milk using dispersive liquid–liquid microextraction coupled with ultrahigh pressure liquid chromatography tandem mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 8645–8652. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, X.J.; Liu, Y.Q.; Yang, H.M.; Guo, Q.L. Determination of aflatoxin M1 in milk by triple quadrupole liquid chromatography-tandem mass spectrometry. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2010, 27, 1261–1265. [Google Scholar] [CrossRef]
- Lachat, L.; Glauser, G. Development and Validation of an Ultra-Sensitive UHPLC–MS/MS Method for Neonicotinoid Analysis in Milk. J. Agric. Food Chem. 2018, 66, 8639–8646. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Meng, W.; Sun, W.; Li, D.; Yu, Z.; Tong, L.; Zhao, Y. Simultaneous qualitative and quantitative analysis of 21 mycotoxins in Radix Paeoniae Alba by ultra-high performance liquid chromatography quadrupole linear ion trap mass spectrometry and QuEChERS for sample preparation. J. Chromatogr. B 2016, 1031, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Li, D.; Wang, X.; Yang, S.; Qian, Y.; Qiu, J. Simultaneous determination of amantadine and rimantadine in feed by liquid chromatography-Qtrap mass spectrometry with information-dependent acquisition. Anal. Bioanal. Chem. 2018, 410, 5555–5565. [Google Scholar] [CrossRef]
- Hou, J.; Xie, W.; Jie, L.; Zhu, Z.; He, J. LC-MS/MS determination of residual amounts of 11 mycotoxins in milk. Phys. Test. Chem. Anal. Part B Chem. Anal. 2015, 51, 600–604. [Google Scholar]
- Han, C.; Li, X.; Lu, H.; Su, L.; Zhang, Q. Determination of Steroid Hormones in Milk by Isotope Dilution-Ultra Performance Liquid Chromatography-Tandem Mass Spectrometry. 2018. Available online: https://en.cnki.com.cn/Article_en/CJFDTOTAL-SPAJ201807041.htm (accessed on 17 January 2022).
- Zhang, L.; Sun, J.; Wang, Y.; Wu, H.; Yu, S. Simultaneous determination of six zeranols in milk samples by immunoaffinity solid phase extraction coupled with ultra-performance convergence chromatography-tandem mass spectrometry. Chin. J. Chrom. 2018, 36, 566–572. [Google Scholar]
- Zhang, X.; Cai, X. Rapid simultameous determination of 53β-lactam antibiotics and their metabolites in milk by ultra-perfomance liquid chromatography coupled with triple quadrupole mass spectrometry. Chin. J. Chrom. 2014, 32, 693–701. [Google Scholar] [CrossRef]
- Ning, L.; Zhang, Y.; Lin, T.; Wang, J.; Liu, H. Analysis of sulfonamide, quinolones, steroid hormones and tetracyclines residues in milk by ultra performance liquid chromatography-mass spectrometry. J. Instrum. Anal. 2016, 35, 714–718. [Google Scholar]
- Accioni, F.; Caballero-Casero, N.; Garcia-Gomez, D.; Rubio, S. Restricted access volatile supramolecular solvents for single-step extraction/cleanup of benzimidazole anthelmintic drugs in milk prior to LC-MS/MS. J. Agric. Food Chem. 2019, 67, 520–530. [Google Scholar] [CrossRef]
- Chiesa, L.M.; Nobile, M.; Panseri, S.; Biolatti, B.; Cannizzo, F.T.; Pavlovic, R.; Arioli, F. A Liquid Chromatography-Tandem Mass Spectrometry Method for the Detection of Antimicrobial Agents from Seven Classes in Calf Milk Replacers: Validation and Application. J. Agric. Food Chem. 2016, 64, 2635–2640. [Google Scholar] [CrossRef]
- Tian, H.; Zhou, X.; Chen, C.; He, Y.; Yu, H.; Zheng, X. Simultaneous Determination of Phenobarbital, Pentobarbital, Amobarbital and Secobarbital in Raw Milk via Liquid Chromatography with Electron Spray Ionization Tandem Mass Spectrometry. Korean J. Food Sci. Anim. Resour. 2017, 37, 847–854. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Contaminant | Concentration (μg/kg) | MRL (μg/kg) | Detection Rate (%) | Source |
|---|---|---|---|---|---|
| 1 | sulfamethazine | 1.79 | 100 (China) | 0.71 | supermarket |
| 2 | cloxacillin | 9.20–36.14 | 30 (China) | 3.57 | dairy farm |
| 7.12–69.70 | 4.29 | supermarket | |||
| 3 | aflatoxin M1 | 0.17, 0.24 | 0.5 (China) | 1.43 | dairy farm |
| 4 | fipronil sulfone | 0.08 | 8 (EU) 1 | 0.71 | dairy farm |
| 5 | imidacloprid | 6.24 | 100 (EU) | 0.71 | supermarket |
| 6 | acetamiprid | 2.36–12.24 | 200 (China) | 2.14 | dairy farm |
| No. | Country | Contaminant | Concentration | MRL | Reference |
|---|---|---|---|---|---|
| (μg/kg) | (μg/kg) | ||||
| 1 | China | AFB1 | 0.8, 1.3 | [45] | |
| 2 | China | AFM1 | 0.046–0.237 | 0.5 (China) | [14] |
| ochratoxin A | 0.058–0.084 | ||||
| zearalenone | 0.028–0.046 | ||||
| α-zearalenol | 0.045–0.074 | ||||
| 3 | China | hydrocortisone | 0.14–1.02 | 10 (China) | [46] |
| 4 | China | zearalenone | 0.085 | [47] | |
| 5 | China | cefminox | 5.6 | [48] | |
| cefradine | 2.8 | ||||
| 6 | China | sulfadimethoxine | 21.5 | 100 (China) * | [49] |
| sulfamerazine | 10.6 | 100 (China) * | |||
| enrofloxacin | 15.3 | 100 (China) | |||
| 7 | China | sulfadiazine | 0.3–9.7 | 100 (China) * | [31] |
| sulfamerazine | 4.9–7.4 | 100 (China) * | |||
| 8 | China | AFM1 | 19.2 | 0.5 (China) | [12] |
| AFM2 | 3.91 | ||||
| 9 | India | sulfamerazine | 1.2–18.2 | [2] | |
| cloxacillin | |||||
| ciprofloxacin | |||||
| enrofloxacin | |||||
| oxytetracycline | |||||
| fenbendazole | |||||
| oxfendazole | |||||
| 10 | Italy | albendazole | 0.11–0.52 | 100 (EU) | [50] |
| fenbendazole sulfone | 0.32–1.21 | 10 (EU) | |||
| oxfendazole | 0.25–1.83 | 10 (EU) | |||
| febantel | 0.13–0.28 | ||||
| 11 | Italy | amoxicillin | 1.26 | 4 (EU) | [51] |
| marbofloxacin | 0.52–0.91 | 100 (EU) | |||
| 12 | Italy | penicillin G | 1.51–236 | 4 (EU) | [35] |
| dihydrostreptomycin | 5.85–197 | 200 (EU) | |||
| 13 | Brazil | tilmicosin | >MRL | 4 (EU) | [28] |
| cloxacillin | 30 (EU) | ||||
| ceftiofur | 100 (EU) | ||||
| 14 | Switzerland | thiamethoxam | 0.002–0.0085 | [42] | |
| clothianidin | 0.004–0.013 | ||||
| imidacloprid | 0.0025–0.0065 | 100 (EU) | |||
| acetamiprid | 0.0005–0.0095 | 200 (EU) | |||
| thiacloprid | 0.0001–0.0002 | ||||
| 15 | Brazil | AFM1 | 0.16–0.48 | 0.5 (Brazil) | [16] |
| 16 | Spain | enrofloxacin | 9.3–15.6 | 100 (EU) | [5] |
| ciprofloxacin | 5.6–32.4 | 100 (EU) | |||
| marbofloxacin | 4.4, 11.3 | 100 (EU) | |||
| amoxicillin | 2.1–18.6 | 4 (EU) | |||
| penicillin G | 2.4–5.3 | 4 (EU) | |||
| cephalonium | 4.6 | ||||
| dicloxacillin | 3.5 | 30 (EU) | |||
| 17 | Malaysia | AFM1 | 0.004, 0.01 | [15] | |
| 18 | China | ochratoxin A | 0.37, 1.59 | [39] | |
| zearalenol | 3.25 | ||||
| AFB1 | 0.21, 0.52 | ||||
| AFM1 | 0.01 | 0.5 (China) | |||
| 19 | China | ofloxacin | 13.1–36 | 100 (China) | [7] |
| enrofloxacin | 14.2–24 | 100 (China) | |||
| ciprofloxacin | 14–44 | 100 (China) | |||
| 20 | Spain | danofloxacin | 0.7–1.5 | [26] | |
| 21 | Italy | cephapirin | 3–6 | 60 (EU) | [33] |
| cefazolin | 9–87 | 50 (EU) | |||
| oxytetracycline | 4 | 50 (EU) | |||
| penicillin G | 2–10 | 4 (EU) | |||
| cefoperazone | 4 | 50 (EU) | |||
| dicloxacillin | 4 | 30 (EU) | |||
| desacetyl cephapirin | 4, 8 | 50 (EU) | |||
| ampicillin | 2–7 | 4 (EU) | |||
| cloxacillin | 2, 32 | 30 (EU) | |||
| cefacetrile | 10 | 125 (EU) | |||
| rifaximin | 11 | 60 (EU) | |||
| lincomycin | 2 | 150 (EU) | |||
| 22 | China | phenobarbital | 8 | [52] | |
| pentobarbital | 10.8 | ||||
| amobarbital | 12.5 | ||||
| secobarbital | 7.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, Q.; Qiu, J.; Zhang, L.; Liao, G.; Jia, Y.; Qian, Y. Multiclass Comparative Analysis of Veterinary Drugs, Mycotoxins, and Pesticides in Bovine Milk by Ultrahigh-Performance Liquid Chromatography–Hybrid Quadrupole–Linear Ion Trap Mass Spectrometry. Foods 2022, 11, 331. https://doi.org/10.3390/foods11030331
Jia Q, Qiu J, Zhang L, Liao G, Jia Y, Qian Y. Multiclass Comparative Analysis of Veterinary Drugs, Mycotoxins, and Pesticides in Bovine Milk by Ultrahigh-Performance Liquid Chromatography–Hybrid Quadrupole–Linear Ion Trap Mass Spectrometry. Foods. 2022; 11(3):331. https://doi.org/10.3390/foods11030331
Chicago/Turabian StyleJia, Qi, Jing Qiu, Lin Zhang, Guangqin Liao, Yanbo Jia, and Yongzhong Qian. 2022. "Multiclass Comparative Analysis of Veterinary Drugs, Mycotoxins, and Pesticides in Bovine Milk by Ultrahigh-Performance Liquid Chromatography–Hybrid Quadrupole–Linear Ion Trap Mass Spectrometry" Foods 11, no. 3: 331. https://doi.org/10.3390/foods11030331
APA StyleJia, Q., Qiu, J., Zhang, L., Liao, G., Jia, Y., & Qian, Y. (2022). Multiclass Comparative Analysis of Veterinary Drugs, Mycotoxins, and Pesticides in Bovine Milk by Ultrahigh-Performance Liquid Chromatography–Hybrid Quadrupole–Linear Ion Trap Mass Spectrometry. Foods, 11(3), 331. https://doi.org/10.3390/foods11030331