
Screening and Regulation Mechanism of Key Transcription Factors of Penicillium expansum Infecting Postharvest Pears by ATAC-Seq Analysis



Abstract
1. Introduction
2. Materials and Methods
2.1. Fruits
2.2. Pathogen
2.3. Sample Preparation
2.4. ATAC-seq and Analysis
2.4.1. Clean Reads Filtering
2.4.2. Irreproducible Discovery Rate (IDR)
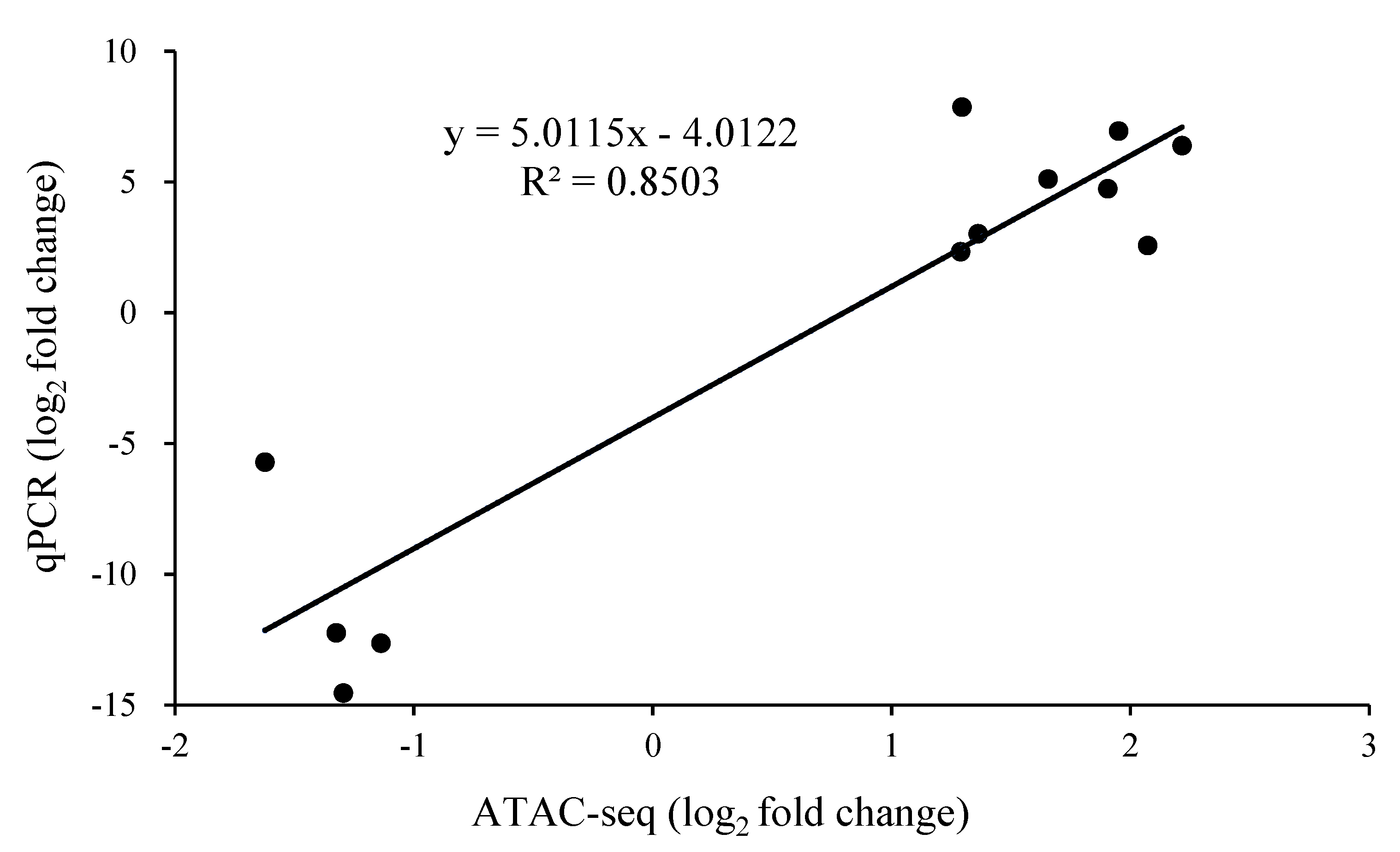
2.5. Verification of the Relative Expression Levels of Differentially Expressed Peak-Related Genes in P. expansum by RT-qPCR
3. Results
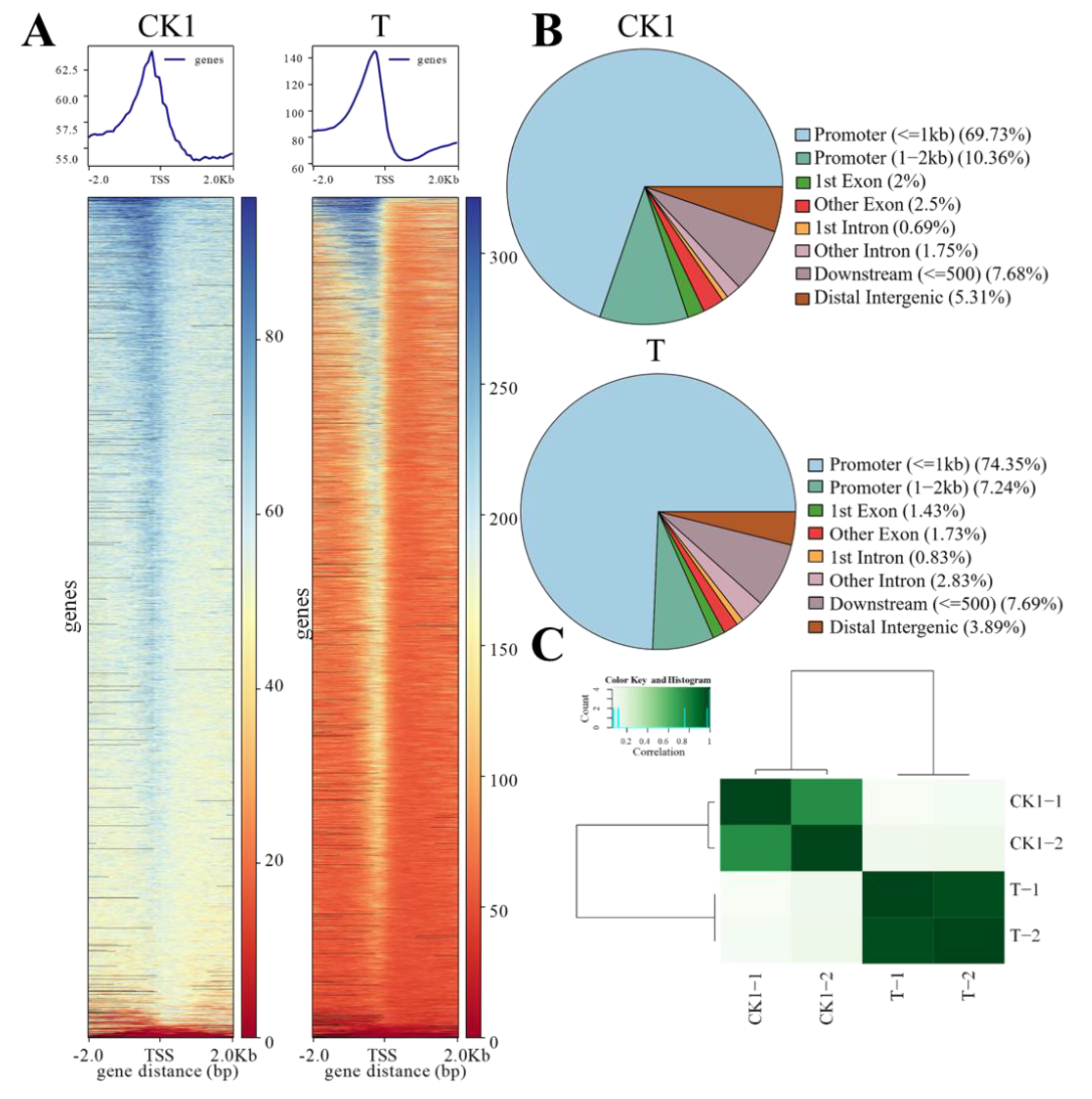
3.1. Chromatin Accessibility Landscape
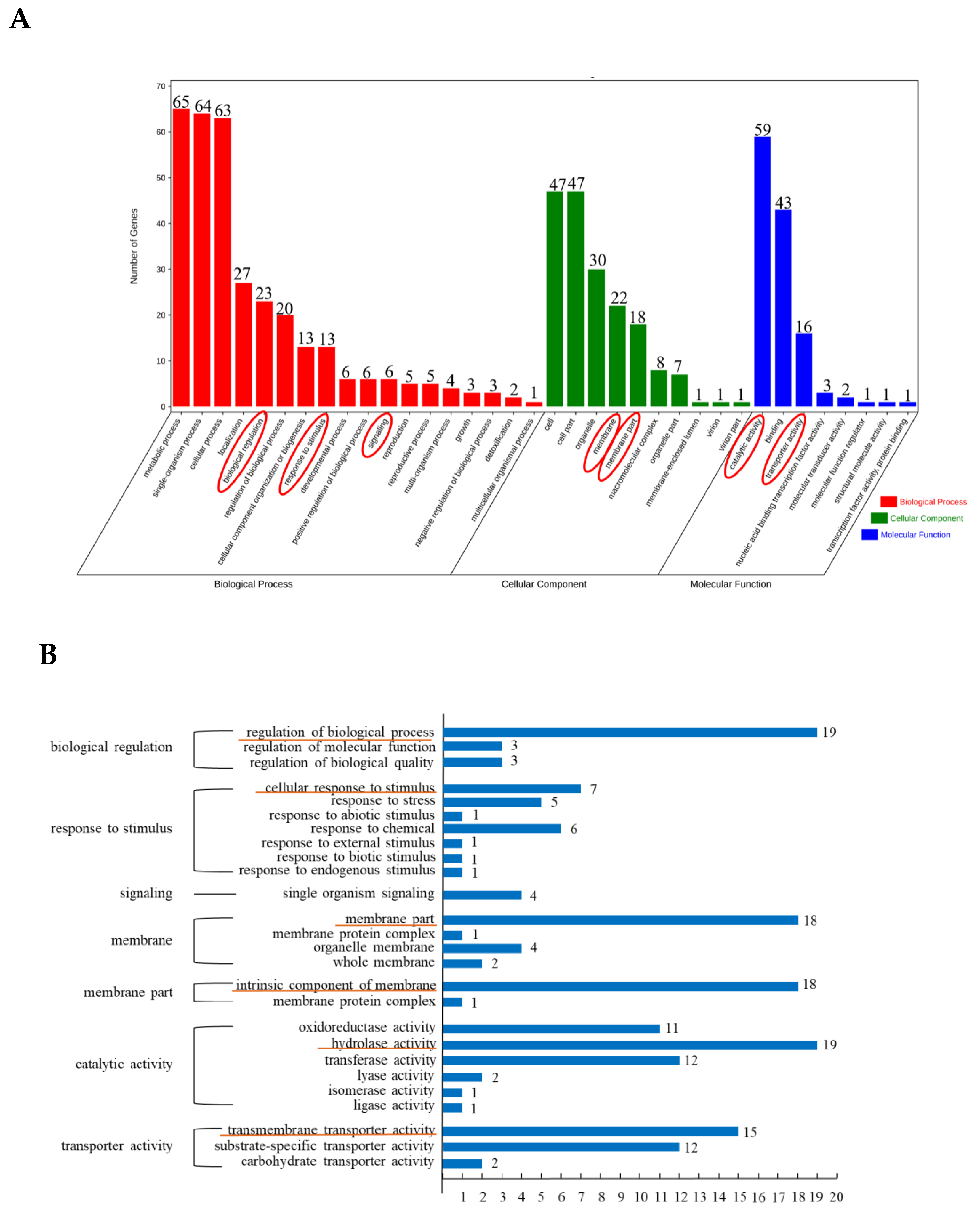
3.2. GO Enrichment Analysis of Differentially Expressed Peak-Related Genes in P. expansum
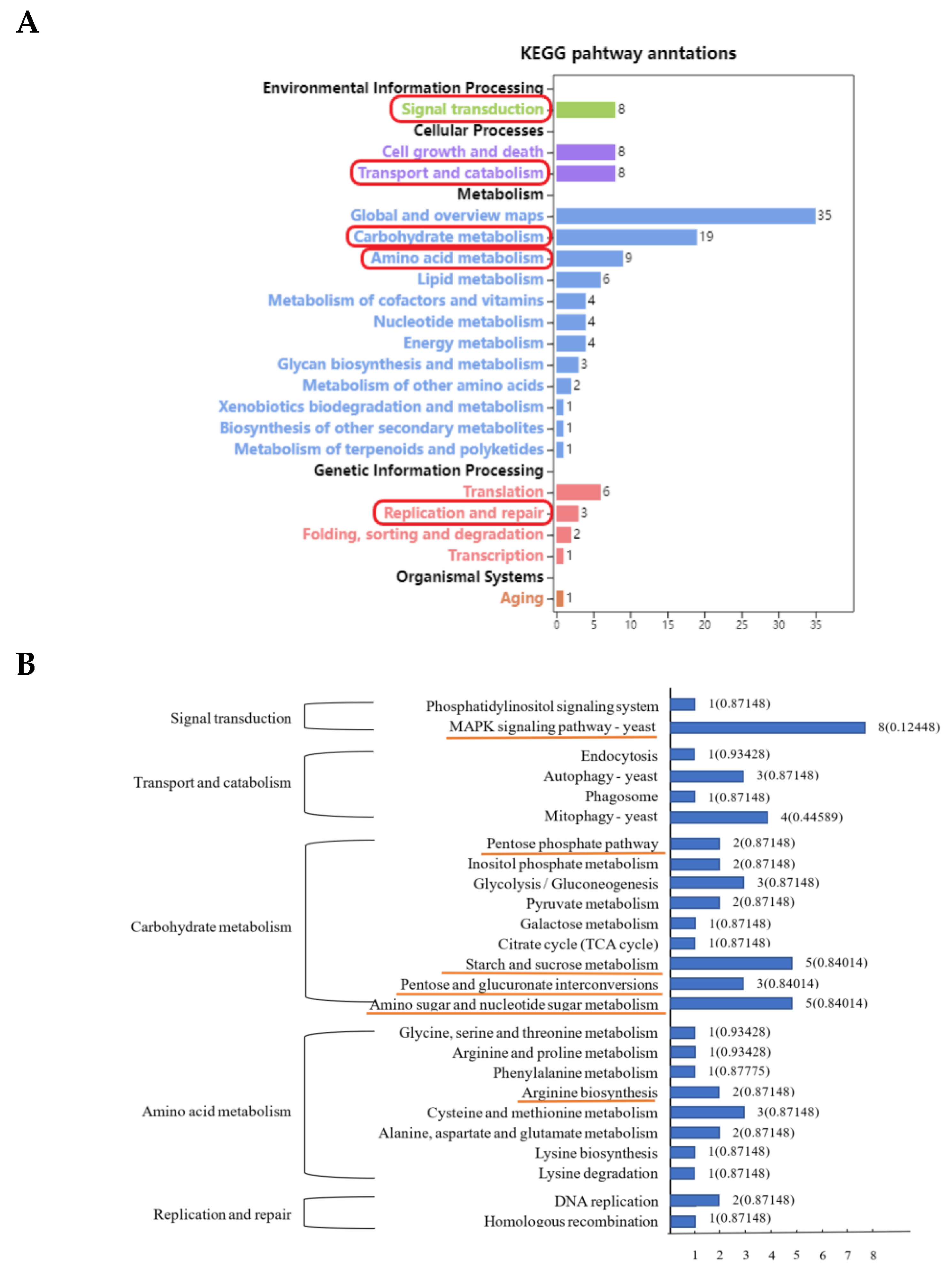
3.3. KEGG Enrichment Analysis of Differentially Expressed Peak-Related Genes in P. expansum
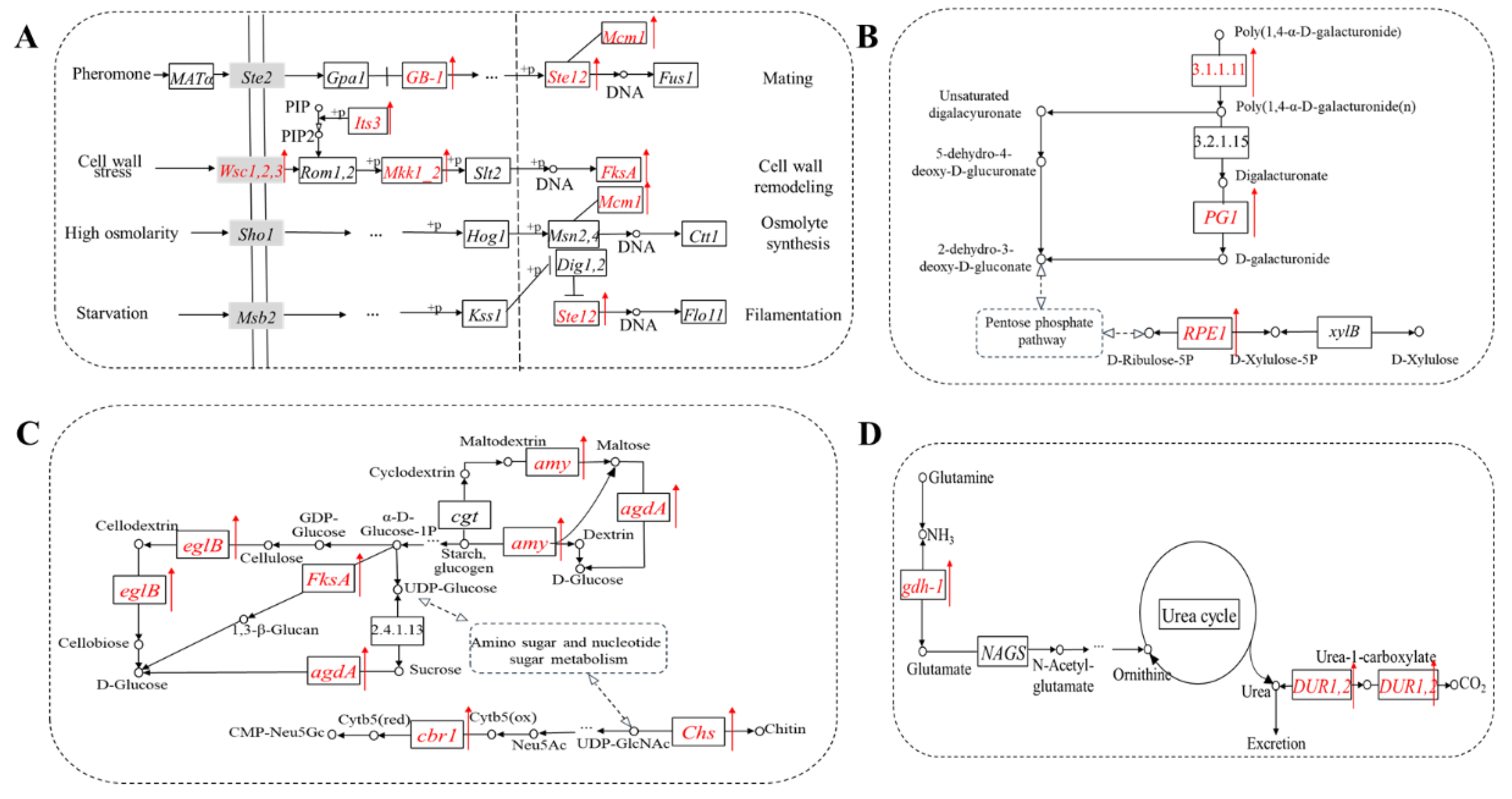
3.4. TF Motif Analysis of Differentially Expressed Peak-Related Genes
3.5. The Expression Level of Differentially Expressed Peak-Related Genes
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dou, Y.; Hu, P.H.; Tian, L.S.; Wu, C.B. Control and resistance induction mechanism of polylysine to postharvest Penicillium in pear fruit. Jiangsu Agric. Sci. 2022, 50, 177–182. [Google Scholar]
- Ianiri, G.; Pinedo, C.; Fratianni, A.; Panfili, G.; Castoria, R. Patulin degradation by the biocontrol yeast Sporobolomyces sp. is an inducible process. Toxins 2017, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Yang, Q.; Boateng, N.A.S.; Ahima, J.; Dou, Y.; Zhang, H. Ultrastructure observation and transcriptome analysis of Penicillium expansum invasion in postharvest pears. Postharvest Biol. Technol. 2020, 165, 111198. [Google Scholar] [CrossRef]
- Tian, S.; Torres, R.; Ballester, A.R.; Li, B.; Vilanova, L.; González-Candelas, L. Molecular aspects in pathogen-fruit interactions: Virulence and resistance. Postharvest Biol. Technol. 2016, 122, 11–21. [Google Scholar] [CrossRef]
- Miedes, E.; Lorences, E.P. Changes in cell wall pectin and pectinase activity in apple and tomato fruits during Penicillium expansum infection. J. Sci. Food Agric. 2010, 86, 1359–1364. [Google Scholar] [CrossRef]
- Levin, E.; Kishore, A.; Ballester, A.R.; Raphael, G.; Feigenberg, O.; Liu, Y.; Norelli, J.; Gonzalez-Candelas, L.; Wisniewski, M.; Droby, S. Identification of pathogenicity-related genes and the role of a subtilisin-related peptidase S8 (PePRT) in authophagy and virulence of Penicillium expansum on apples. Postharvest Biol. Technol. 2019, 149, 209–220. [Google Scholar] [CrossRef]
- Touhami, N.; Soukup, S.T.; Schmidt-Heydt, M.; Kulling, S.E.; Geisen, R. Citrinin as an accessory establishment factor of P. expansum for the colonization of apples. Int. J. Food Microbiol. 2018, 266, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Torres, P.; Vilanova, L.; Ballester, A.R.; López-Pérez, M.; Teixidó, N.; Viñas, I.; Usall, J.; González-Candelas, L.; Torres, R. Unravelling the contribution of the Penicillium expansum PeSte12 transcription factor to virulence during apple fruit infection. Food Microbiol. 2018, 69, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.G.; Jiao, M.; Guo, J.; Liu, P.; Tan, C.L.; Yang, Q.; Zhang, Y.; Thomas Voegele, R.; Kang, Z.S.; Guo, J. A novel MADS-box transcription factor PstMCM1-1 is responsible for full virulence of Puccinia striiformis f. sp. tritici. Environ. Microbiol 2018, 20, 1452–1463. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nussbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.C.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef]
- Li, Q.; Brown, J.B.; Huang, H.; Bickel, P.J. Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat. 2011, 5, 1752–1779. [Google Scholar] [CrossRef]
- Stark, R.; Brown, G. DiffBind: Differential Binding Analysis of ChIP-Seq Peak Data, R package version; R Foundation for Statistical Computing: Cambridge, UK, 2011. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.D.; Keyhani, N.O.; Yu, X.D.; He, Z.J.; Luo, Z.B.; Pei, Y.; Zhang, Y.J. The MAP kinase Bbslt2 controls growth, conidiation, cell wall integrity, and virulence in the insect pathogenic fungus Beauveria bassiana. Fungal Genet. Biol. 2012, 49, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.B.; Pande, K.; French, S.D.; Tuch, B.B.; Noble, S.M. An Iron Homeostasis Regulatory Circuit with Reciprocal Roles in Candida albicans Commensalism and Pathogenesis. Cell Host Microbe 2011, 10, 118–135. [Google Scholar] [CrossRef]
- Gsaller, F.; Hortschansky, P.; Beattie, S.R.; Klammer, V.; Tuppatsch, K.; Lechner, B.E.; Rietzschel, N.; Werner, E.R.; Vogan, A.A.; Chung, D.; et al. The Janus transcription factor HapX controls fungal adaptation to both iron starvation and iron excess. EMBO J. 2014, 33, 2261–2276. [Google Scholar] [CrossRef]
- Errede, B.; Ammerer, G. STE12, a protein involved in cell-type-specific transcription and signal transduction in yeast, is part of protein-DNA complexes. Genet Dev. 1989, 3, 1349. [Google Scholar] [CrossRef]
- Garcia-Sanchez, M.A.; Martin-Rodrigues, N.; Ramos, B.; de Vega-Bartol, J.J.; Perlin, M.H.; Diaz-Minguez, J.M. fost12, the Fusarium oxysporum hornolog of the transcription factor Ste12, is upregulated during plant infection and required for virulence. Fungal Genet Biol. 2010, 47, 216–225. [Google Scholar] [CrossRef]
- Rispail, N.; Pietro, A.D. Fusarium oxysporum Ste12 Controls Invasive Growth and Virulence Downstream of the Fmk1 MAPK Cascade. Mol. Plant Microbe Interact. 2009, 22, 830. [Google Scholar] [CrossRef] [PubMed]
- Vilanova, L.; Teixidó, N.; Torres, R.; Usall, J.; Viñas, I.; Sánchez-Torres, P. Relevance of the transcription factor PdSte12 in Penicillium digitatum conidiation and virulence during citrus fruit infection. Int. J. Food Microbiol. 2016, 235, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Schamber, A.; Leroch, M.; Diwo, J.; Mendgen, K.; Hahn, M. The role of mitogen-activated protein (MAP) kinase signalling components and the Ste12 transcription factor in germination and pathogenicity of Botrytis cinerea. Mol. Plant Pathol. 2010, 11, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento-Villamil, J.L.; Prieto, P.; Klosterman, S.J.; García-Pedrajas, M.D. Characterization of two homeodomain transcription factors with critical but distinct roles in virulence in the vascular pathogen Verticillium dahliae. Mol. Plant Pathol. 2018, 19, 986–1004. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.Q.; Li, P.; Wu, M.; Hao, Z.M.; Gong, X.D.; Zhang, X.Y.; Tian, L.; Zhang, P.; Wang, Y.; Cao, Z.Y.; et al. StSTE12 is required for the pathogenicity of Setosphaeria turcica by regulating appressorium development and penetration. Microbiol. Res. 2014, 169, 817–823. [Google Scholar] [CrossRef]
- Althoefer, H.; Schleiffer, A.; Wassmann, K.; Nordheim, A.; Ammerer, G. Mcm1 is required to coordinate G2-specific transcription in Saccharomyces cerevisiae. Mol. Cell. Biol. 1995, 15, 5917–5928. [Google Scholar] [CrossRef]
- Mead, J.; Bruning, A.R.; Gill, M.K.; Steiner, A.M.; Acton, T.B.; Vershon, A.K. Interactions of the Mcm1 MADS box protein with cofactors that regulate mating in yeast. Mol. Cell. Biol. 2002, 22, 4607–4621. [Google Scholar] [CrossRef]
- Yang, C.; Liu, H.Q.; Li, G.T.; Liu, M.G.; Yun, Y.Z.; Wang, C.F.; Ma, Z.H.; Xu, J.R. The MADS-box transcription factor FgMcm1 regulates cell identity and fungal development in Fusarium graminearum. Environ. Microbiol. 2015, 17, 2762–2776. [Google Scholar] [CrossRef]
- Zhou, X.Y.; Liu, W.D.; Wang, C.F.; Xu, Q.J.; Wang, Y.; Ding, S.L.; Xu, J.R. A MADS-box transcription factor MoMcm1 is required for male fertility, microconidium production and virulence in Magnaporthe oryzae. Mol. Microbiol. 2011, 80, 33–53. [Google Scholar] [CrossRef]
- Qu, X.Y.; Yu, B.D.; Liu, J.L.; Zhang, X.H.; Li, G.H.; Zhang, D.J.; Li, L.; Wang, X.L.; Wang, L.; Chen, J.Y.; et al. MADS-box transcription factor SsMADS is involved in regulating growth and virulence in Sclerotinia sclerotiorum. Int. J. Mol. Sci. 2014, 15, 8049–8062. [Google Scholar] [CrossRef]
- Pan, X.W.; Heitman, J. Sok2 regulates yeast pseudohyphal differentiation via a transcription factor cascade that regulates cell-cell adhesion. Mol. Cell. Biol. 2000, 20, 8364–8372. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Philip, B.; Levin, D.E. Wsc1 and Mid2 are cell surface sensors for cell wall integrity signaling that act through Rom2, a guanine nucleotide exchange factor for Rho1. Mol. Cell. Biol. 2001, 21, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Gualtieri, T.; Ragni, E.; Mizzi, L.; Fascio, U.; Popolo, L. The cell wall sensor Wsc1p is involved in reorganization of actin cytoskeleton in response to hypo-osmotic shock in Saccharomyces cerevisiae. Yeast 2004, 21, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Rodicio, R.; Buchwald, U.; Schmitz, H.P.; Heinisch, J.J. Dissecting sensor functions in cell wall integrity signaling in Kluyveromyces lactis. Fungal Genet Biol. 2008, 45, 422–435. [Google Scholar] [CrossRef]
- Futagami, T.; Nakao, S.; Kido, Y.; Oka, T.; Kajiwara, Y.; Takashita, H.; Omori, T.; Furukawa, K.; Goto, M. Putative stress sensors WscA and WscB are involved in hypo-osmotic and acidic pH stress tolerance in Aspergillus nidulans. Eukaryot. Cell 2011, 10, 1504–1515. [Google Scholar] [CrossRef] [PubMed]
- Maddi, A.; Dettman, A.; Fu, C.; Seiler, S.; Free, S.J. WSC-1 and HAM-7 are MAK-1 MAP kinase pathway sensors required for cell wall integrity and hyphal fusion in Neurospora crassa. PLoS ONE 2012, 7, e42374. [Google Scholar] [CrossRef][Green Version]
- Le Cam, B.; Massiot, P.; Rouxel, F. Cell wall polysaccharide-degrading enzymes produced by isolates of Mycocentrospora acerina differing in aggressiveness on carrot. Physiol. Mol. Plant Pathol. 1994, 44, 187–198. [Google Scholar] [CrossRef]
- ten Have, A.; Mulder, W.; Visser, J.; van Kan, J.A. The endopolygalacturonase gene Bcpg1 is required for full virulence of Botrytis cinerea. Mol. Plant Microbe 1998, 11, 1009–1016. [Google Scholar] [CrossRef]
- Tenberge, K.B.; Homann, V.; Oeser, B.; Tudzynski, P. Structure and expression of two polygalacturonase genes of Claviceps purpurea oriented in tandem and cytological evidence for pectinolytic enzyme activity during infection of rye. Phytopathology 1996, 86, 1084–1097. [Google Scholar] [CrossRef]
- Oeser, B.; Heidrich, P.M.; Müller, U.; Tudzynski, P.; Tenberge, K.B. Polygalacturonase is a pathogenicity factor in the Claviceps purpurea/rye interaction. Fungal Genet Biol. 2002, 36, 176–186. [Google Scholar] [CrossRef]
- Shieh, M.T.; Brown, R.L.; Whitehead, M.P.; Cary, J.W.; Cotty, P.J.; Cleveland, T.E.; Dean, R.A. Molecular genetic evidence for the involvement of a specific polygalacturonase, P2c, in the invasion and spread of Aspergillus flavus in cotton bolls. Appl. Environ. Microbiol. 1997, 63, 3548. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Symbol | Primer | Sequence(5′-3′) | Length (mer) | Tm (°C) |
|---|---|---|---|---|---|
| ncbi_27683293 | sebA | XM_016747872.1-F | CACAGCCTTGAAGACTCGGATGAC | 24 | 60.2 |
| XM_016747872.1-R | TGCTACGCTTCTTGATCTTGGGTTC | 25 | 59.3 | ||
| ncbi_27675759 | Pma2 | XM_016740340.1-F | CTGGATTCTCGCTGCCTGGATTG | 23 | 60.9 |
| XM_016740340.1-R | AGTGGAGCAGGTTGTTGTCGTTATC | 25 | 59.2 | ||
| ncbi_27674750 | isp4 | XM_016739331.1-F | CGGAGGAGATTGTCACGCATGTC | 23 | 60.7 |
| XM_016739331.1-R | CACCGCAATGGAAGGGCTTACC | 22 | 61.7 | ||
| ncbi_27683189 | WSC | XM_016747768.1-F | AACAGACCTCGAAGAGCAAGAACAC | 25 | 59.1 |
| XM_016747768.1-R | CCAGAAGAAGATAGCACCGCAGAG | 24 | 59.9 | ||
| ncbi_27683152 | fksA | XM_016747731.1-F | TGCCTTCTGGTTCTTCACTGCTTAC | 25 | 59.2 |
| XM_016747731.1-R | CGGCGAGATATTTGGGCGAGTC | 22 | 61.1 | ||
| ncbi_27682174 | PG1 | XM_016746754.1-F | CGAGCAGGATTATCAGAACGGTAGC | 25 | 59.6 |
| XM_016746754.1-R | CGCAGAGAATGTAGACGGGAATAGC | 25 | 59.6 | ||
| ncbi_27677161 | Ste12 | XM_016741742.1-F | CAGGAACTCGCTCGCACTTACC | 22 | 60.9 |
| XM_016741742.1-R | CTGGATGTTGTACGGTGGTCTGATC | 25 | 59.6 | ||
| ncbi_27682273 | Mkk1_2 | XM_016746853.1-F | TCGCACCACATCTGTCGTTACTATG | 25 | 58.7 |
| XM_016746853.1-R | GATGCTGTCCAGACTTCCACCTTC | 24 | 60.1 | ||
| ncbi_27683634 | adhB | XM_016748213.1-F | GAATTTGGCAATTTGGGAGGCTGTC | 25 | 59.3 |
| XM_016748213.1-R | TGTTGAGAATGGCGTGGCTAGTTAG | 25 | 59 | ||
| ncbi_27676242 | catA | XM_016740823.1-F | CGTCAGCTACCCGCAGAAACAC | 22 | 61.4 |
| XM_016740823.1-R | CGGCGAGAGCGAGTTGTAGAATAC | 24 | 59.9 | ||
| ncbi_27678339 | cetA | XM_016742920.1-F | CATCAAGATGTCCACCAGCGAGAG | 24 | 60.1 |
| XM_016742920.1-R | GGTGAAGCCCGACTTGACAAACTC | 24 | 60.8 | ||
| ncbi_27674565 | YFL054C | XM_016739146.1-F | CATTGTTGAAGCCGCCACATTAGC | 24 | 59.6 |
| XM_016739146.1-R | GAAGAACGCAGTCGCAGGATGG | 22 | 61.5 | ||
| ncbi_27673157 | DOA4 | XM_016737738.1-F | GGAAGACTCGCTGACGGAAGAAC | 23 | 60.4 |
| XM_016737738.1-R | TTCAGGGAACCGACGCAAGAAAG | 23 | 60.1 |
| TF | TF Description | Gene ID | Symbol | Gene Description |
|---|---|---|---|---|
| HAP3 | Heteromeric CCAAT-binding factors | ncbi_27674657 | its3 | 1-phosphatidylinositol-4-phosphate 5-kinase |
| ncbi_27677161 | Ste12 | transcription factor STE12 | ||
| ncbi_27679022 | MCM1 | pheromone receptor transcription factor | ||
| ncbi_27682441 | agdA | alpha-glucosidase | ||
| SOK2 | APSES-type DNA-binding domain | ncbi_27674657 | its3 | 1-phosphatidylinositol-4-phosphate 5-kinase |
| ncbi_27682445 | DUR1,2 | urea carboxylase / allophanate hydrolase | ||
| ncbi_27683189 | WSC | cell wall integrity and stress response component | ||
| SIP4 | C6 zinc cluster factors | ncbi_27679542 | RPE1 | ribulose-phosphate 3-epimerase |
| ncbi_27683189 | WSC | cell wall integrity and stress response component | ||
| YPR196W | C6 zinc cluster factors | ncbi_27678364 | eglB | endoglucanase |
| ncbi_27677161 | Ste12 | transcription factor STE12 | ||
| ECM22 | C6 zinc cluster factors | ncbi_27679542 | RPE1 | ribulose-phosphate 3-epimerase |
| ncbi_27683189 | WSC | cell wall integrity and stress response component | ||
| YLR278C | C6 zinc cluster factors | ncbi_27679542 | RPE1 | ribulose-phosphate 3-epimerase |
| ncbi_27683189 | WSC | cell wall integrity and stress response component | ||
| SUT2 | C6 zinc cluster factors | ncbi_27679542 | RPE1 | ribulose-phosphate 3-epimerase |
| STP3 | C2H2 zinc finger factors | ncbi_27672954 | WSC | cell wall integrity and stress response component |
| YNR063W | C6 zinc cluster factors | ncbi_27677161 | Ste12 | transcription factor STE12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, L.; Shu, Y.; Quan, S.; Dhanasekaran, S.; Zhang, X.; Zhang, H. Screening and Regulation Mechanism of Key Transcription Factors of Penicillium expansum Infecting Postharvest Pears by ATAC-Seq Analysis. Foods 2022, 11, 3855. https://doi.org/10.3390/foods11233855
Zhao L, Shu Y, Quan S, Dhanasekaran S, Zhang X, Zhang H. Screening and Regulation Mechanism of Key Transcription Factors of Penicillium expansum Infecting Postharvest Pears by ATAC-Seq Analysis. Foods. 2022; 11(23):3855. https://doi.org/10.3390/foods11233855
Chicago/Turabian StyleZhao, Lina, Yuling Shu, Sihao Quan, Solairaj Dhanasekaran, Xiaoyun Zhang, and Hongyin Zhang. 2022. "Screening and Regulation Mechanism of Key Transcription Factors of Penicillium expansum Infecting Postharvest Pears by ATAC-Seq Analysis" Foods 11, no. 23: 3855. https://doi.org/10.3390/foods11233855
APA StyleZhao, L., Shu, Y., Quan, S., Dhanasekaran, S., Zhang, X., & Zhang, H. (2022). Screening and Regulation Mechanism of Key Transcription Factors of Penicillium expansum Infecting Postharvest Pears by ATAC-Seq Analysis. Foods, 11(23), 3855. https://doi.org/10.3390/foods11233855