

Environment-Related Genes Analysis of Limosilactobacillus fermentum Isolated from Food and Human Gut: Genetic Diversity and Adaption Evolution

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Genome Sequence
2.2. Average Nucleotide Identity Values, Pan-, and Core-Genome, and Phylogenetic Analyses
2.3. Clusters of Orthologous Groups (COGs) Analysis
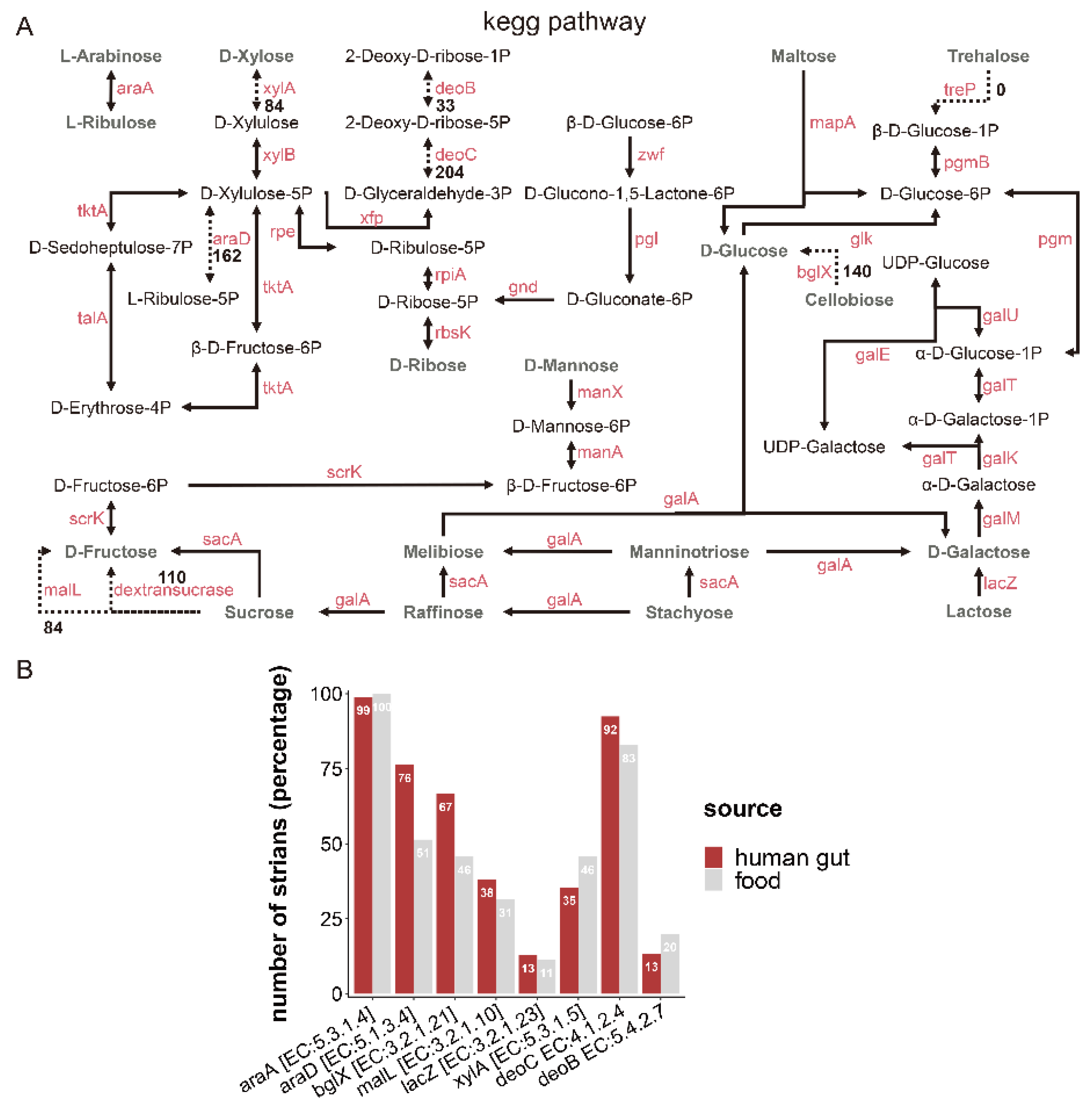
2.4. Carbohydrate Metabolism
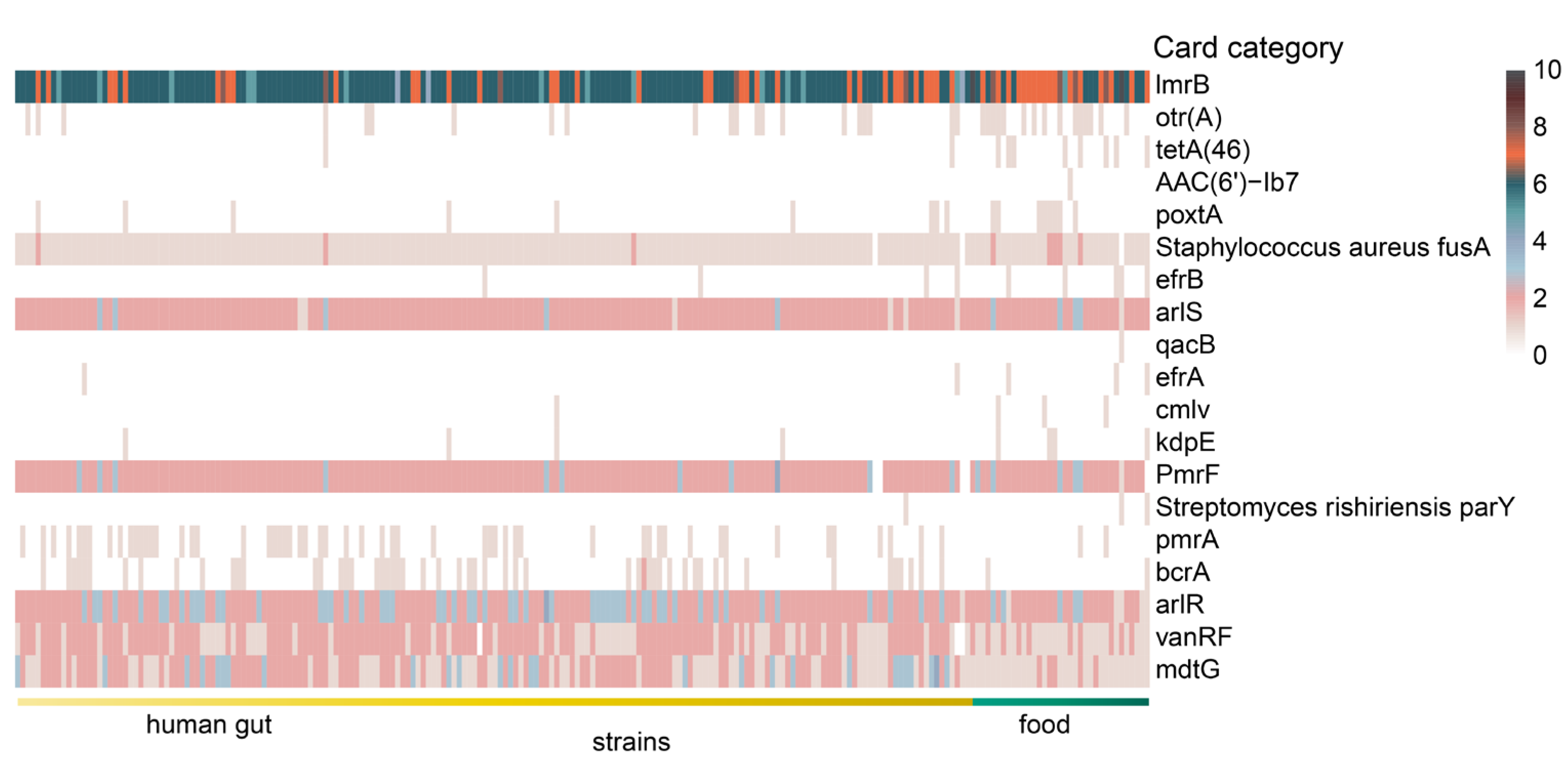
2.5. Antibiotic Resistance Genes (ARGs)
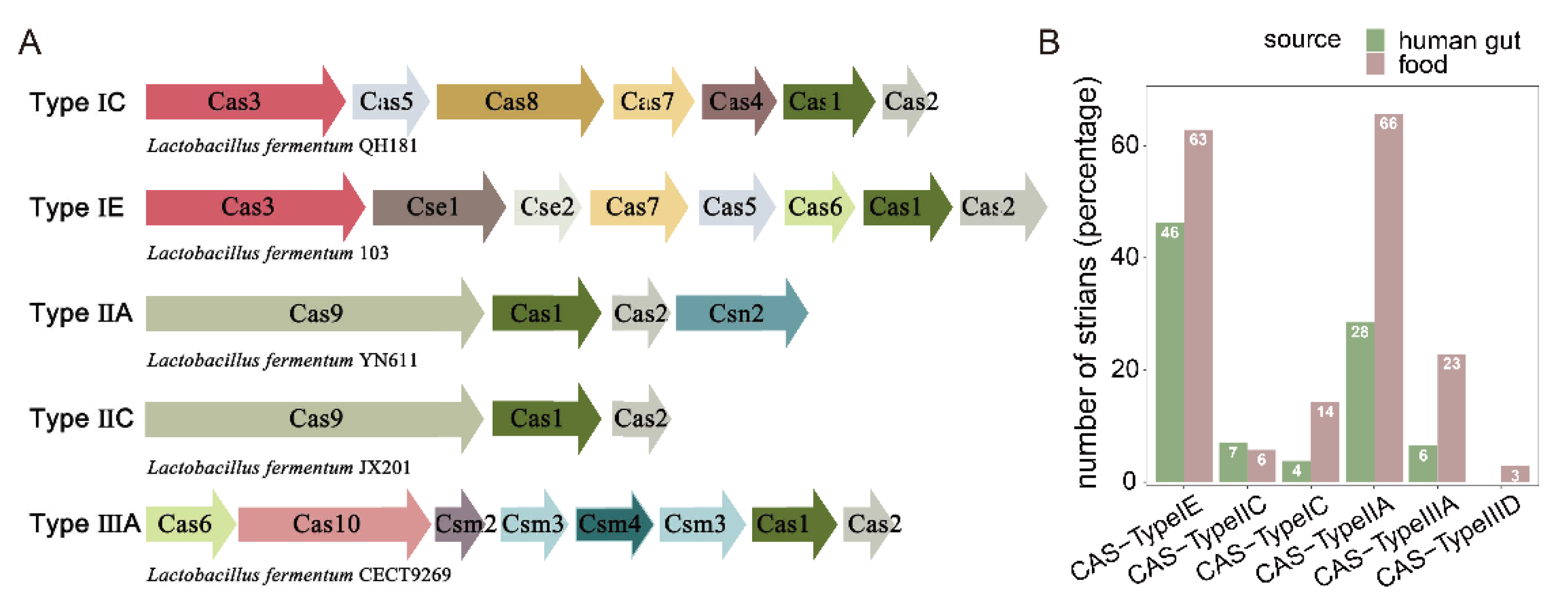
2.6. CRISPR-Cas Systems
2.7. Prophage Identification
2.8. Statistical Analysis
2.9. Data Deposition
3. Results
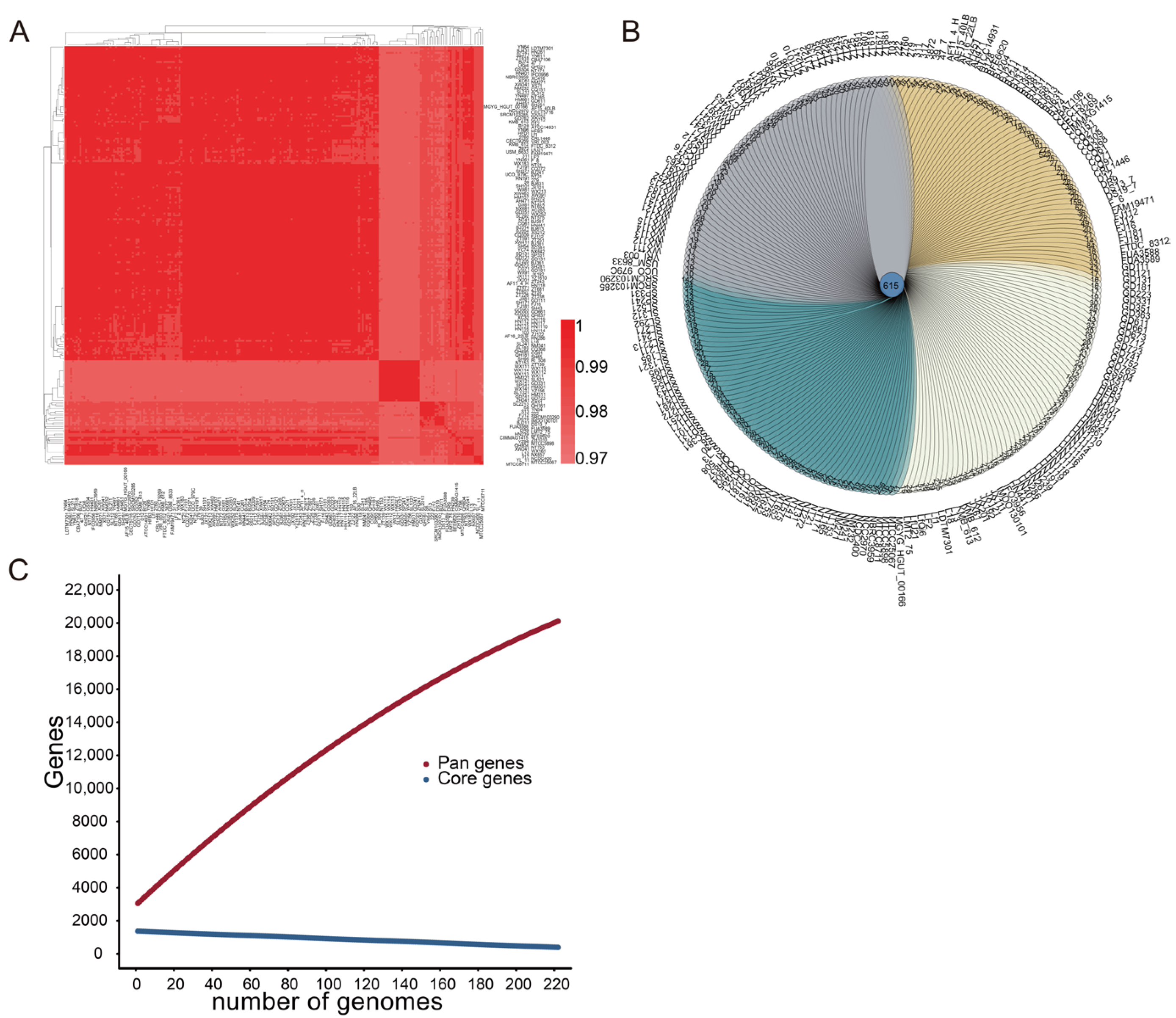
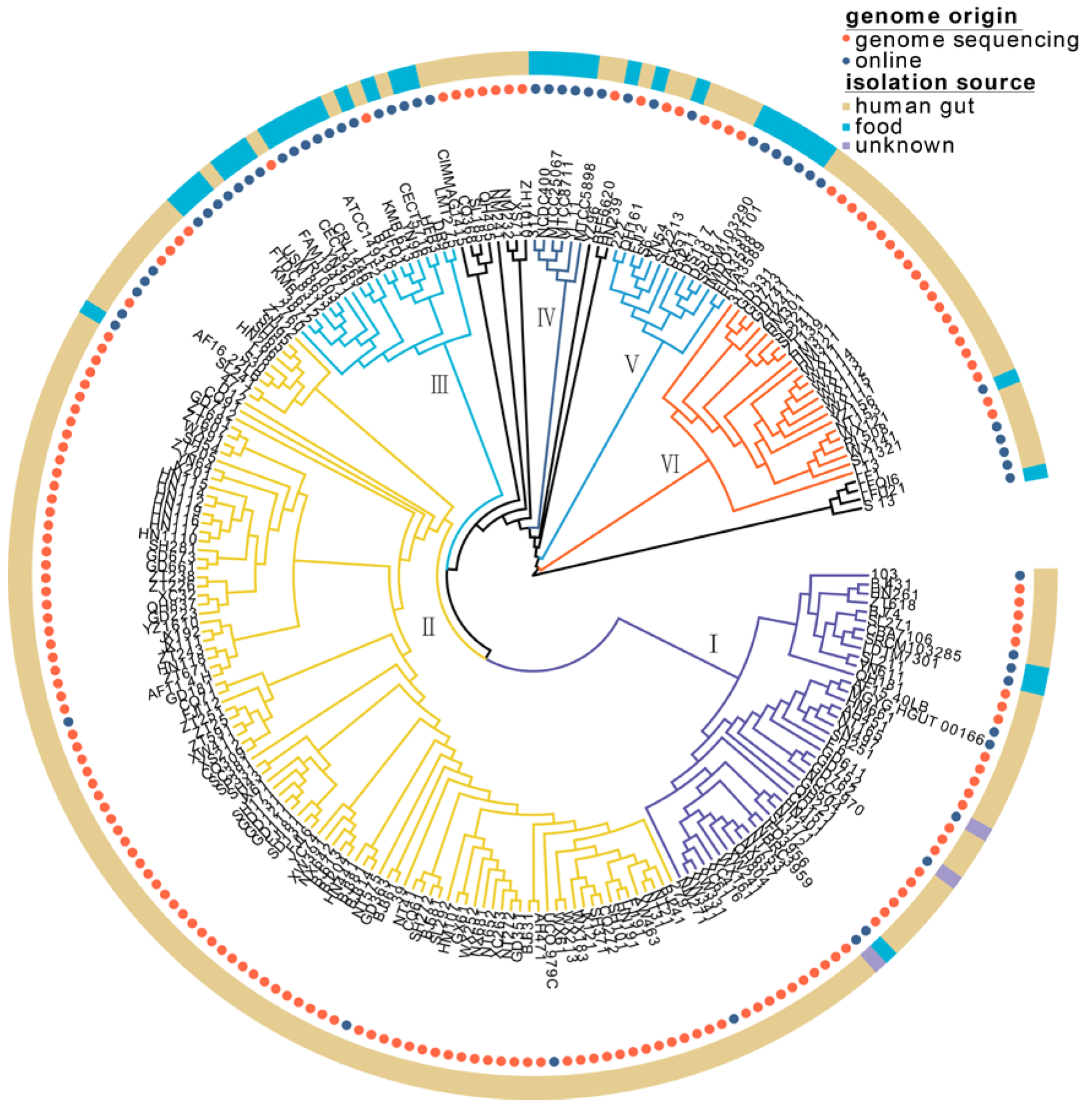
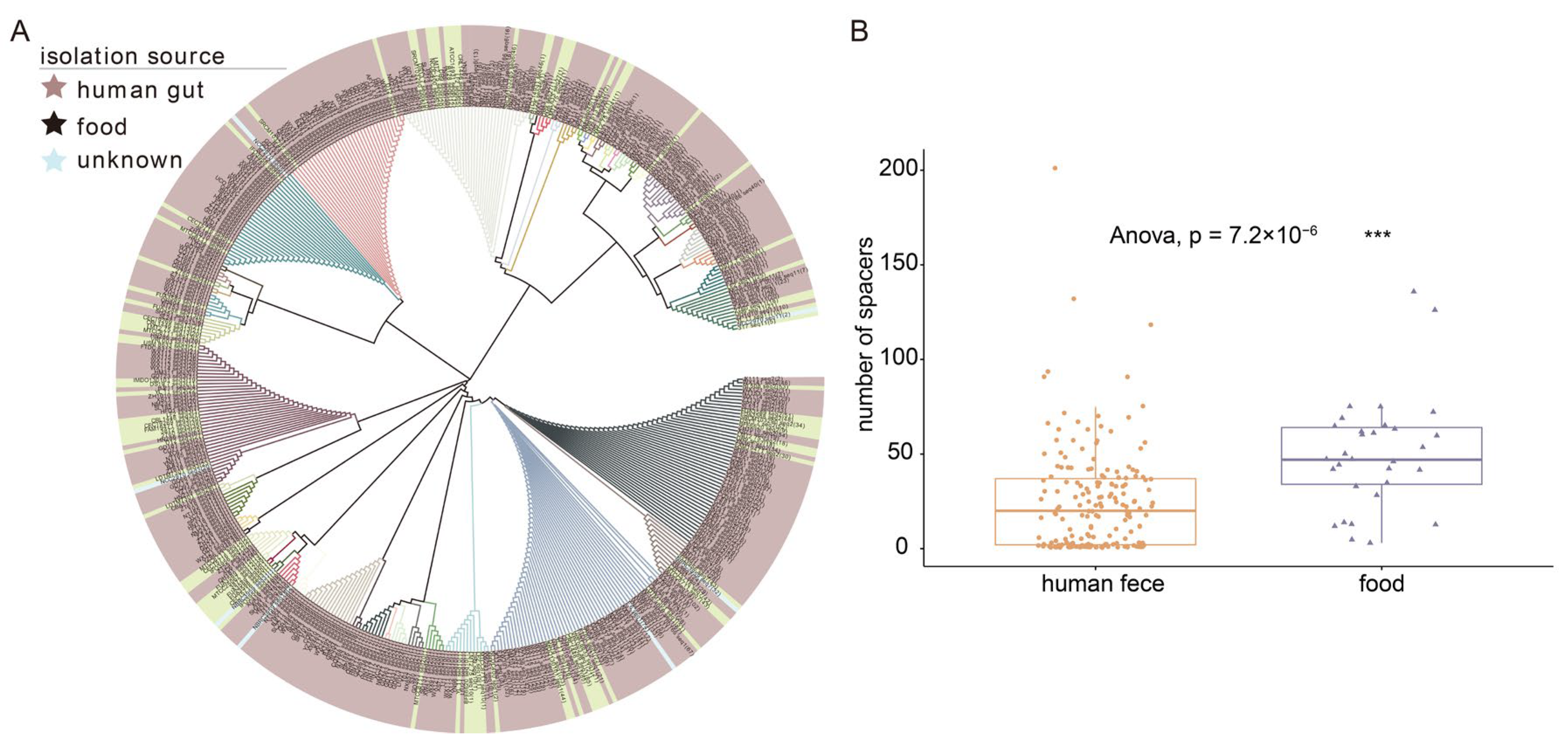
3.1. Genetic Diversity and Phylogenetic Analysis of 224 L. fermentum Strains
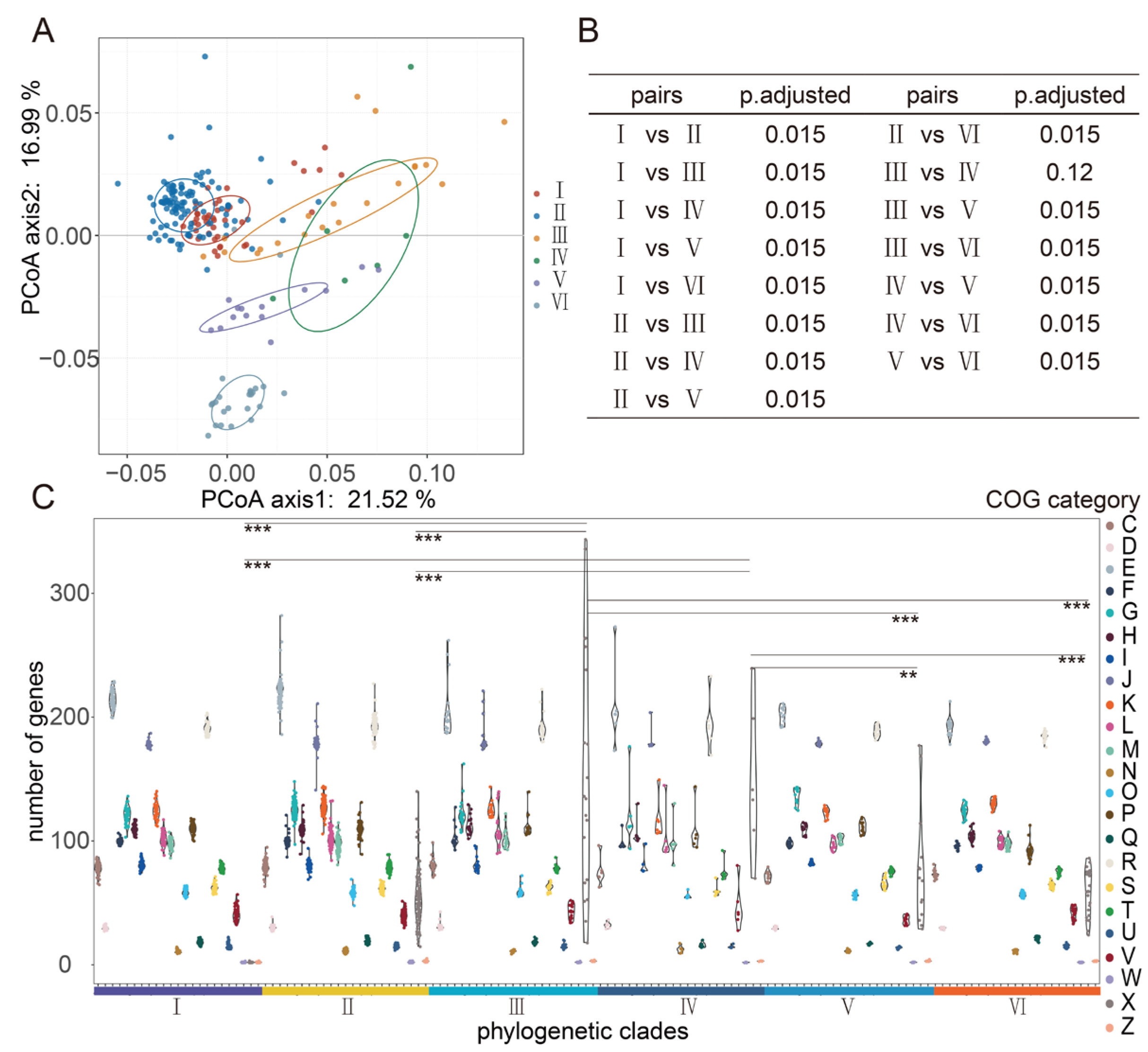
3.2. Analysis of Clusters of Orthologous Groups (COGs) in L. fermentum Strains
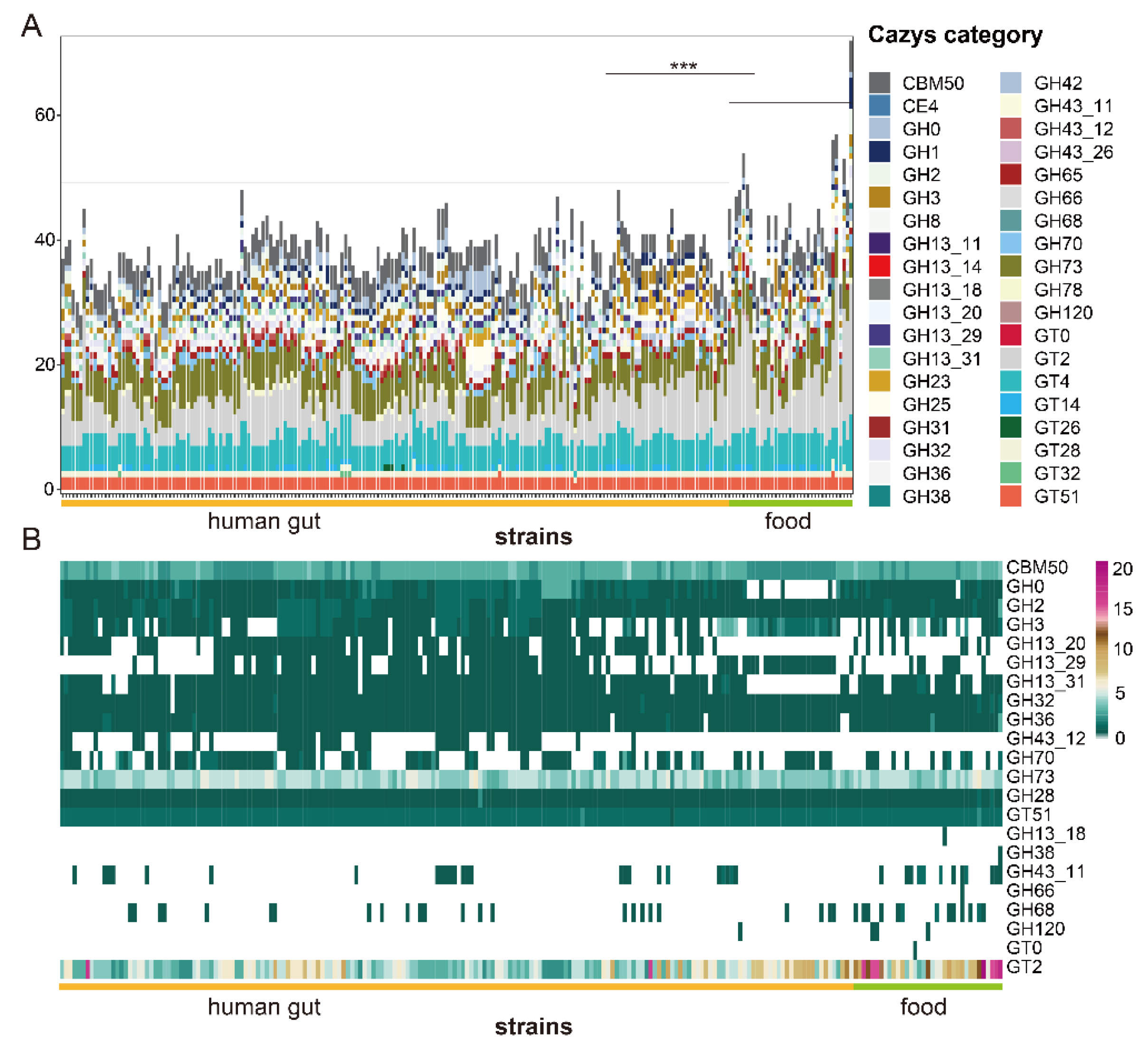
3.3. Identification of Carbohydrate Metabolism in L. fermentum Strains
3.4. Characteristic of Antibiotic Resistance Genes in L. fermentum Strains
3.5. Identification of CRISPR-Cas Systems in L. fermentum Strains
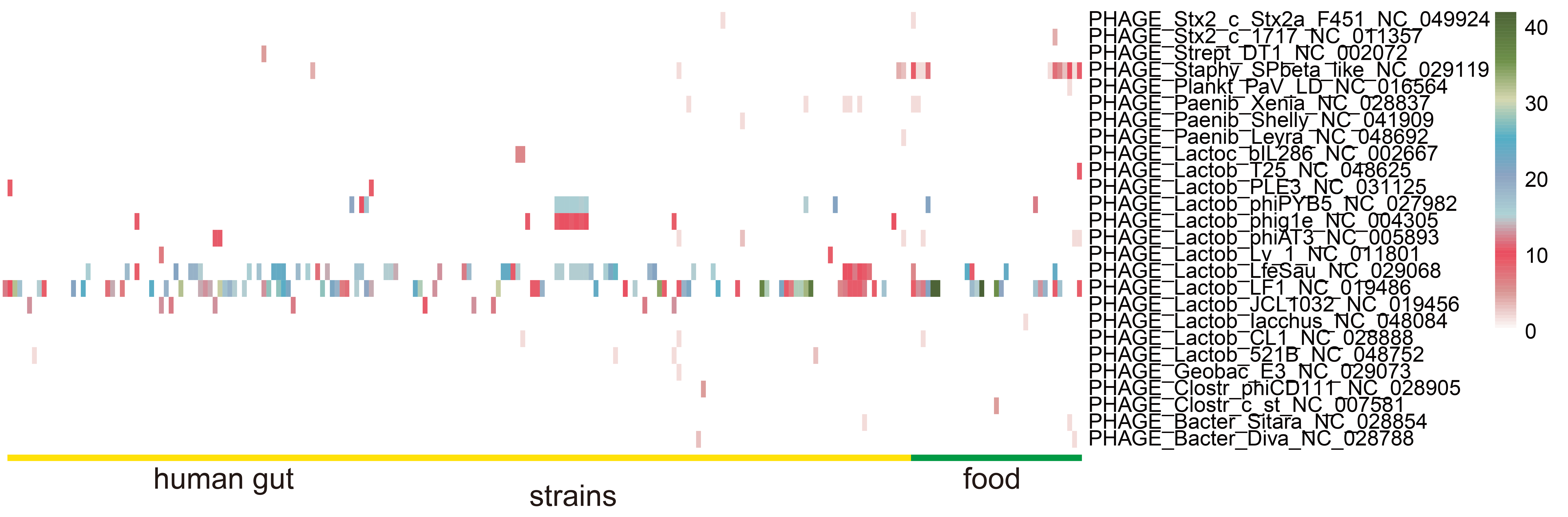
3.6. Identification of Prophages in L. fermentum Strains
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pederson, C.S. The Gas-producing Species of the Genus Lactobacillus. J. Bacteriol. 1938, 35, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Hammes, W.P.; Hertel, C. Bergey’s Manual of Systematics of Archaea and Bacteria; Wiley: Hoboken, NJ, USA, 2015. [Google Scholar]
- Swain, M.R.; Anandharaj, M.; Ray, R.C.; Parveen Rani, R. Fermented Fruits and Vegetables of Asia: A Potential Source of Probiotics. Biotechnol. Res. Int. 2014, 1, 250424. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Verón, H.; Contreras, L.; Isla, M.I. An overview of plant-autochthonous microorganisms and fermented vegetable foods. Food Sci. Hum. Wellness 2020, 9, 112–123. [Google Scholar] [CrossRef]
- Zhao, Y.; Yu, L.; Tian, F.; Zhao, J.; Zhang, H.; Chen, W.; Zhai, Q. An optimized culture medium to isolate Lactobacillus fermentum strains from the human intestinal tract. Food Funct. 2021, 12, 6740–6754. [Google Scholar] [CrossRef]
- Duar, R.M.; Lin, X.B.; Zheng, J.; Martino, M.E.; Grenier, T.; Perez-Munoz, M.E.; Leulier, F.; Ganzle, M.; Walter, J. Lifestyles in transition: Evolution and natural history of the genus Lactobacillus. FEMS Microbiol. Rev. 2017, 41, 27–48. [Google Scholar] [CrossRef]
- Zhao, Y.; Hong, K.; Zhao, J.; Zhang, H.; Zhai, Q.; Chen, W. Lactobacillus fermentum and its potential immunomodulatory properties. J. Funct. Foods 2019, 56, 21–32. [Google Scholar] [CrossRef]
- Rodas, A.M.; Ferrer, S.; Pardo, I. Polyphasic study of wine Lactobacillus strains: Taxonomic implications. Int. J. Syst. Evol. Microbiol. 2005, 55, 197–207. [Google Scholar] [CrossRef]
- Zheng, J.; Wittouck, S.; Salvetti, E.; Franz, C.; Harris, H.M.B.; Mattarelli, P.; O’Toole, P.W.; Pot, B.; Vandamme, P.; Walter, J.; et al. A taxonomic note on the genus Lactobacillus: Description of 23 novel genera, emended description of the genus Lactobacillus Beijerinck 1901, and union of Lactobacillaceae and Leuconostocaceae. Int. J. Syst. Evol. Microbiol. 2020, 70, 2782–2858. [Google Scholar] [CrossRef]
- Pang, H.; Qin, G.; Tan, Z.; Li, Z.; Wang, Y.; Cai, Y. Natural populations of lactic acid bacteria associated with silage fermentation as determined by phenotype, 16S ribosomal RNA and recA gene analysis. Syst. Appl. Microbiol. 2011, 34, 235–241. [Google Scholar] [CrossRef]
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA sequencing at 40: Past, present and future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, C.; Yu, L.; Tian, F.; Zhao, J.; Zhang, H.; Chen, W.; Zhai, Q. Phylogenetic and comparative genomic analysis of Lactobacillus fermentum Strains and the Key Genes Related to their Intestinal Anti-inflammatory Effects. Engineering 2021, in press. [CrossRef]
- Dan, T.; Liu, W.; Song, Y.; Xu, H.; Menghe, B.; Zhang, H.; Sun, Z. The evolution and population structure of Lactobacillus fermentum from different naturally fermented products as determined by multilocus sequence typing (MLST). BMC Microbiol. 2015, 15, 107. [Google Scholar] [CrossRef]
- Li, Q.; Gänzle, M.G. Host-adapted lactobacilli in food fermentations: Impact of metabolic traits of host adapted lactobacilli on food quality and human health. Curr. Opin. Food Sci. 2020, 31, 71–80. [Google Scholar] [CrossRef]
- Ao, X.; Zhang, X.; Zhang, X.; Shi, L.; Zhao, K.; Yu, J.; Dong, L.; Cao, Y.; Cai, Y. Identification of lactic acid bacteria in traditional fermented yak milk and evaluation of their application in fermented milk products. J. Dairy Sci. 2012, 95, 1073–1084. [Google Scholar] [CrossRef]
- Huang, F.; Hong, R.; Zhang, R.; Yi, Y.; Dong, L.; Liu, L.; Jia, X.; Ma, Y.; Zhang, M. Physicochemical and biological properties of longan pulp polysaccharides modified by Lactobacillus fermentum fermentation. Int. J. Biol. Macromol. 2019, 125, 232–237. [Google Scholar] [CrossRef]
- Zhang, X.; Kong, B.; Xiong, Y.L. Production of cured meat color in nitrite-free Harbin red sausage by Lactobacillus fermentum fermentation. Meat Sci. 2007, 77, 593–598. [Google Scholar]
- Falasconi, I.; Fontana, A.; Patrone, V.; Rebecchi, A.; Duserm Garrido, G.; Principato, L.; Callegari, M.L.; Spigno, G.; Morelli, L. Genome-Assisted Characterization of Lactobacillus fermentum, Weissella cibaria, and Weissella confusa Strains Isolated from Sorghum as Starters for Sourdough Fermentation. Microorganisms 2020, 8, 1388. [Google Scholar] [CrossRef]
- Oguntoyinbo, F.A.; Cho, G.S.; Trierweiler, B.; Kabisch, J.; Rosch, N.; Neve, H.; Bockelmann, W.; Frommherz, L.; Nielsen, D.S.; Krych, L.; et al. Fermentation of African kale (Brassica carinata) using L. plantarum BFE 5092 and L. fermentum BFE 6620 starter strains. Int. J. Food Microbiol. 2016, 238, 103–112. [Google Scholar]
- Li, S.; Tao, Y.; Li, D.; Wen, G.; Zhou, J.; Manickam, S.; Han, Y.; Chai, W.S. Fermentation of blueberry juices using autochthonous lactic acid bacteria isolated from fruit environment: Fermentation characteristics and evolution of phenolic profiles. Chemosphere 2021, 276, 130090. [Google Scholar] [CrossRef]
- Wu, J.; Tian, Y.; Wu, Z.; Weng, P.; Zhang, X. Effects of pretreatment with dimethyl dicarbonate on the quality characteristics of fermented Huyou juice and storage stability. J. Food Process Preserv. 2021, 45, e15343. [Google Scholar]
- Badel, S.; Bernardi, T.; Michaud, P. New perspectives for Lactobacilli exopolysaccharides. Biotechnol. Adv. 2011, 29, 54–66. [Google Scholar] [CrossRef]
- Macori, G.; Cotter, P.D. Novel insights into the microbiology of fermented dairy foods. Curr. Opin. Biotechnol. 2018, 49, 172–178. [Google Scholar] [CrossRef]
- Barrangou, R.; Notebaart, R.A. CRISPR-Directed Microbiome Manipulation across the Food Supply Chain. Trends Microbiol. 2019, 27, 489–496. [Google Scholar] [CrossRef]
- Paulino do Nascimento, L.C.; Lacerda, D.C.; Ferreira, D.J.S.; de Souza, E.L.; de Brito Alves, J.L. Limosilactobacillus fermentum, Current Evidence on the Antioxidant Properties and Opportunities to be Exploited as a Probiotic Microorganism . Probiotics Antimicrob Proteins 2022, 14, 960–979. [Google Scholar]
- Lehri, B.; Seddon, A.M.; Karlyshev, A.V. Lactobacillus fermentum 3872 as a potential tool for combatting Campylobacter jejuni infections. Virulence 2017, 8, 1753–1760. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, X.; Wang, H.; Yang, Z.; Li, J.; Suo, H. Prevent Effects of Lactobacillus Fermentum HY01 on Dextran Sulfate Sodium-Induced Colitis in Mice. Nutrients 2017, 9, 545. [Google Scholar] [CrossRef]
- Kullisaar, T.; Zilmer, K.; Salum, T.; Rehema, A.; Zilmer, M. The use of probiotic L. fermentum ME-3 containing Reg’Activ Cholesterol sment for 4 weeks has a positive influence on blood lipoprotein profiles and inflammatory cytokines: An open-label preliminary study. Nutr. J. 2016, 15, 93. [Google Scholar] [CrossRef]
- Barone, R.; Rappa, F.; Macaluso, F.; Caruso Bavisotto, C.; Sangiorgi, C.; Di Paola, G.; Tomasello, G.; Di Felice, V.; Marciano, V.; Farina, F.; et al. Alcoholic Liver Disease: A Mouse Model Reveals Protection by Lactobacillus fermentum. Clin. Transl. Gastroenterol. 2016, 7, e138. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, Y.; Ye, L.; Wang, C. The anti-cancer effects and mechanisms of lactic acid bacteria exopolysaccharides in vitro: A review. Carbohydr. Polym. 2021, 253, 117308. [Google Scholar] [CrossRef] [PubMed]
- Riglar, D.T.; Silver, P.A. Engineering bacteria for diagnostic and therapeutic applications. Nat. Rev. Microbiol. 2018, 16, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Craven, S.S.; Sula, N.F.; Adhikari, A.A.; Nicholas, D.B.; Mina, A.I.; Banks, A.S.; Lynn, B.; Sloan, D. A selective gut bacterial bile salt hydrolase alters host metabolism. eLife Sci. 2018, 7, e37182. [Google Scholar] [CrossRef] [PubMed]
- Sanders, M.E.; Merenstein, D.J.; Reid, G.; Gibson, G.R.; Rastall, R.A. Probiotics and prebiotics in intestinal health and disease: From biology to the clinic. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.J.; Allen, B.M.; Hiam, K.J.; Dodd, D.; Van Treuren, W.; Higginbottom, S.; Nagashima, K.; Fischer, C.R.; Sonnenburg, J.L.; Spitzer, M.H.; et al. Depletion of microbiome-derived molecules in the host using Clostridium genetics. Science 2019, 366, 1309. [Google Scholar] [CrossRef]
- Morita, H.; Toh, H.; Fukuda, S.; Horikawa, H.; Oshima, K.; Suzuki, T.; Murakami, M.; Hisamatsu, S.; Kato, Y.; Takizawa, T.; et al. Comparative genome analysis of Lactobacillus reuteri and Lactobacillus fermentum reveal a genomic island for reuterin and cobalamin production. DNA Res. 2008, 15, 151–161. [Google Scholar] [CrossRef]
- Brandt, J.; Albertsen, M. Investigation of detection limits and the influence of DNA extraction and primer choice on the observed microbial communities in drinking water samples using 16S rRNA gene amplicon sequencing. Front. Microbiol. 2018, 9, 2140. [Google Scholar] [CrossRef]
- Wang, S.; Yang, B.; Ross, R.P.; Stanton, C.; Zhao, J.; Zhang, H.; Chen, W. Comparative Genomics Analysis of Lactobacillus ruminis from Different Niches. Genes 2020, 11, 70. [Google Scholar] [CrossRef]
- Ferreiro, A.; Crook, N.; Gasparrini, A.J.; Dantas, G. Multiscale Evolutionary Dynamics of Host-Associated Microbiomes. Cell 2018, 172, 1216–1227. [Google Scholar] [CrossRef]
- Good, B.H.; McDonald, M.J.; Barrick, J.E.; Lenski, R.E.; Desai, M.M. The dynamics of molecular evolution over 60,000 generations. Nature 2017, 551, 45–50. [Google Scholar] [CrossRef]
- Rook, G.; Bäckhed, F.; Levin, B.R.; McFall-Ngai, M.J.; McLean, A.R. Evolution, human-microbe interactions, and life history plasticity. Lancet 2017, 390, 521–530. [Google Scholar] [CrossRef]
- Zhang, C.; Gong, W.; Li, Z.; Gao, D.; Gao, Y. Research progress of gut flora in improving human wellness. Food Sci. Hum. Wellness 2019, 8, 102–105. [Google Scholar] [CrossRef]
- Youngblut, N.D.; Reischer, G.H.; Walters, W.; Schuster, N.; Walzer, C.; Stalder, G.; Ley, R.E.; Farnleitner, A.H. Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades. Nat. Commun. 2019, 10, 2200. [Google Scholar] [CrossRef]
- Filannino, P.; Di Cagno, R.; Gobbetti, M. Metabolic and functional paths of lactic acid bacteria in plant foods: Get out of the labyrinth. Curr. Opin. Biotechnol. 2018, 49, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Batstone, R.T.; O’Brien, A.M.; Harrison, T.L.; Frederickson, M.E. Experimental evolution makes microbes more cooperative with their local host genotype. Science 2020, 370, 476–478. [Google Scholar] [CrossRef]
- Verce, M.; De Vuyst, L.; Weckx, S. Comparative genomics of Lactobacillus fermentum suggests a free-living lifestyle of this lactic acid bacterial species. Food Microbiol. 2020, 89, 103448. [Google Scholar] [CrossRef]
- Yu, J.; Zhao, J.; Song, Y.; Zhang, J.; Yu, Z.; Zhang, H.; Sun, Z. Comparative genomics of the herbivore gut symbiont Lactobacillus reuteri reveals genetic diversity and lifestyle adaptation. Front. Microbiol. 2018, 9, 1151. [Google Scholar] [CrossRef]
- Pennisi, E. No microbiome is an island, survey reveals. Science 2019, 365, 851. [Google Scholar] [CrossRef]
- Pasolli, E.; De Filippis, F.; Mauriello, I.E.; Cumbo, F.; Walsh, A.M.; Leech, J.; Cotter, P.D.; Segata, N.; Ercolini, D. Large-scale genome-wide analysis links lactic acid bacteria from food with the gut microbiome. Nat. Commun. 2020, 11, 2610. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Wolf, Y.I.; Makarova, K.S.; Roberto, V.A.; David, L.; Koonin, E.V. COG database update: Focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 2020, 49, D274–D281. [Google Scholar] [CrossRef]
- Carr, V.R.; Shkoporov, A.; Hill, C.; Mullany, P.; Moyes, D.L. Probing the Mobilome: Discoveries in the Dynamic Microbiome. Trends Microbiol. 2021, 29, 158–170. [Google Scholar] [CrossRef]
- Wibowo, M.C.; Yang, Z.; Borry, M.; Hubner, A.; Huang, K.D.; Tierney, B.T.; Zimmerman, S.; Barajas-Olmos, F.; Contreras-Cubas, C.; Garcia-Ortiz, H.; et al. Reconstruction of ancient microbial genomes from the human gut. Nature 2021, 594, 234–239. [Google Scholar] [CrossRef]
- Luo, H.; Gao, F.; Lin, Y. Evolutionary conservation analysis between the essential and nonessential genes in bacterial genomes. Sci. Rep. 2015, 5, 13210. [Google Scholar] [CrossRef] [PubMed]
- Armet, A.M.; Deehan, E.C.O.; Sullivan, A.F.; Mota, J.F.; Field, C.J.; Prado, C.M.; Lucey, A.J.; Walter, J. Rethinking healthy eating in light of the gut microbiome. Cell Host Microbe 2022, 30, 764–785. [Google Scholar] [CrossRef]
- Armstrong, Z.; Mewis, K.; Liu, F.; Morgan-Lang, C.; Scofield, M.; Durno, E.; Chen, H.M.; Mehr, K.; Withers, S.G.; Hallam, S.J. Metagenomics reveals functional synergy and novel polysaccharide utilization loci in the Castor canadensis fecal microbiome. ISME J 2018, 12, 2757–2769. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, L.; Ross, P.; Zhao, J.; Zhang, H.; Chen, W. Comparative genomics of Lactobacillus crispatus from the gut and vagina reveals genetic diversity and lifestyle adaptation. Genes 2020, 11, 360. [Google Scholar] [CrossRef]
- Mao, B.; Yin, R.; Li, X.; Cui, S.; Zhang, H.; Zhao, J.; Chen, W. Comparative Genomic Analysis of Lactiplantibacillus plantarum Isolated from Different Niches. Genes 2021, 12, 241. [Google Scholar] [CrossRef]
- Hehemann, J.H.; Correc, G.; Barbeyron, T.; Helbert, W.; Czjzek, M.; Michel, G. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature 2010, 464, 908–912. [Google Scholar] [CrossRef]
- Martino, M.E.; Joncour, P.; Leenay, R.; Gervais, H.; Shah, M.; Hughes, S.; Gillet, B.; Beisel, C.; Leulier, F. Bacterial Adaptation to the Host’s Diet Is a Key Evolutionary Force Shaping Drosophila-Lactobacillus Symbiosis. Cell Host Microbe 2018, 24, 109–119.e6. [Google Scholar] [CrossRef]
- Jiang, D.; Cheng, Z.; Chen, X.; Dong, F.; Xu, J.; Liu, X.; Wu, X.; Pan, X.; An, X.; Zheng, Y. Occurrences of eight common-used pesticide adjuvants in ten vegetable species and implications for dietary intake in North China. Food Chem. 2021, 347, 128984. [Google Scholar] [CrossRef]
- Sommer, M.O.A.; Munck, C.; Toft-Kehler, R.V.; Andersson, D.I. Prediction of antibiotic resistance: Time for a new preclinical paradigm? Nat. Rev. Microbiol. 2017, 15, 689–696. [Google Scholar] [CrossRef]
- Debroas, D.; Siguret, C. Viruses as key reservoirs of antibiotic resistance genes in the environment. ISME J. 2019, 13, 2856–2867. [Google Scholar] [CrossRef]
- Lee, K.; Kim, D.W.; Lee, D.H.; Kim, Y.S.; Bu, J.H.; Cha, J.H.; Thawng, C.N.; Hwang, E.M.; Seong, H.J.; Sul, W.J.; et al. Mobile resistome of human gut and pathogen drives anthropogenic bloom of antibiotic resistance. Microbiome 2020, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Horvath, P.; Barrangou, R.J.S. CRISPR/Cas, the Immune System of Bacteria and Archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Harris, H.M.; McCann, A.; Guo, C.; Argimon, S.; Zhang, W.; Yang, X.; Jeffery, I.B.; Cooney, J.C.; Kagawa, T.F.; et al. Expanding the biotechnology potential of lactobacilli through comparative genomics of 213 strains and associated genera. Nat. Commun. 2015, 6, 8322. [Google Scholar] [CrossRef] [PubMed]
- Deveau, H.; Garneau, J.E.; Moineau, S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu. Rev. Microbiol. 2010, 64, 475–493. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR-Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef]
- Hidalgo-Cantabrana, C.; Sanozky-Dawes, R.; Barrangou, R. Insights into the Human Virome Using CRISPR Spacers from Microbiomes. Viruses 2018, 10, 479. [Google Scholar] [CrossRef]
- Yoon, B.H.; Chang, H.I. Complete genomic sequence of the Lactobacillus temperate phage LF1. Arch. Virol. 2011, 156, 1909–1912. [Google Scholar] [CrossRef]
- Miller-Ensminger, T.; Mormando, R.; Maskeri, L.; Shapiro, J.W.; Wolfe, A.J.; Putonti, C. Introducing Lu-1, a Novel Lactobacillus jensenii Phage Abundant in the Urogenital Tract. PLoS ONE 2020, 15, e0234159. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Yu, L.; Tian, F.; Zhao, J.; Zhang, H.; Chen, W.; Xue, Y.; Zhai, Q. Environment-Related Genes Analysis of Limosilactobacillus fermentum Isolated from Food and Human Gut: Genetic Diversity and Adaption Evolution. Foods 2022, 11, 3135. https://doi.org/10.3390/foods11193135
Zhao Y, Yu L, Tian F, Zhao J, Zhang H, Chen W, Xue Y, Zhai Q. Environment-Related Genes Analysis of Limosilactobacillus fermentum Isolated from Food and Human Gut: Genetic Diversity and Adaption Evolution. Foods. 2022; 11(19):3135. https://doi.org/10.3390/foods11193135
Chicago/Turabian StyleZhao, Yan, Leilei Yu, Fengwei Tian, Jianxin Zhao, Hao Zhang, Wei Chen, Yuzheng Xue, and Qixiao Zhai. 2022. "Environment-Related Genes Analysis of Limosilactobacillus fermentum Isolated from Food and Human Gut: Genetic Diversity and Adaption Evolution" Foods 11, no. 19: 3135. https://doi.org/10.3390/foods11193135
APA StyleZhao, Y., Yu, L., Tian, F., Zhao, J., Zhang, H., Chen, W., Xue, Y., & Zhai, Q. (2022). Environment-Related Genes Analysis of Limosilactobacillus fermentum Isolated from Food and Human Gut: Genetic Diversity and Adaption Evolution. Foods, 11(19), 3135. https://doi.org/10.3390/foods11193135