
Whole-Genome Analysis of Staphylococcus aureus Isolates from Ready-to-Eat Food in Russia
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Determination of Antibiotic Susceptibility
2.2. Sample Collection, DNA Isolation, Sequencing, and Genome Assembly
2.3. Data Processing
3. Results
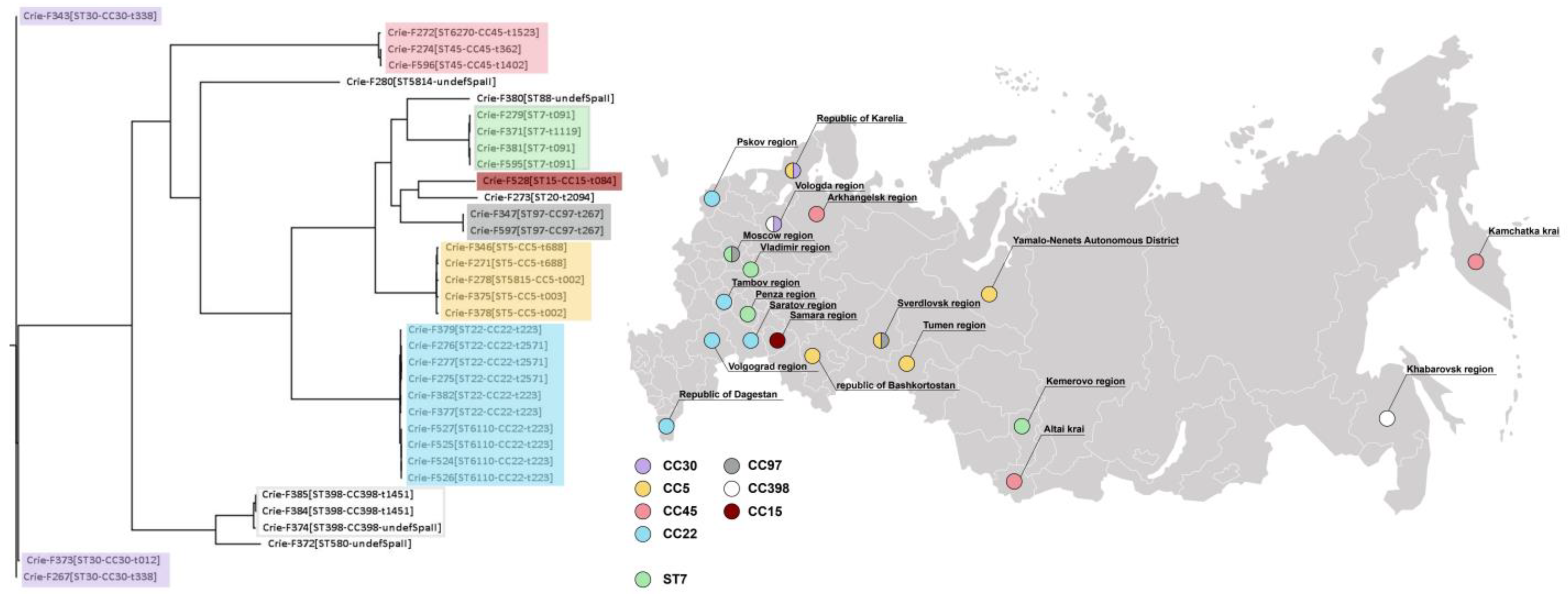
3.1. Isolate Typing
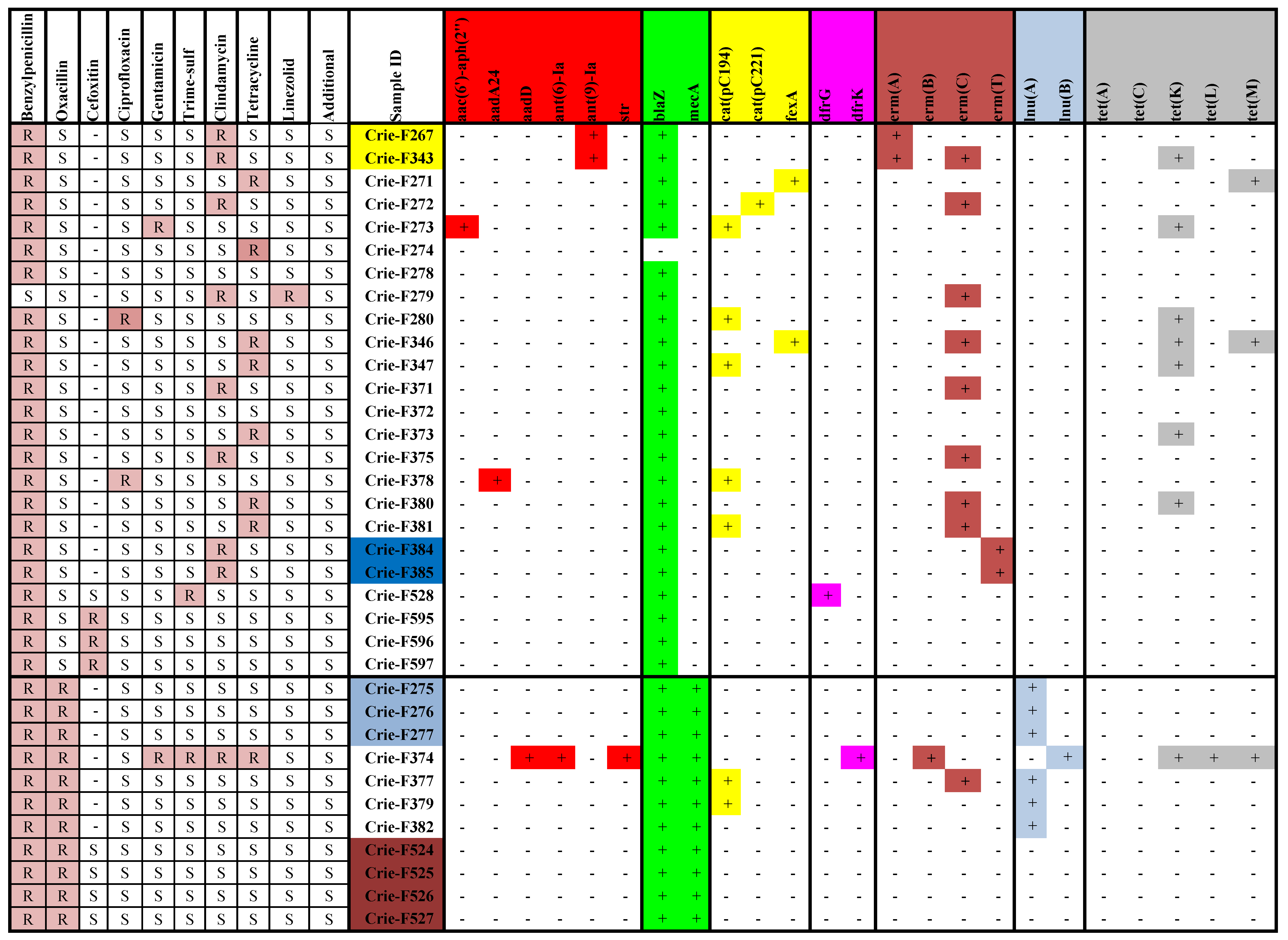
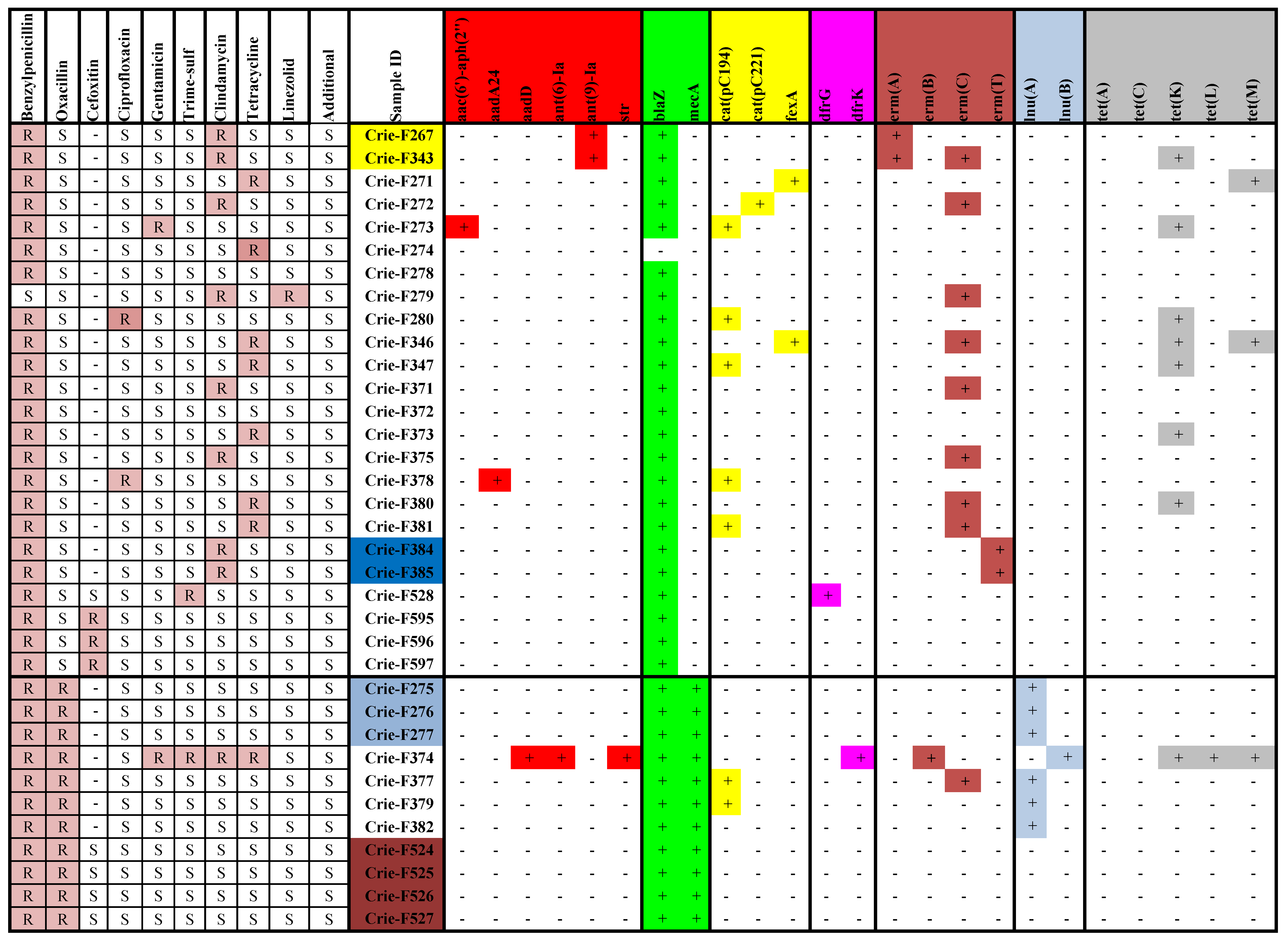
3.2. Antimicrobial Resistance Phenotypes and Genotypes
3.3. Virulence Gene Profiles
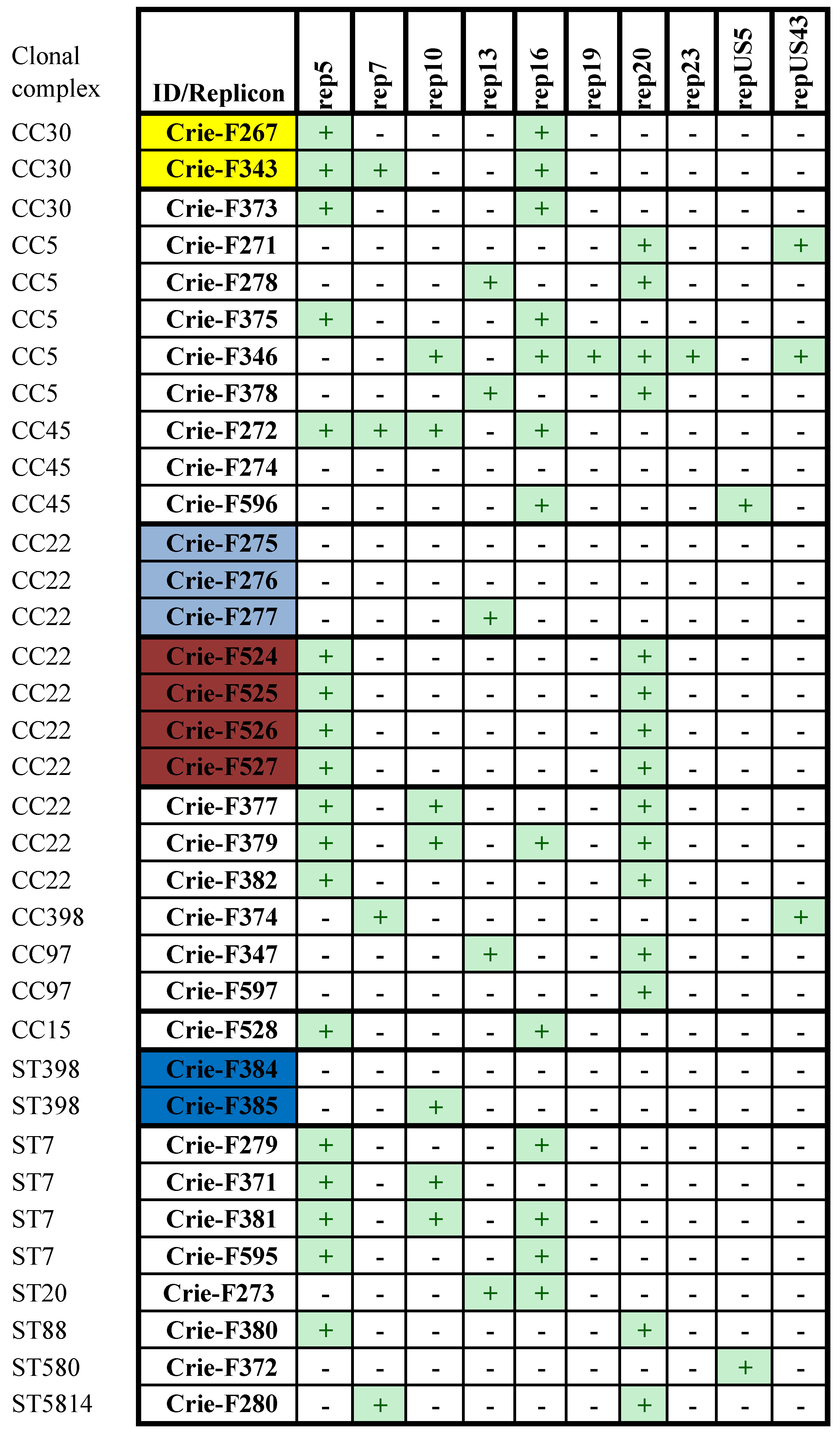
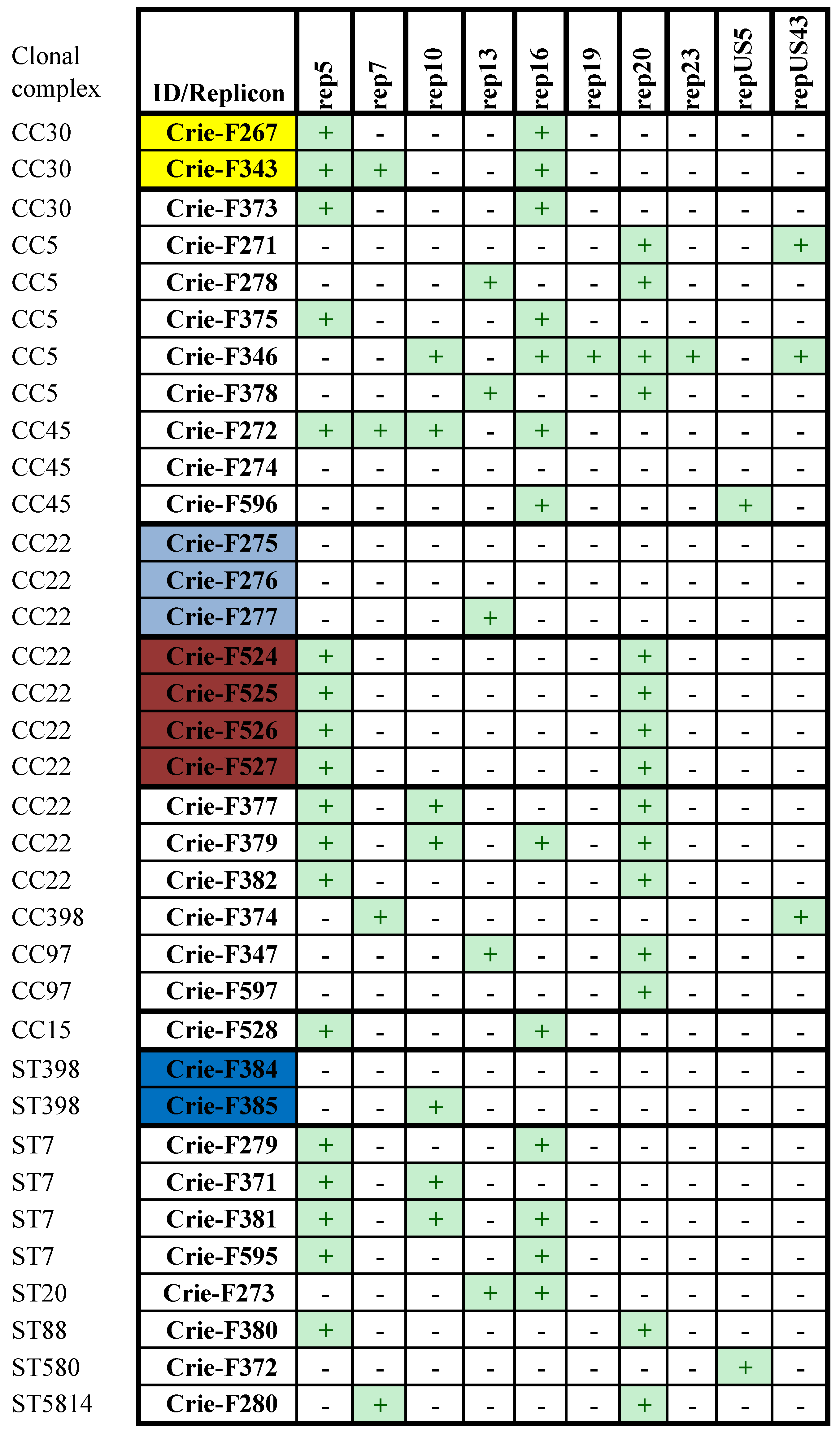
3.4. Plasmid Sequences
3.5. CRISPR/Cas Systems
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Food and Agriculture Organization of the United Nations. Available online: http://www.fao.org/food-systems/en/ (accessed on 25 April 2022).
- Sergelidis, D.; Angelidis, A. Methicillin-resistant Staphylococcus aureus: A controversial food-borne pathogen. Lett. Appl. Microbiol. 2017, 64, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Acheson, D. Iatrogenic High-Risk Populations and Foodborne Disease. Infect. Dis. Clin. N. Am. 2013, 27, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Feil, E.J.; Cooper, J.E.; Grundmann, H.; Robinson, D.A.; Enright, M.C.; Berendt, T.; Peacock, S.J.; Smith, J.M.; Murphy, M.; Spratt, B.G.; et al. How Clonal Is Staphylococcus aureus? J. Bacteriol. 2003, 185, 3307–3316. [Google Scholar] [CrossRef] [PubMed]
- Fetsch, A.; Johler, S. Staphylococcus aureus as a Foodborne Pathogen. Curr. Clin. Microbiol. Rep. 2018, 5, 88–96. [Google Scholar] [CrossRef]
- Murray, R.J. Recognition and management of Staphylococcus aureus toxin-mediated disease. Intern. Med. J. 2005, 35, S106–S119. [Google Scholar] [CrossRef]
- European Food Safety Authority and European Centre for Disease Prevention and Control. The European Union summary report on trends and sources of zoonoses, zoonotic agents and foodborne outbreaks in 2015. EFSA J. 2016, 11, e04634. [Google Scholar] [CrossRef]
- Chajecka-Wierzchowska, W.; Zadernowska, A.; Nalepa, B.; Sierpi´nska, M.; Łaniewska-Trokenheim, L. Retail Ready-to-Eat Food as a Potential Vehicle for Staphylococcus spp. Harboring Antibiotic Resistance Genes. J. Food Prot. 2014, 77, 993–998. [Google Scholar] [CrossRef]
- Yang, X.; Yu, S.; Wu, Q.; Zhang, J.; Wu, S.; Rong, D. Multilocus Sequence Typing and Virulence-Associated Gene Profile Analysis of Staphylococcus aureus Isolates from Retail Ready-to-Eat Food in China. Front. Microbiol. 2018, 9, 197. [Google Scholar] [CrossRef]
- Argudín, M.A.; Mendoza, M.C.; González-Hevia, M.A.; Bances, M.; Guerra, B.; Rodicio, M.R. Genotypes, Exotoxin Gene Content, and Antimicrobial Resistance of Staphylococcus aureus Strains Recovered from Foods and Food Handlers. Appl. Environ. Microbiol. 2012, 78, 2930–2935. [Google Scholar] [CrossRef]
- Luo, K.; Shao, F.; Kamara, K.N.; Chen, S.; Zhang, R.; Duan, G.; Yang, H. Molecular characteristics of antimicrobial resistance and virulence determinants of Staphylococcus aureus isolates derived from clinical infection and food. J. Clin. Lab. Anal. 2018, 32, e22456. [Google Scholar] [CrossRef] [Green Version]
- Lv, G.; Jiang, R.; Zhang, H.; Wang, L.; Li, L.; Gao, W.; Zhang, H.; Pei, Y.; Wei, X.; Dong, H.; et al. Molecular Characteristics of Staphylococcus aureus From Food Samples and Food Poisoning Outbreaks in Shijiazhuang, China. Front. Microbiol. 2021, 12, 1436. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Huang, J.; Zhang, F.; Wu, Q.; Zhang, J.; Pang, R.; Zeng, H.; Yang, X.; Chen, M.; Wang, J.; et al. Prevalence and Characterization of Food-Related Methicillin-Resistant Staphylococcus aureus (MRSA) in China. Front. Microbiol. 2019, 10, 304. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, A.; Niederhauser, I.; Johler, S. Virulence and Resistance Gene Profiles of Staphylococcus aureus Strains Isolated from Ready-to-Eat Foods. J. Food Prot. 2014, 77, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tang, T.; Stegger, M.; Dalsgaard, A.; Liu, T.; Leisner, J.J. Characterization of antimicrobial-resistant Staphylococcus aureus from retail foods in Beijing, China. Food Microbiol. 2021, 93, 103603. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Baker, M.; Hu, Y.; Xu, J.; Yang, D.; Maciel-Guerra, A.; Xue, N.; Li, H.; Yan, S.; Li, M.; et al. Whole-Genome Sequencing and Machine Learning Analysis of Staphylococcus aureus from Multiple Heterogeneous Sources in China Reveals Common Genetic Traits of Antimicrobial Resistance. mSystems 2021, 6, e0118520. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Shelenkov, A.; Mikhaylova, Y.; Yanushevich, Y.; Samoilov, A.; Petrova, L.; Fomina, V.; Gusarov, V.; Zamyatin, M.; Shagin, D.; Akimkin, V. Molecular Typing, Characterization of Antimicrobial Resistance, Virulence Profiling and Analysis of Whole-Genome Sequence of Clinical Klebsiella pneumoniae Isolates. Antibiotics 2020, 9, 261. [Google Scholar] [CrossRef]
- Shelenkov, A.; Petrova, L.; Zamyatin, M.; Mikhaylova, Y.; Akimkin, V. Diversity of International High-Risk Clones of Acinetobacter baumannii Revealed in a Russian Multidisciplinary Medical Center during 2017–2019. Antibiotics 2021, 10, 1009. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef]
- Goerke, C.; Esser, S.; Kummel, M.; Wolz, C. Staphylococcus aureus strain designation by agr and cap polymorphism typing and delineation of agr diversification by sequence analysis. Int. J. Med. Microbiol. 2005, 295, 67–75. [Google Scholar] [CrossRef]
- Schürch, A.; Arredondo-Alonso, S.; Willems, R.; Goering, R. Whole genome sequencing options for bacterial strain typing and epidemiologic analysis based on single nucleotide polymorphism versus gene-by-gene–based approaches. Clin. Microbiol. Infect. 2018, 24, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Sieber, R.N.; Skov, R.L.; Nielsen, J.; Schulz, J.; Price, L.B.; Aarestrup, F.M.; Larsen, A.R.; Stegger, M.; Larsen, J. Drivers and Dynamics of Methicillin-Resistant Livestock-Associated Staphylococcus aureus CC398 in Pigs and Humans in Denmark. mBio 2018, 9, e02142-18. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.R.; Fouts, D.E.; Archer, G.L.; Mongodin, E.F.; DeBoy, R.T.; Ravel, J.; Paulsen, I.T.; Kolonay, J.F.; Brinkac, L.; Beanan, M.; et al. Insights on Evolution of Virulence and Resistance from the Complete Genome Analysis of an Early Methicillin-Resistant Staphylococcus aureus Strain and a Biofilm-Producing Methicillin-Resist. Staphylococcus Epidermidis Strain. J. Bacteriol. 2005, 187, 2426–2438. [Google Scholar] [CrossRef] [PubMed]
- Harper, L.; Balasubramanian, D.; Ohneck, E.A.; Sause, W.E.; Chapman, J.; Mejia-Sosa, B.; Lhakhang, T.; Heguy, A.; Tsirigos, A.; Ueberheide, B.; et al. Staphylococcus aureus Responds to the Central Metabolite Pyruvate to Regulate Virulence. mBio 2018, 9, e02272-17. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Wang, B.; Tao, X.; Hu, Q.; Cui, Z.; Zhang, J.; Lin, Y.; You, Y.; Shi, X.; Grundmann, H. Characterization of Staphylococcus aureus Strains Associated with Food Poisoning in Shenzhen, China. Appl. Environ. Microbiol. 2012, 78, 6637–6642. [Google Scholar] [CrossRef]
- Ferry, T.; Thomas, D.; Genestier, A.-L.; Bes, M.; Lina, G.; Vandenesch, F.; Etienne, J. Comparative Prevalence of Superantigen Genes in Staphylococcus aureus Isolates Causing Sepsis with and Without Septic Shock. Clin. Infect. Dis. 2005, 41, 771–777. [Google Scholar] [CrossRef]
- Xu, S.X.; Gilmore, K.J.; Szabo, P.A.; Zeppa, J.J.; Baroja, M.L.; Haeryfar, S.M.; McCormick, J.K. Superantigens Subvert the Neutrophil Response to Promote Abscess Formation and Enhance Staphylococcus aureus Survival In Vivo. Infect. Immun. 2014, 82, 3588–3598. [Google Scholar] [CrossRef]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin. Microbiol. Rev. 2018, 31, e00088-17. [Google Scholar] [CrossRef]
- Projan, S.J.; Novick, R. Comparative analysis of five related staphylococcal plasmids. Plasmid 1988, 19, 203–221. [Google Scholar] [CrossRef]
- Jensen, L.; Garcia-Migura, L.; Valenzuela, A.; Løhr, M.; Hasman, H.; Aarestrup, F. A classification system for plasmids from enterococci and other Gram-positive bacteria. J. Microbiol. Methods 2010, 80, 25–43. [Google Scholar] [CrossRef]
- Zhao, X.; Yu, Z.; Xu, Z. Study the Features of 57 Confirmed CRISPR Loci in 38 Strains of Staphylococcus aureus. Front. Microbiol. 2018, 9, 1591. [Google Scholar] [CrossRef] [PubMed]
- Deurenberg, R.H.; Stobberingh, E.E. The evolution of Staphylococcus aureus. Infect. Genet. Evol. 2008, 8, 747–763. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, H.; Aanensen, D.M.; Van Den Wijngaard, C.C.; Spratt, B.G.; Harmsen, D.; Friedrich, A.W.; the European Staphylococcal Reference Laboratory Working Group. Geographic Distribution of Staphylococcus aureus Causing Invasive Infections in Europe: A Molecular-Epidemiological Analysis. PLoS Med. 2010, 7, e1000215. [Google Scholar] [CrossRef] [PubMed]
- Monaco, M.; de Araujo, F.P.; Cruciani, M.; Coccia, E.M.; Pantosti, A. Worldwide Epidemiology and Antibiotic Resistance of Staphylococcus aureus. Poxviruses 2017, 409, 21–56. [Google Scholar] [CrossRef]
- Wyllie, D.; Paul, J.; Crook, D. Waves of trouble: MRSA strain dynamics and assessment of the impact of infection control. J. Antimicrob. Chemother. 2011, 66, 2685–2688. [Google Scholar] [CrossRef]
- Kraushaar, B.; Ballhausen, B.; Leeser, D.; Tenhagen, B.-A.; Käsbohrer, A.; Fetsch, A. Antimicrobial resistances and virulence markers in Methicillin-resistant Staphylococcus aureus from broiler and turkey: A molecular view from farm to fork. Veter Microbiol. 2017, 200, 25–32. [Google Scholar] [CrossRef]
- Back, S.H.; Eom, H.S.; Lee, H.H.; Lee, G.Y.; Park, K.T.; Yang, S.-J. Livestock-associated methicillin-resistant Staphylococcus aureus in Korea: Antimicrobial resistance and molecular characteristics of LA-MRSA strains isolated from pigs, pig farmers, and farm environment. J. Veter Sci. 2020, 21, e2. [Google Scholar] [CrossRef]
- Fitzgerald, J.R. Livestock-associated Staphylococcus aureus: Origin, evolution and public health threat. Trends Microbiol. 2012, 20, 192–198. [Google Scholar] [CrossRef]
- Fetsch, A.; Etter, D.; Johler, S. Livestock-Associated Meticillin-Resistant Staphylococcus aureus—Current Situation and Impact from a One Health Perspective. Curr. Clin. Microbiol. Rep. 2021, 8, 103–113. [Google Scholar] [CrossRef]
- Goerge, T.; Lorenz, M.B.; van Alen, S.; Hübner, N.-O.; Becker, K.; Köck, R. MRSA colonization and infection among persons with occupational livestock exposure in Europe: Prevalence, preventive options and evidence. Veter Microbiol. 2017, 200, 6–12. [Google Scholar] [CrossRef]
- Mellmann, A.; Weniger, T.; Berssenbrügge, C.; Rothgänger, J.; Sammeth, M.; Stoye, J.; Harmsen, D. Based Upon Repeat Pattern (BURP): An algorithm to characterize the long-term evolution of Staphylococcus aureus populations based on spa polymorphisms. BMC Microbiol. 2007, 7, 98. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Stegger, M.; Dalsgaard, A.; Leisner, J.J. Bacterial content and characterization of antibiotic resistant Staphylococcus aureus in Danish sushi products and association with food inspector rankings. Int. J. Food Microbiol. 2019, 305, 108244. [Google Scholar] [CrossRef] [PubMed]
- Rinsky, J.L.; Nadimpalli, M.; Wing, S.; Hall, D.; Baron, D.; Price, L.B.; Larsen, J.; Stegger, M.; Stewart, J.; Heaney, C.D. Livestock-Associated Methicillin and Multidrug Resistant Staphylococcus aureus is Present among Industrial, Not Antibiotic-Free Livestock Operation Workers in North Carolina. PLoS ONE 2013, 8, e67641. [Google Scholar] [CrossRef] [PubMed]
- Wattinger, L.; Stephan, R.; Layer, F.; Johler, S. Comparison of Staphylococcus aureus isolates associated with food intoxication with isolates from human nasal carriers and human infections. Eur. J. Clin. Microbiol. 2012, 31, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Monecke, S.; Luedicke, C.; Slickers, P.; Ehricht, R. Molecular epidemiology of Staphylococcus aureus in asymptomatic carriers. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 1159–1165. [Google Scholar] [CrossRef] [PubMed]
- Shehreen, S.; Chyou, T.-Y.; Fineran, P.C.; Brown, C.M. Genome-wide correlation analysis suggests different roles of CRISPR-Cas systems in the acquisition of antibiotic resistance genes in diverse species. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180384. [Google Scholar] [CrossRef]
- Palmer, K.L.; Gilmore, M.S. Multidrug-resistant enterococci lack CRISPR-cas. mBio 2010, 1, e00227-10. [Google Scholar] [CrossRef]
- Wheatley, R.M.; MacLean, R.C. CRISPR-Cas systems restrict horizontal gene transfer in Pseudomonas aeruginosa. ISME J. 2020, 15, 1420–1433. [Google Scholar] [CrossRef]
- Tyumentseva, M.; Mikhaylova, Y.; Prelovskaya, A.; Tyumentsev, A.; Petrova, L.; Fomina, V.; Zamyatin, M.; Shelenkov, A.; Akimkin, V. Genomic and Phenotypic Analysis of Multidrug-Resistant Acinetobacter baumannii Clinical Isolates Carrying Different Types of CRISPR/Cas Systems. Pathogens 2021, 10, 205. [Google Scholar] [CrossRef]
- Marraffini, L.A.; Sontheimer, E.J. CRISPR Interference Limits Horizontal Gene Transfer in Staphylococci by Targeting DNA. Science 2008, 322, 1843–1845. [Google Scholar] [CrossRef] [Green Version]
- Oun, S.; Redder, P.; Didier, J.-P.; François, P.; Corvaglia, A.-R.; Buttazzoni, E.; Giraud, C.; Girard, M.; Schrenzel, J.; Linder, P. The CshA DEAD-box RNA helicase is important for quorum sensing control in Staphylococcus aureus. RNA Biol. 2013, 10, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Cruz-López, E.A.; Rivera, G.; Cruz-Hernández, M.A.; Martínez-Vázquez, A.V.; Castro-Escarpulli, G.; Flores-Magallón, R.; Vázquez-Cisneros, K.; Cruz-Pulido, W.L.; Bocanegra-García, V. Identification and Characterization of the CRISPR/Cas System in Staphylococcus aureus Strains from Diverse Sources. Front. Microbiol. 2021, 12, 656996. [Google Scholar] [CrossRef] [PubMed]
- Lozano, C.; García-Migura, L.; Aspiroz, C.; Zarazaga, M.; Torres, C.; Aarestrup, F.M. Expansion of a Plasmid Classification System for Gram-Positive Bacteria and Determination of the Diversity of Plasmids in Staphylococcus aureus Strains of Human, Animal, and Food Origins. Appl. Environ. Microbiol. 2012, 78, 5948–5955. [Google Scholar] [CrossRef] [PubMed]
- Malachowa, N.; DeLeo, F.R. Mobile genetic elements of Staphylococcus aureus. Cell Mol. Life Sci. 2010, 67, 3057–3071. [Google Scholar] [CrossRef]
- Jensen, S.O.; Lyon, B.R. Genetics of antimicrobial resistance in Staphylococcus aureus. Future Microbiol. 2009, 4, 565–582. [Google Scholar] [CrossRef]
- Wall, B.A.; Mateus, A.; Marshall, L.; Pfeiffer, D.U. Drivers, Dynamics and Epidemiology of Antimicrobial Resistance in Animal Production; Food and Agriculture Organization of the United Nations: Rome, Italy, 2016. [Google Scholar]
- Shaidullina, E.; Shelenkov, A.; Yanushevich, Y.; Mikhaylova, Y.; Shagin, D.; Alexandrova, I.; Ershova, O.; Akimkin, V.; Kozlov, R.; Edelstein, M. Antimicrobial Resistance and Genomic Characterization of OXA-48- and CTX-M-15-co-Producing Hypervirulent Klebsiella pneumoniae ST23 Recovered from Nosocomial Outbreak. Antibiotics 2020, 9, 862. [Google Scholar] [CrossRef]
- Zhou, W.; Jin, Y.; Liu, X.; Chen, Y.; Shen, P.; Xiao, Y. Comparison of Genetic Features and Evolution of Global and Chinese Strains of Community-Associated Methicillin-Resistant Staphylococcus aureus ST22. Microbiol. Spectr. 2022, 10, e0203721. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikhaylova, Y.; Shelenkov, A.; Chernyshkov, A.; Tyumentseva, M.; Saenko, S.; Egorova, A.; Manzeniuk, I.; Akimkin, V. Whole-Genome Analysis of Staphylococcus aureus Isolates from Ready-to-Eat Food in Russia. Foods 2022, 11, 2574. https://doi.org/10.3390/foods11172574
Mikhaylova Y, Shelenkov A, Chernyshkov A, Tyumentseva M, Saenko S, Egorova A, Manzeniuk I, Akimkin V. Whole-Genome Analysis of Staphylococcus aureus Isolates from Ready-to-Eat Food in Russia. Foods. 2022; 11(17):2574. https://doi.org/10.3390/foods11172574
Chicago/Turabian StyleMikhaylova, Yulia, Andrey Shelenkov, Aleksey Chernyshkov, Marina Tyumentseva, Stepan Saenko, Anna Egorova, Igor Manzeniuk, and Vasiliy Akimkin. 2022. "Whole-Genome Analysis of Staphylococcus aureus Isolates from Ready-to-Eat Food in Russia" Foods 11, no. 17: 2574. https://doi.org/10.3390/foods11172574
APA StyleMikhaylova, Y., Shelenkov, A., Chernyshkov, A., Tyumentseva, M., Saenko, S., Egorova, A., Manzeniuk, I., & Akimkin, V. (2022). Whole-Genome Analysis of Staphylococcus aureus Isolates from Ready-to-Eat Food in Russia. Foods, 11(17), 2574. https://doi.org/10.3390/foods11172574