Abstract
Staphylococcus aureus (S. aureus) has a strong tolerance to high salt stress. It is a major reason as to why the contamination of S. aureus in salted food cannot be eradicated. To elucidate its response and survival mechanisms, changes in the morphology, biofilm formation, virulence, transcriptome, and metabolome of S. aureus were investigated. IsaA positively regulates and participates in the formation of biofilm. Virulence was downregulated to reduce the depletion of nonessential cellular functions. Inositol phosphate metabolism was downregulated to reduce the conversion of functional molecules. The MtsABC transport system was downregulated to reduce ion transport and signaling. Aminoacyl-tRNA biosynthesis was upregulated to improve cellular homeostasis. The betaine biosynthesis pathway was upregulated to protect the active structure of proteins and nucleic acids. Within a 10% NaCl concentration, the L-proline content was upregulated to increase osmotic stability. In addition, 20 hub genes were identified through an interaction analysis. The findings provide theoretical support for the prevention and control of salt-tolerant bacteria in salted foods.
1. Introduction
In traditional food processing systems, salting methods are often used to prevent food spoilage [1]. Previous studies demonstrated that an increased salt concentration in food can effectively reduce the water activity of food-borne pathogens, thereby inhibiting their growth [2]. Therefore, the water activity of salted foods is usually regulated by the addition of salt. This method can reduce the metabolic activity of microorganisms, so as to better control the spoilage rate of food [3]. Staphylococcus aureus (S. aureus) is considered to be the most important pathogen in salted food processing, and commonly causes food poisoning [4]. However, the health standards of various countries limit the detection of S. aureus in food, meaning that there are still many batches of food that exceed the prescribed limit [5]. S. aureus has a strong tolerance to high salt stress. It is major reason as to why the contamination of S. aureus in salted food cannot be eradicated [1]. Therefore, both the prevention and control of S. aureus present important research topics in food safety.
S. aureus is widely distributed and demonstrates strong adaptability to the environment [6]. It can survive in soil, air, food, and skin and has resistance to drying, heat, salt, alkali, and low temperatures [7]. In addition, when S. aureus is permitted to grow in foods, it produces a large number of toxic extracellular proteins, enterotoxins, and hemolysins, which are destructive to the intestinal tract and can seriously endanger human safety and health [8,9]. The ability of S. aureus to quickly adapt to environmental changes is also one of the reasons for its high pathogenicity [10].
S. aureus can adapt and survive in a salt environment. It was previously demonstrated that biofilm is an extracellular polymeric matrix secreted by certain microorganisms, such as S. aureus, and its production is well correlated with NaCl concentrations [11]. Recent research revealed that variations in NaCl concentrations can induce the increased production of biofilm in S. aureus, accompanied by the increased expression of genes related to biofilm, such as the icaA gene [12]. Furthermore, biofilm formation can provide S. aureus with a protective environment that prevents direct contact between salt and the bacterial cell membrane, allowing bacteria to maintain a growth state in adverse environments [13]. Additionally, numerous studies indicate that many small molecules referred to as compatible solutes, such as glutamine, proline and glycine betaine, can intracellularly accumulate in S. aureus due to increased biosynthesis or uptake, and decreased degradation modes, resulting in an improvement in osmotolerance [14,15].
S. aureus demonstrates a strong tolerance to high salt. Ming et al. conducted a preliminary study on the growth of S. aureus when subjected to high salinity stress based on proteomics [16]. However, the regulation mechanism of gene expression and the regulation of metabolites remain unclear. The adaptive changes of S. aureus after high salinity stress have not been systematically and comprehensively studied.
Generally, the salt concentration in salted food needs to be higher than 15% to inhibit bacterial growth. At present, the research on the mechanism of S. aureus salt tolerance is mainly limited to the low salt concentration range [12,15,17]. Therefore, in this study, we simulated salt stress with culture conditions of 0% NaCl, 10% NaCl, and 20% NaCl. Transcriptomic and metabolomic analyses were integrated to understand the responses and survival mechanisms of S. aureus under high salinity stress. This study presents genes, metabolites, and pathways that can be used as potential targets for preventing S. aureus contamination. The purpose of the study is to provide theoretical support for the prevention and control of salt-tolerant bacteria in salted food.
2. Materials and Methods
2.1. Strain and Culture Conditions
Commercial S. aureus ATCC 27217 was purchased from the Chinese Center of Industrial Culture Collection. The strain was incubated in a nutrient broth (NB; Guoyao, China) medium at 37 °C with shaking (140 rpm). For the experiment, equal volumes of the cells were inoculated in a 100 mL NB medium supplemented with 0% NaCl (control group), 10% NaCl, and 20% NaCl for 48 h. Three replicates were performed for each concentration culture. Bacterial liquids under different salt concentration stresses were used for subsequent experiments. Cell growth was monitored every 4 h. The optical density (OD) at 600 nm was used to monitor the growth of S. aureus in different conditions.
2.2. Scanning Electron Microscopy (SEM)
The harvested cells (12,000 rpm, 2 min) were fixed with 2.5% glutaraldehyde at 4 °C for 2 h and washed three times with PBS buffer. Then, different concentrations of ethanol (30%, 50%, 70%, 90%, 100%) were used for the gradient dehydration of cells, and acetone was dehydrated twice. Finally, the suspension of acetone and cells was dropped on a coverslip to dry and fix. Gold coating was performed with a Zhongke model BS2 surface treatment machine. Images were captured with a Hitachi model S3000N scanning electron microscope. A total of 9 images of the 3 samples were obtained with SEM.
2.3. Biofilm Analysis
The volume of bacterial liquid under different salt concentration stresses was adjusted to obtain a final cell count of 5 × 106 CFU/mL in the NB medium. Then, the 3MTM PetrifilmTM Rapid S. aureus Count Plate method was used for cell counting. The bacterial solution (1 mL) was collected by centrifugation at 12,000 rpm for 2 min. First, the cells were washed three times with PBS buffer and fixed in 2.5% glutaraldehyde at 4 °C for 1.5 h. Then, cells were washed again and S. aureus was stained with FITC-labeled Concanavalin A (FITC-ConA; Sigma, St. Louis, MO, USA) for 30 min and Propidium Iodide (PI; Sigma, St. Louis, MO, USA) for 15 min at 4 °C. Finally, coverslips were mounted with 20 μL of the Antifade Mounting Medium (P0126, Beyotime, Shanghai, China). Biofilm formation was observed under confocal laser scanning microscopy (CLSM) immediately after tableting.
FITC-ConA and PI were used for fluorescence staining in the dark. Extracellular polymeric substances (EPSs) bind with FITC-ConA to emit green fluorescence (λex = 488 nm and λem = 520 nm), and damaged or dead cell DNA bind with PI to emit red light (λex = 543 nm and λem = 572 nm). Images were captured with the CLSM Zeiss model LSM880 microscope.
The Easy3D function of the IMARIS 9.0 software package was used for the 3D reconstruction of the biofilm structure. The thicknesses of the biofilms were determined using the confocal stack images. Z stack represents the thickness of biofilm. Biofilm thickness measurements were performed with Zen black 2.1 software. Three replicates were performed for each sample. The Biofilm thickness was statistically analyzed using ANOVA.
2.4. Hemolysis Assay
S. aureus was inoculated into an NB medium with different conditions (0% NaCl, 10% NaCl, and 20% NaCl) and cultured with shaking at 140 rpm for 48 h at 37 °C. There were three replicates for each treatment. The NB liquid medium containing bacteria (1 × 108 cells) was centrifuged at 12,000 rpm for 5 min at 4 °C. The supernatant was aseptically filtered (0.22-μm pores), and 500 μL of the filtered sample was poured into a centrifuge tube. Then, 25 μL of sterile defibrinated sheep blood was added, shaken gently to mix, and incubated at 37 °C for 2 h. After centrifugation at 3000 rpm for 1 min, 200 μL of the supernatant was utilized, and the absorption at 450 nm (OD450) was measured. A statistical analysis of the hemolysis data was performed using ANOVA.
2.5. Coagulase Assay
To prepare the coagulase assay, 0.5 mL of sterile normal saline was withdrawn using a pipette and added to a vial of freeze-dried plasma (rabbit plasma) and shaken until fully dissolved. Then, 0.3 mL of the stress-treated bacterial solution (0% NaCl, 10% NaCl, and 20% NaCl) was added to the dissolved rabbit plasma. After mixing evenly, it was placed in an incubator at 37 °C for static culture. It was observed once every 0.5 h for a total of 6 h. Three replicates were performed for each treatment. If coagulation occurred (a clot or coagulation with a volume exceeding one-half the total volume is positive), the plasma coagulase of the tested bacterial solution was considered to be positive. Otherwise, it was considered to be negative.
2.6. Transcriptome Analysis
Two samples from each treatment were selected for the transcriptome analysis. Total RNA extraction, library construction, and transcriptome sequencing were performed. Total RNA was extracted using the TRIzol reagent (Takara Bio, Otsu, Japan) according to the manufacturer’s instruction and its purity and quantity were tested using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Subsequently, rRNA was removed with the Ribo-Zero™ Magnetic Kit b’j (Illumina). mRNA was fragmented into small pieces. Random hexamer primers were used for the cDNA synthesis. Finally, sequencing was carried out using the Illumina Hiseq 4000 platform (Majorbio Bio-Pharm Technology Co., Ltd., Shanghai, China) [18].
The fragments per kilobase of transcript per million fragments (FPKM) were used to calculate gene expression levels [19]. FDR ≤ 0.05 and |log2FC| ≥ 1 were set as the thresholds for determining significantly differentially expressed genes (DEGs). Furthermore, Gene Ontology (GO) enrichment and the Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of the DEGs were analyzed using the R package. An adjusted p value ≤ 0.05 was used as a threshold to determine significant enrichment. The transcriptomic data were deposited in the NCBI Sequence Read Archive under the following accession code: PRJNA764353.
2.7. Quantitative Real-Time PCR Experimental Validation
Quantitative real-time PCR (qRT–PCR) was adopted to detect the expression of 10 selected DEGs to validate the Illumina sequencing results. The Primer 5 program was used to design the gene-specific primers. The primer sequences are listed in Table S1. The 20 µL reaction system contained 10 μL 2 × SYBR® Premix Ex TaqTM II, 0.4 μL of each of the forward and reverse primers, 1 μL of the cDNA template, and 7.4 μL of nuclease-free water. The cycling conditions were performed as follows: pre-denaturation at 95 °C for 10 s; denaturation at 95 °C for 10 s; annealing at 55 °C for 10 s; extension at 72 °C for 20 s, in 40 cycles; and three replicates were performed for each DEG. The relative expression of DEGs was calculated according to the 2−∆∆Ct method with 16S rDNA used as the housekeeping gene. A statistical analysis of the relative expression was performed using ANOVA.
2.8. Metabolome Analysis
Sample preparation and extraction were performed as follows: bacterial cells were fully ground in liquid nitrogen and 60% methanol was added to extract metabolites. Then, the extracts were nitrogen-dried and freeze-dried. After drying, the samples were successively mixed with methoxyamine pyridine hydrochloride (Aladdin, China), bis(trimethylsilyl)trifluoroacetamide (BSTFA, Aladdin, China), and Chlorotrimethylsilane (TMCS, Aladdin, China). Finally, heptane was added to complete derivatization. Three samples were selected from each treatment condition for GC–MS [20].
GC–MS and SIMCA-P + ver14 software were utilized to identify and analyze the metabolites [21]. An orthogonal partial least squares discriminant analysis (OPLS-DA) was employed to maximize the metabolome differences between the two samples. Metabolites with a variable importance in the projection (VIP) value > 1 and p value ≤ 0.05 were regarded as differential metabolites (DMs). MetaboAnalyst 5.0 was used to perform the KEGG analysis.
2.9. Correlation Analysis of Transcriptome and Metabolome
Cytoscape software was utilized to visualize the relationships between the metabolome and transcriptome based on Spearman’s correlation coefficients. A p value ≤ 0.05 was set as the threshold, and the network relationship diagram was drawn. Then, the hub genes were screened. In addition, the hub genes were analyzed using GO and KEGG functional annotation.
3. Results
3.1. High Salinity Stress Affects Cell Growth and Morphology
As the salinity increased, the growth activity of cells decreased gradually (Figure 1A). The inhibitory effect of high salt on cell growth was greater than that of medium salt. In this study, the bacterial cells survived in the 20% NaCl group. This demonstrated the high salt tolerance of S. aureus ATCC 27217.
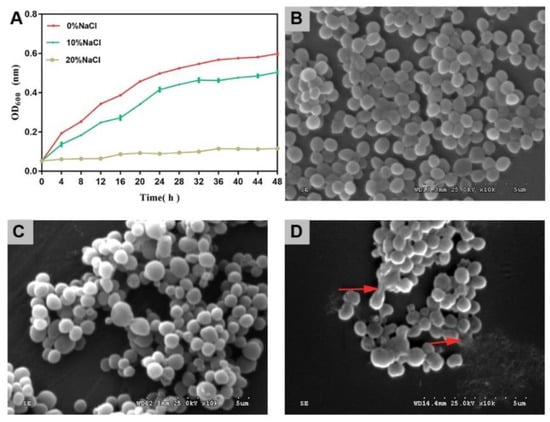
Figure 1.
Cell growth and SEM images of S. aureus during salinity changes. (A) Growth curves. The error bars indicate the standard deviation (N = 3). (B) SEM image of S. aureus in control group. (C) SEM image of S. aureus in 10% NaCl group. (D) SEM image of S. aureus in 20% NaCl group. ×10K-fold.
The control group cells presented with a spherical shape and smooth surface, which is a typical feature of S. aureus cells (Figure 1B). After treatment with 10% NaCl, most cells showed little change in morphology (Figure 1C). After treatment with 20% NaCl, the cytomembrane of a few of the cells ruptured, and the contents of the cells were released (Figure 1D).
With an increased salinity, the growth activity of S. aureus was further inhibited, and the cell morphology was destroyed.
3.2. High Salinity Stress Affects Biofilm Formation
CLSM images of bacterial cells labeled with FITC-ConA and PI are shown in Figure 2A–C (representative schematic diagrams). The distribution of the cells and the biofilm were assessed. Green fluorescence and red fluorescence were indicative of biofilm and cell DNA, respectively. After treatment with increasing salinity, the biofilm thickness increased and then decreased. Furthermore, significant differences were identified between the adjacent groups (Figure 2D). These results suggest that an increased salt concentration promoted biofilm formation in the low salt range (<10% NaCl), while an increased salt concentration inhibited biofilm formation in the high salt range (>10% NaCl).
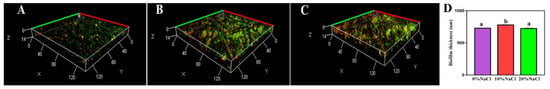
Figure 2.
CLSM images of treated S. aureus biofilm cells. (A) Control group; (B) 10% NaCl group; (C) 20% NaCl group; (D) thickness of biofilm.
3.3. High Salinity Stress Affects Virulence
To determine whether NaCl affects the virulence of S. aureus ATCC 27217, we measured hemolytic activity and coagulase expression. After the salt stress intensity increased, the hemolytic activity showed a continuous downward trend, and the differences between different treatment groups were significant (Figure 3). Our results showed that high salinity stress had a significant inhibitory effect on the hemolytic activity of S. aureus.
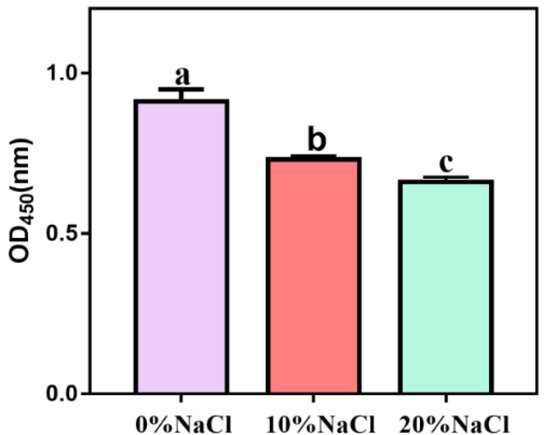
Figure 3.
Hemolytic activity of S. aureus during change in salinity.
The coagulase results showed that the control group underwent coagulation first, followed by the 10% NaCl group; however, the 20% NaCl group did not form a coagulation state until the sixth hour (Table S2). Different NaCl concentrations inhibited the expression of S. aureus coagulase in a dose-dependent manner, thereby reducing the invasiveness of S. aureus.
3.4. Transcriptome Responses to Salinity Stress
A total of 1007, 1150 and 172 DEGs were identified in the 0% NaCl vs. 10% NaCl comparison, 0% NaCl vs. 20% NaCl comparison, and 10% NaCl vs. 20% NaCl comparison, respectively, (Figure 4). Compared with the 0% NaCl group, both the 10% NaCl group and the 20% NaCl group mobilized a large number of DEGs to respond to salinity stress. Moreover, the number of DEGs in the 0% NaCl vs. 20% NaCl comparison was higher than that in the 0% NaCl vs. 10% NaCl comparison. It demonstrated that S. aureus requires the mobilization of more DEGs to adapt and survive under higher salt stress.
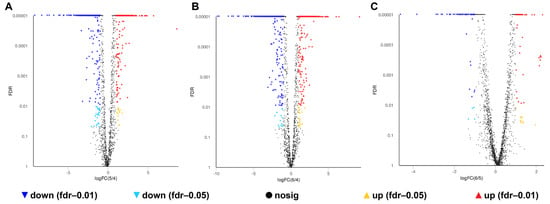
Figure 4.
Volcano plot of genes. The red and green data points indicate upregulated and downregulated DEGs, respectively. (A): 0% NaCl vs. 10% NaCl; (B): 0% NaCl vs. 20% NaCl; (C): 10% NaCl vs. 20% NaCl.
The following four change patterns were observed after the salt concentration gradually increased (Figure 5): increase/increase, increase/decrease, decrease/decrease, and decrease/increase. The results showed that 6, 4, 72, and 32 DEGs were identified in each pattern. Detailed annotation information is listed in Table S3. Therefore, we believe these DEGs are candidate target genes in the salt stress response.
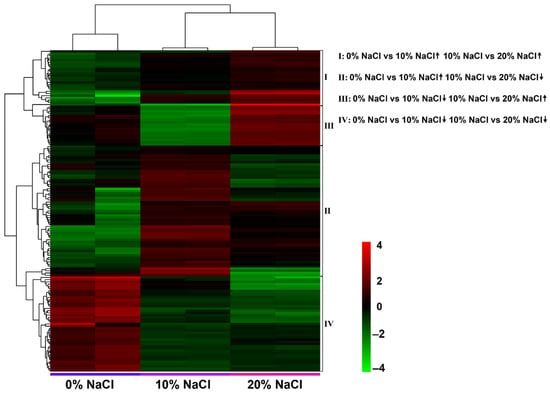
Figure 5.
DEGs related to salt stress identified with pairwise comparison of four expression patterns. Heatmap of the four expression patterns of DEGs after a gradual increase in salt concentration.
In total, nine predicted KEGG pathways were enriched significantly (Table S4). They include S. aureus infection, aminoacyl-tRNA biosynthesis, ribosome, fatty acid degradation, monobactam biosynthesis, butanoate metabolism, benzoate degradation, tyrosine metabolism, purine metabolism, glycine, serine, and threonine metabolism. These pathways were mainly involved in amino acid metabolism, translation, infectious diseases, purine metabolism, carbohydrate metabolism, and lipid metabolism.
3.5. qRT–PCR Validation
The mRNA transcription levels of ten DEGs were quantified to further verify the accuracy of RNA sequencing. Figure 6 shows that the expression levels of the selected DEGs were highly consistent with those suggested by the RNA-Seq results. Therefore, the qRT–PCR results confirmed the reliability and accuracy of the transcriptome data.
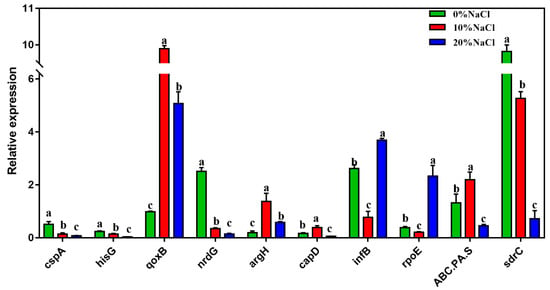
Figure 6.
The relative expression of the validation genes. The Y-axis represents the expression of selected genes. Different letters correspond to statistically significant differences (p < 0.05) between groups.
3.6. Metabolomic Responses to High Salinity Stress
In this analysis, a total of 75 metabolites were identified under high salinity stress (Table S5). The differences between samples were assessed by performing a principal component analysis (PCA). As seen in Figure 7A, endogenous metabolites of the three treatment groups were located in three different quadrants, implying that there were obvious differences among the samples. In total, 37 DMs were screened after OPLS-DA (Table S6). The DMs were subjected to a KEGG pathway analysis to identify important contributions in response to high salt stress. The metabolic pathways in the different groups are displayed in Figure 7B–D. These pathways mainly involve inositol phosphate metabolism; glycine, serine, and threonine metabolism; glyoxylate and dicarboxylate metabolism; alanine, aspartate, and glutamate metabolism; glutathione metabolism; sulfur metabolism; the TCA cycle; glycolysis/gluconeogenesis; and pyruvate metabolism (Table S7). Therefore, the metabolic results suggest that signal response, amino acid regulation, and carbohydrate metabolism regulation are the main pathways by which S. aureus ATCC 27217 adapts to high salinity stress.
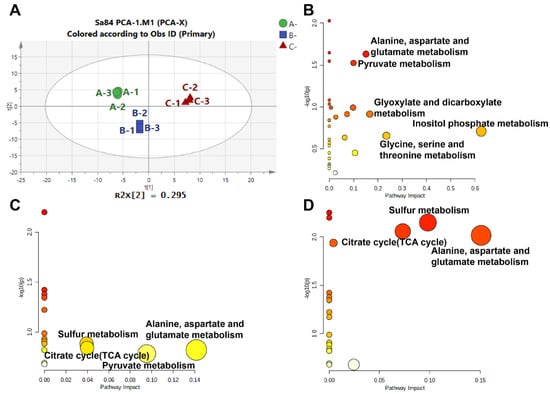
Figure 7.
Overview of the metabolome analysis results. (A) PCA score plot of the overall metabolites expression; (B) KEGG pathways in 0% NaCl vs. 10% NaCl comparison; (C) KEGG pathways in 10% NaCl vs. 20% NaCl comparison; (D) KEGG pathways in 0% NaCl vs. 20% NaCl comparison.
3.7. Integrated Analysis of Transcriptome and Metabolomic Data
As mentioned above, 53 key candidate genes and 37 DMs were identified. Then, transcriptome and metabolomic data were combined to obtain a Spearman correlation network of S. aureus ATCC 27217 in conditions causing high salinity stress. As a result, 90 nodes were connected in the network with 654 edges in the image obtained with Cytoscape. By analyzing the edges between more than 15 nodes, 20 hub genes (ABC.CD. P, adhP, arlR, atl, clpL, crtO, E2.3.1., fib, K07078, lctP, LDH, lysC, mtsA, mtsB, mtsC, ohyA, opuD, pflA, pstS, and spoVG) responding to salt stress were identified (Figure 8A). Moreover, these genes were all observed to be in a continuous downward adjustment mode. In total, 19 hub genes were assigned to the following three functional categories: 49 biological processes (BP), 14 cellular components (CC) and 23 molecular functions (MF) (Figure 8B). Most hub genes were categorized into a single-organism process, catalytic activity, cellular process, and metabolic process. In addition, 11 hub genes were assigned to 30 KEGG pathways (Figure 8C). These pathways mainly involve the transfer system (ABC transporters, two-component system) and carbon and nitrogen metabolism (glycolysis/gluconeogenesis, fatty acid degradation, pyruvate metabolism, tyrosine metabolism, cysteine, and methionine metabolism, lysine biosynthesis, glycine, serine, and threonine metabolism). Overall, S. aureus showed adaptive changes after undergoing high salinity stress. These hub genes require in-depth functional verification and provide the basis for further research.
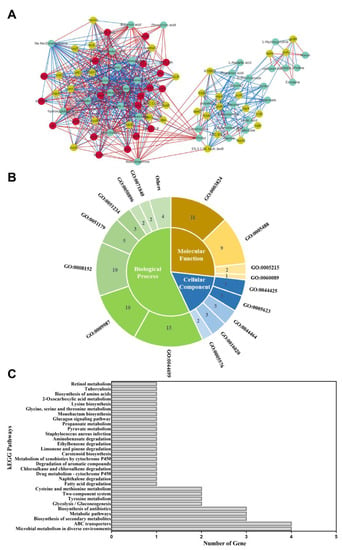
Figure 8.
The results of the integration analysis. (A) Relational Graph of key candidate genes and DMs. Green node represents DM, yellow node represents candidate gene, and red node represents hub gene. The red and green lines represent positive and negative correlations, respectively. The thicker the line, the larger the correlation coefficient, and the thinner the line, the smaller the correlation coefficient; (B) GO functional classification; Biological processes, molecular functions, and cellular components associated with hub genes; (C) Pathway analysis of hub genes. The X-axis represents the number of annotated genes. The Y-axis represents the pathway terms.
4. Discussion
S. aureus can sense exogenous salt-stimulated signaling and then activate the stress response, changing its metabolic and physiological states, thereby improving its ability to survive [22]. In this study, phenotypic identification was combined with transcriptomic and metabolomic analyses to comprehensively elucidate this mechanism. We found that biofilm formation, virulence, transfer system, and osmotic regulation were the main changes that occurred in S. aureus ATCC 27217 under high salinity stress.
4.1. Biofilm Formation
EPS is the main component of biofilm and contributes to the stability of cells [23]. Biofilms are recalcitrant to harsh environments and can protect bacteria from severe conditions by acting as “protective clothing” [24]. Autolysin IsaA modulates cell wall homeostasis as a protein implicated in biofilm formation [25]. In the increase/decrease change pattern, we found that IsaA was a key candidate target gene in response to salt stress. From the characterization results in our study, we observed that the amount of biofilm formation increased and then decreased, which is consistent with the change in the expression of IsaA. Lopes et al. [26] revealed that the deletion of IsaA led to a significant decrease in biofilm formation. Therefore, we speculate that when S. aureus ATCC 27217 is subjected to salt stress, IsaA positively regulates and participates in biofilm formation.
4.2. Virulence
As an exotoxin, hemolysin is an important virulence factor that causes S. aureus infection, and it is also the leading cause of red blood cell hemolysis [27]. Plasma coagulase is an aggressive enzyme whose role is to deposit and coagulate fibrin in the plasma on the surface of bacteria to prevent phagocytosis by phagocytes [28]. The hemolysin and coagulase produced by S. aureus can cause sepsis and purulent infections in humans and, in severe cases, may be fatal [29]. This study found that high salinity stress inhibited the hemolytic activity and expression of coagulase in S. aureus ATCC 27217; that is, high salinity stress inhibited the expression of virulence factors, and this inhibition occurred in a dose-dependent manner.
Staphylococcal enterotoxins represent a group of toxin proteins and the most common cause of staphylococcal food poisoning [30]. They are the main factors that contribute to vomiting and diarrhea. Staphylococcal enterotoxins have strong heat resistance, so they are difficult to remove by heating [31]. In the RNA-seq results, we found that the staphylococcal enterotoxins gene set was significantly down-regulated by 2.1-fold (0% NaCl vs. 20% NaCl). It can be seen that an increase in salinity reduces the expression of set. Sihto et al. [32] studied the expression of staphylococcal enterotoxin genes under NaCl stress at the mRAN level, and demonstrated that NaCl stress led to a decrease in the expression of staphylococcal enterotoxin genes in S. aureus. This is consistent with the result we obtained in our study. It can be seen that under high salt stress, the down-regulation of the enterotoxin gene represents a decline in its virulence.
Atl plays a key role in bacterial adhesion, internalization, and biofilm formation [33]. Porayath et al. [34] emphasized that atl is one of the vital proteins in staphylococci. Atl assists staphylococci in binding to many kinds of host cell components during colonization and infection. Efb, a fibrinogen-binding protein secreted by S. aureus, is an important virulence factor in staphylococcal infection [35]. Efb attracts fibrinogen to the surface of the bacteria, thereby providing S. aureus with the ability to cause both superficial and deep infections [36,37]. In this study, the sequential downregulation of the hub genes atl and efb indicated the reduced ability of bacteria to colonize and infect. In the significant metabolic pathway, the number of downregulated genes (twenty-five) mapped to S. aureus infection was greater than the number of upregulated genes (four); therefore, we inferred that this metabolic pathway was downregulated. Masters et al. [38] demonstrated that a large number of virulence factors and toxins are produced by S. aureus during infection. Therefore, we suggest that the downregulation of S. aureus infection pathway is also one of the reasons for the downregulation of virulence.
When microorganisms have to engage in strong metabolic alterations for survival under severe stress, they downregulate the effort directed to cellular functions which are not of core significance for survival, such as virulence [39]. Herein, we believe that virulence was downregulated to reduce the depletion of nonessential cellular functions to survive in a highly salt-stressed environment.
4.3. Transfer System
DM myo-inositol (VIP = 1.27395) was downregulated significantly (p < 0.01) in 0% NaCl vs. 10% NaCl. Simultaneously, the impact score (impact score = 0.625) of the inositol phosphate metabolism was the highest in this group. The transformation of various phosphoinositide molecules occurs in this pathway. Myo-inositol is the only base molecule involved in the conversion of all phosphoinositide molecules, so a decrease in inositol content leads to the downregulation of this pathway (Figure 9). Previously, researchers reported that phosphoinositides participate in various life-related activities, such as stress resistance, DNA repair, signal response, and RNA transport [40,41,42]. These molecules play various functions in different metabolic pathways. Taken together, we speculate that S. aureus ATCC 27217 reduces the conversion rate of functional molecules by downregulating inositol phosphate metabolism, thereby reducing the unnecessary activation of molecular functions to alleviate the survival crisis caused by high salt stress.
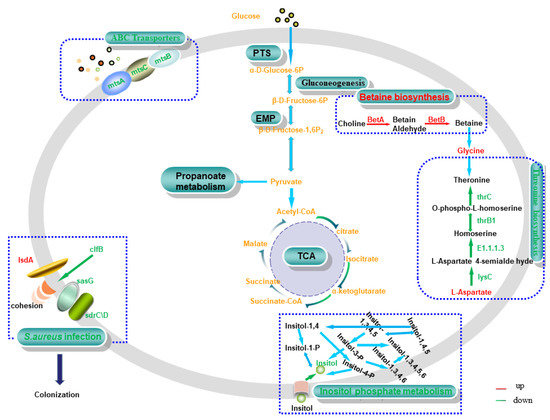
Figure 9.
Relevant pathways in S. aureus. Red and green represent upregulated and downregulated expression, respectively. Matched genes include choline dehydrogenase (betA), betaine-aldehyde dehydrogenase (betB), manganese ABC transporter substrate-binding protein (mtsA), membrane protein ABC transporter permease (mtsB), phosphonate ABC transporter ATP-binding protein (mtsC), clumping factor B (clfB), heme transporter IsdA (isdA), Ser-Asp rich fibrinogen-binding protein (sdrC/D), surface protein G (sasG), aspartate kinase (lysC), homoserine dehydrogenase (E1.1.1.3), homoserine kinase (thrB1), and threonine synthase (thrC).
MtsABC is a typical ABC transporter system composed of lipoprotein mtsA, ATP-binding protein mtsB, and hydrophobic integral membrane protein mtsC [43]. In this study, the expression of the hub genes mtsA, mtsB, and mtsC was continuously downregulated as the concentration of salt stress increased (Figure 9). ABC transporters regulate the homeostasis of ions in bacteria, and participate in nutrient absorption, signal molecule output, and the elimination of toxic compounds. They play an essential role in cellular defense [44]. Thus, this finding suggests that the MtsABC transport system maintains intracellular homeostasis in S. aureus ATCC 27217 by reducing ion transport and signaling, thereby protecting cells from high salt damage.
In S. aureus ATCC 27217, 17 DEGs were associated with the aminoacyl-tRNA biosynthesis pathway, and 16 DEGs were upregulated. Therefore, the aminoacyl-tRNA biosynthesis pathway showed an overall upward activation trend. This pathway can transport amino acids to promote protein synthesis. It ensures that gene sequences can be accurately transcribed and translated into proteins, thereby maintaining cellular homeostasis [45]. From the pathway analysis results, we also found that salt stress leads to abnormal amino acid metabolism. Hence, S. aureus ATCC 27217 improved cellular homeostasis by upregulating aminoacyl-tRNA biosynthesis, ensuring the rigor and diversity of its life forms and resisting salt stress.
4.4. Osmotic Adjustment
The accumulation of compatible substances and osmotic adaptation are critical processes by which cells regulate high osmotic pressure [46,47]. In glycine, serine and threonine metabolism, two DEGs (betA and betB) were mapped to the betaine biosynthesis pathway. Additionally, both DEGs were upregulated, so we concluded that the upregulation of the betaine biosynthesis pathway was activated (Figure 9). Ming et al. [16] demonstrated that the upregulation of betA and betB in S. aureus regulates betaine synthesis and transformation, thereby increasing osmotic stability. Brown et al. [48] suggested that this osmoregulation is capable of protecting the active structure of proteins and nucleic acids. It was also previously reported in foodborne bacteria such as Vibrio parahaemolyticus [49], and Listeria monocytogenes [50]. All of these results indicate the upregulation of the betaine biosynthesis pathway can regulate osmotic pressure and protect S. aureus ATCC 27217 intracellular biomacromolecules against salt stress.
Previous studies reported that after undergoing low-concentration salt stress, S. aureus accumulates a great amount of L-proline for osmotic regulation [51,52]. This mechanism was also reported in foodborne bacteria such as Staphylococcus saprophyticus [53], Vibrio parahaemolyticus [48] and Listeria monocytogenes [54]. In the present study, the content of DM L-proline was significantly increased in the environment of 10% NaCl, which was consistent with previous studies. However, after the bacteria were transferred to the 20% NaCl environment, the L-proline content decreased. We speculate that the decrease in the L-proline content may be due to the high salt (20%NaCl) damage caused to the cell structure, resulting in the release of proline and further contents outside the cell. Eventually, the normal metabolism of proline becomes disturbed. The results of this study provide a scientific basis for the further exploration of the changes in the metabolic system of S. aureus in a high osmotic environment.
In the present study, although we employed an omics-combined characterization approach, the mechanism of S. aureus survival in salted foods was investigated from a genetic and metabolic perspective. However, the findings were focused on S. aureus ATCC 27217, and it is unclear whether they are applicable to other S. aureus. In addition, two biological replicates were performed for the transcriptome and three biological replicates for the metabolome. Nevertheless, there is still a deficiency in the number of replicates performed for the samples.
5. Conclusions
Through a comprehensive analysis of the transcriptome and metabolome, we provided valuable information about the regulatory network of S. aureus ATCC 27217 in response to high salinity stress. Virulence metabolism and signaling pathways were downregulated to reduce the consumption of non-essential cellular functions. The synthesis pathway of osmotic protective substances was upregulated to protect the stability of intracellular life substances and osmotic pressure. The aminoacyl-tRNA biosynthesis pathway was upregulated to maintain the rigor and diversity of cellular life forms. The regulation of these metabolic pathways was an essential method by which S. aureus was able to self-protect against salinity stress. They could serve as potential targets to help prevent bacterial infections. The results of this work will provide additional knowledge on the biological strategies employed by salt-tolerant bacteria in salted foods.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/foods11101503/s1, Table S1: Primers used in this study. Table S2: Effect of salt stress on coagulase expression. Table S3: Details of four expression change patterns of the DEGs with increasing NaCl concentrations. Table S4: Statistical analysis showing significant KEGG pathways in the transcriptome. Table S5: Details of intracellular metabolites of S. aureus ATCC 27217 identified using GC–MS. Concentrations are presented as the means ± SD, and “-” represents uncertain endogenous metabolites. Table S6: Details of the DMs between different comparison groups. Table S7: Statistical analysis showing the KEGG pathways in the metabolome.
Author Contributions
Conceptualization, R.W. and X.S.; project administration, T.M.; visualization, J.Z.; writing—original draft, Y.F.; writing—review and editing, C.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Key R&D Program of China (2019YFD0900101), by the Scientific Research Project of Jilin Provincial Department of Education (JJKH20210531KJ), and by the K.C. Wong Magna Fund in Ningbo University.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are openly available in the NCBI Sequence Read Archive under accession code PRJNA764353.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kim, N.H.; Cho, T.J.; Rhee, M.S. Sodium Chloride Does Not Ensure Microbiological Safety of Foods: Cases and Solutions. Adv. Appl. Microbiol. 2017, 101, 1–47. [Google Scholar] [PubMed]
- Zwietering, M.H.; Wijtzes, T.; Rombouts, F.M.; Klaas, V.K. A decision support system for prediction of microbial spoilage in foods. J. Ind. Microbiol. Biotechnol. 1993, 12, 324–329. [Google Scholar] [CrossRef]
- Lupín, H.M.; Boeri, R.L.; Moschiar, S.M. Water activity and salt content relationship in moist salted fish products. Int. J. Food Sci. Technol. 1981, 16, 31–38. [Google Scholar] [CrossRef]
- Bencardino, D.; Amagliani, G.; Brandi, G. Carriage of Staphylococcus aureus among food handlers: An ongoing challenge in public health. Food Control 2021, 130, 108362. [Google Scholar] [CrossRef]
- Gordon, A.; Devlieger, D.; Vasan, A.; Bedard, B.J.F.S. Technical considerations for the implementation of food safety and quality systems in developing countries. In Food Safety and Quality Systems in Developing Countries; Academic Press: Cambridge, MA, USA, 2020; Volume 3, pp. 1–40. [Google Scholar]
- Xue, L.; Chen, Y.; Yan, Z.; Lu, W.; Zhu, H.J.I.; Resistance, D. Staphyloxanthin: A potential target for antivirulence therapy. Infect. Drug Resist. 2019, 12, 2151–2160. [Google Scholar] [CrossRef]
- Sergelidis, D.; Abrahim, A.; Papadopoulos, T.; Soultos, N.; Martziou, E. Isolation of methicillin-resistant Staphylococcus spp. from ready-to-eat fish products. Lett. Appl. Microbiol. 2014, 59, 500–506. [Google Scholar] [CrossRef]
- Ma, Y.; Lan, G.; Li, C.; Cambaza, E.M.; Ding, T.J.M.P. Stress tolerance of Staphylococcus aureus with different antibiotic resistance profiles. Microb. Pathog. 2019, 133, 103549. [Google Scholar] [CrossRef]
- Alibayov, B.; Zdeňková, K.; Purkrtová, S.; Demnerová, K.; Karpiskova, R. Detection of some phenotypic and genotypic characteristics of Staphylococcus aureus isolated from food items in the Czech Republic. Ann. Microbiol. 2014, 64, 1587–1596. [Google Scholar] [CrossRef]
- Alreshidi, M.M.; Dunstan, R.H.; Macdonald, M.M.; Smith, N.D.; Gottfries, J.; Roberts, T.K. Metabolomic and proteomic responses of Staphylococcus aureus to prolonged cold stress. J. Proteom. 2015, 121, 44–55. [Google Scholar] [CrossRef]
- Lee, S.; Choi, K.-H.; Yoon, Y. Effect of NaCl on Biofilm Formation of the Isolate from Staphylococcus aureus Outbreak Linked to Ham. Korean J. Food Sci. Anim. Resour. 2014, 34, 257–261. [Google Scholar] [CrossRef]
- Alreshidi, M.M.; Dunstan, R.H.; Macdonald, M.M.; Gottfries, J.; Roberts, T.K. The Uptake and Release of Amino Acids by Staphylococcus aureus at Mid-Exponential and Stationary Phases and Their Corresponding Responses to Changes in Temperature, pH and Osmolality. Front. Microbiol. 2020, 10, 03059. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Brissonnet, F.; Trotier, E.; Briandet, R. The Biofilm Lifestyle Involves an Increase in Bacterial Membrane Saturated Fatty Acids. Front. Microbiol. 2016, 7, 01673. [Google Scholar] [CrossRef] [PubMed]
- Detkova, E.N.; Boltyanskaya, Y.V. Osmoadaptation of haloalkaliphilic bacteria: Role of osmoregulators and their possible practical application. Microbiology 2007, 76, 511–522. [Google Scholar] [CrossRef]
- Vijaranakul, U.; Nadakavukaren, M.; Dejonge, B.; Wilkinson, B.; Jayaswal, R. Increased cell size and shortened peptidoglycan interpeptide bridge of NaCl-stressed Staphylococcus aureus and their reversal by glycine betaine. J. Bacteriol. 1995, 177, 5116–5121. [Google Scholar] [CrossRef] [PubMed]
- Ming, T.; Geng, L.; Feng, Y.; Lu, C.; Zhou, J.; Li, Y.; Zhang, D.; He, S.; Li, Y.; Cheong, L.; et al. iTRAQ-Based Quantitative Proteomic Profiling of Staphylococcus aureus Under Different Osmotic Stress Conditions. Front. Microbiol. 2019, 10, 01082. [Google Scholar] [CrossRef] [PubMed]
- Alreshidi, M.M.; Dunstan, R.H.; Macdonald, M.M.; Smith, N.D.; Gottfries, J.; Roberts, T.K. Amino acids and proteomic acclimation of Staphylococcus aureus when incubated in a defined minimal medium supplemented with 5% sodium chloride. Microbiologyopen 2019, 8, e00772. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hou, L.; Zhang, S.; Kou, X.; Li, R.; Wang, S. Mechanism of S. aureus ATCC 25923 in response to heat stress under different water activity and heating rates. Food Control 2020, 108, 106837. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Ming, T.; Han, J.; Li, Y.; Lu, C.; Qiu, D.; Li, Y.; Zhou, J.; Su, X. A metabolomics and proteomics study of the Lactobacillus plantarum in the grass carp fermentation. BMC Microbiol. 2018, 18, 216. [Google Scholar] [CrossRef]
- Hashim, Z.; Mukai, Y.; Bamba, T.; Fukusaki, E. Metabolic profiling of retrograde pathway transcription factors rtg1 and rtg3 knockout yeast. Metabolites 2014, 4, 580–598. [Google Scholar] [CrossRef]
- Krämer, R. Bacterial stimulus perception and signal transduction: Response to osmotic stress. Chem. Rec. 2010, 10, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y. Cloning of genes affecting capsule expression in Staphylococcus aureus strain M. Mol. Microbiol. 1992, 6, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Wang, Y.; Liu, L.; He, J. Biofilms: The Microbial “Protective Clothing” in Extreme Environments. Int. J. Mol. Sci. 2019, 20, 3423. [Google Scholar] [CrossRef] [PubMed]
- Buchad, H.; Nair, M. The small RNA SprX regulates the autolysin regulator WalR in Staphylococcus aureus. Microbiol. Res. 2021, 250, 126785. [Google Scholar] [CrossRef]
- Lopes, A.A.; Yoshii, Y.; Yamada, S.; Nagakura, M.; Okuda, K.I. Roles of Lytic Transglycosylases in Biofilm Formation and β-Lactam Resistance in Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2019, 63, e01277-19. [Google Scholar] [CrossRef]
- Burnside, K.; Lembo, A.; de Los Reyes, M.; Iliuk, A.; Binhtran, N.T.; Connelly, J.E.; Lin, W.J.; Schmidt, B.Z.; Richardson, A.R.; Fang, F.C.; et al. Regulation of Hemolysin Expression and Virulence of Staphylococcus aureus by a Serine/Threonine Kinase and Phosphatase. PLoS ONE 2010, 5, 1–16. [Google Scholar] [CrossRef]
- Vanassche, T.; Verhaegen, J.; Peetermans, W.E.; Hoylaerts, M.F.; Verhamme, P. Dabigatran inhibits Staphylococcus aureus coagulase activity. J. Clin. Microbiol. 2010, 48, 4248–4250. [Google Scholar] [CrossRef]
- Cunha, M.L.R.S.; Calsolari, R.A.O. Toxigenicity in Staphylococcus aureus and Coagulase-Negative Staphylococci: Epidemiological and Molecular Aspects. Microbiol. Insights 2008, 1, 13–24. [Google Scholar] [CrossRef]
- Shimamura, Y.; Utsumi, M.; Hirai, C.; Nakano, S.; Ito, S.; Tsuji, A.; Ishii, T.; Hosoya, T.; Kan, T.; Ohashi, N.; et al. Binding of Catechins to Staphylococcal Enterotoxin A. Molecules 2018, 23, 1125. [Google Scholar] [CrossRef]
- Balaban, N.; Rasoonly, A. Staphylococcal enterotoxins. Int. J. Food Microbiol. 2000, 61, 1–10. [Google Scholar] [CrossRef]
- Sihto, H.M.; Tasara, T.; Stephan, R.; Johler, S. Temporal expression of the staphylococcal enterotoxin D gene under NaCl stress conditions. FEMS Microbiol. Lett. 2015, 362, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Steinbacher, T.; Peters, G.; Heilmann, C.; Becker, K. The adhesive properties of the Staphylococcus lugdunensis multifunctional autolysin AtlL and its role in biofilm formation and internalization. Int. J. Med. Microbiol. 2015, 305, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Porayath, C.; Suresh, M.K.; Biswas, R.; Nair, B.G.; Mishra, N.; Pal, S. Autolysin mediated adherence of Staphylococcus aureus with Fibronectin, Gelatin and Heparin. Int. J. Biol. Macromol. 2018, 110, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Wade, D.; Palma, M.; Lofving-Arvholm, I.; Sallberg, M.; Silberring, J.; Flock, J.I. Identification of functional domains in Efb, a fibrinogen binding protein of Staphylococcus aureus. Biochem. Biophys. Res. Commun. 1998, 248, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.P.; Kang, M.; Ganesh, V.K.; Ravirajan, D.; Li, B.; Höök, M. Coagulase and Efb of Staphylococcus aureus Have a Common Fibrinogen Binding Motif. Mbio 2016, 7, e01885-15. [Google Scholar] [CrossRef]
- Fehrmann, C.; Jurk, K.; Bertling, A.; Seidel, G.; Heilmann, C. Role for the fibrinogen-binding proteins Coagulase and Efb in the Staphylococcus aureus–Candida interaction. Int. J. Med. Microbiol. IJMM 2013, 303, 230–238. [Google Scholar] [CrossRef]
- Masters, T.L.; Johnson, S.; Patricio, R.J.; Greenwood-Quaintance, K.E.; Cunningham, S.A.; Abdel, M.P.; Chia, N.; Patel, R. Comparative Transcriptomic Analysis of Staphylococcus aureus Associated with Periprosthetic Joint Infection Under in vivo and in vitro Conditions. J. Mol. Diagn. 2021, 23, 986–999. [Google Scholar] [CrossRef]
- Flint, A.; Butcher, J.; Stintzi, A. Stress Responses, Adaptation, and Virulence of Bacterial Pathogens during Host Gastrointestinal Colonization. Microbiol. Spectr. 2016, 4, 385–411. [Google Scholar] [CrossRef]
- Hanakahi, L.A.; Bartlet, M.; Chappell, C.; Pappin, D.; West, S.C. Binding of Inositol Phosphate to DNA-PK and Stimulation of Double-Strand Break Repair. Cell 2000, 102, 721–729. [Google Scholar] [CrossRef]
- Bolger, T.A.; Folkmann, A.W.; Tran, E.J.; Wente, S.R. The mRNA Export Factor Gle1 and Inositol Hexakisphosphate Regulate Distinct Stages of Translation. Cell 2008, 134, 624–633. [Google Scholar] [CrossRef]
- Feng, Y.; Gu, D.; Wang, Z.; Lu, C.; Su, X. Comprehensive Evaluation and Analysis of the Salinity Stress Response Mechanisms Based on Transcriptome and Metabolome of Staphylococcus aureus. Arch. Microbiol. 2021, 204, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Baker, H.; Ge, R.; Sun, H.; He, Q.; Baker, E. Crystal structure and metal binding properties of the lipoprotein MtsA, responsible for iron transport in Streptococcus pyogenes. Biochemistry 2009, 48, 6184–6190. [Google Scholar] [CrossRef] [PubMed]
- Beis, K. Structural basis for the mechanism of ABC transporters. Biochem. Soc. Trans. 2015, 43, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Francklyn, C.S.; Mullen, P. Progress and Challenges in Aminoacyl-tRNA Synthetase-based Therapeutics. J. Biol. Chem. 2019, 294, 5365–5385. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, J.F.; Rahn, T.; Künzel, S.; Keller, A.; Neulinger, S.C. Osmotic Adaptation and Compatible Solute Biosynthesis of Phototrophic Bacteria as Revealed from Genome Analyses. Microorganisms 2021, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Song, W.S.; Kim, S.W.; Jo, S.H.; Lee, J.S.; Jeon, H.J.; Joon, B.; Choi, K.Y.; Yang, Y.H.; Kim, Y.G. Multi-omics characterization of the osmotic stress resistance and protease activities of the halophilic bacterium Pseudoalteromonas phenolica in response to salt stress. RSC Adv. 2020, 10, 23792–23800. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.D. Microbial water stress. Bacteriol. Rev. 1976, 40, 803–846. [Google Scholar] [CrossRef]
- Ongagna-Yhombi, S.Y.; McDonald, N.D.; Boyd, E.F. Deciphering the Role of Multiple Betaine-Carnitine-Choline Transporters in the Halophile Vibrio parahaemolyticus. Appl. Environ. Microbiol. 2015, 81, 351–363. [Google Scholar] [CrossRef]
- Angelidis, A.S.; Smith, G.M. Three Transporters Mediate Uptake of Glycine Betaine and Carnitine by Listeria monocytogenes in Response to Hyperosmotic Stress. Appl. Environ. Microbiol. 2003, 69, 1013–1022. [Google Scholar] [CrossRef]
- Miller, K.J.; Zelt, S.C.; Bae, J.H. Glycine betaine and proline are the principal compatible solutes of Staphylococcus aureus. Curr. Microbiol. 1991, 23, 131–137. [Google Scholar] [CrossRef]
- Townsend, D.E.; Wilkinson, B.J. Proline transport in Staphylococcus aureus: A high-affinity system and a low-affinity system involved in osmoregulation. J. Bacteriol. 1992, 174, 2702–2710. [Google Scholar] [CrossRef] [PubMed]
- Deutch, C.E. L-Proline nutrition and catabolism in Staphylococcus saprophyticus. Antonie Van Leeuwenhoek 2011, 99, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Bayles, D.O.; Wilkinson, B.J. Osmoprotectants and cryoprotectants for Listeria monocytogenes. Lett. Appl. Microbiol. 2000, 30, 23–27. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).