A Combined Metabolomic and Metagenomic Approach to Discriminate Raw Milk for the Production of Hard Cheese


 ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Extraction of Milk Metabolites
2.3. Untargeted Metabolomics Based on UHPLC-QTOF Mass Spectrometry
2.4. DNA Extraction and Sequencing
2.5. Sequencing Data Processing and Statistical Analysis
2.6. Multivariate Analysis on Metabolomics-Based Data
3. Results and Discussion
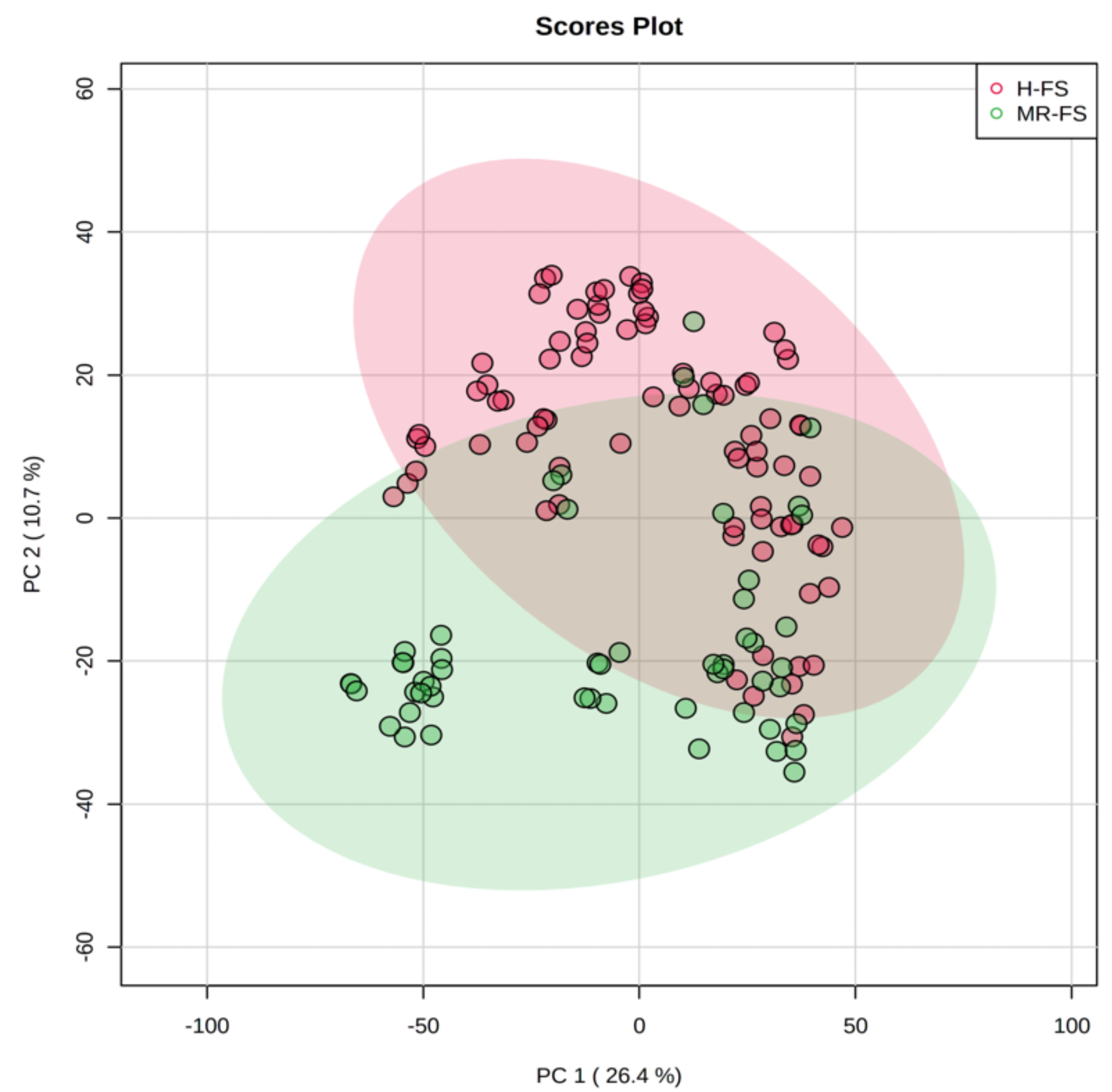
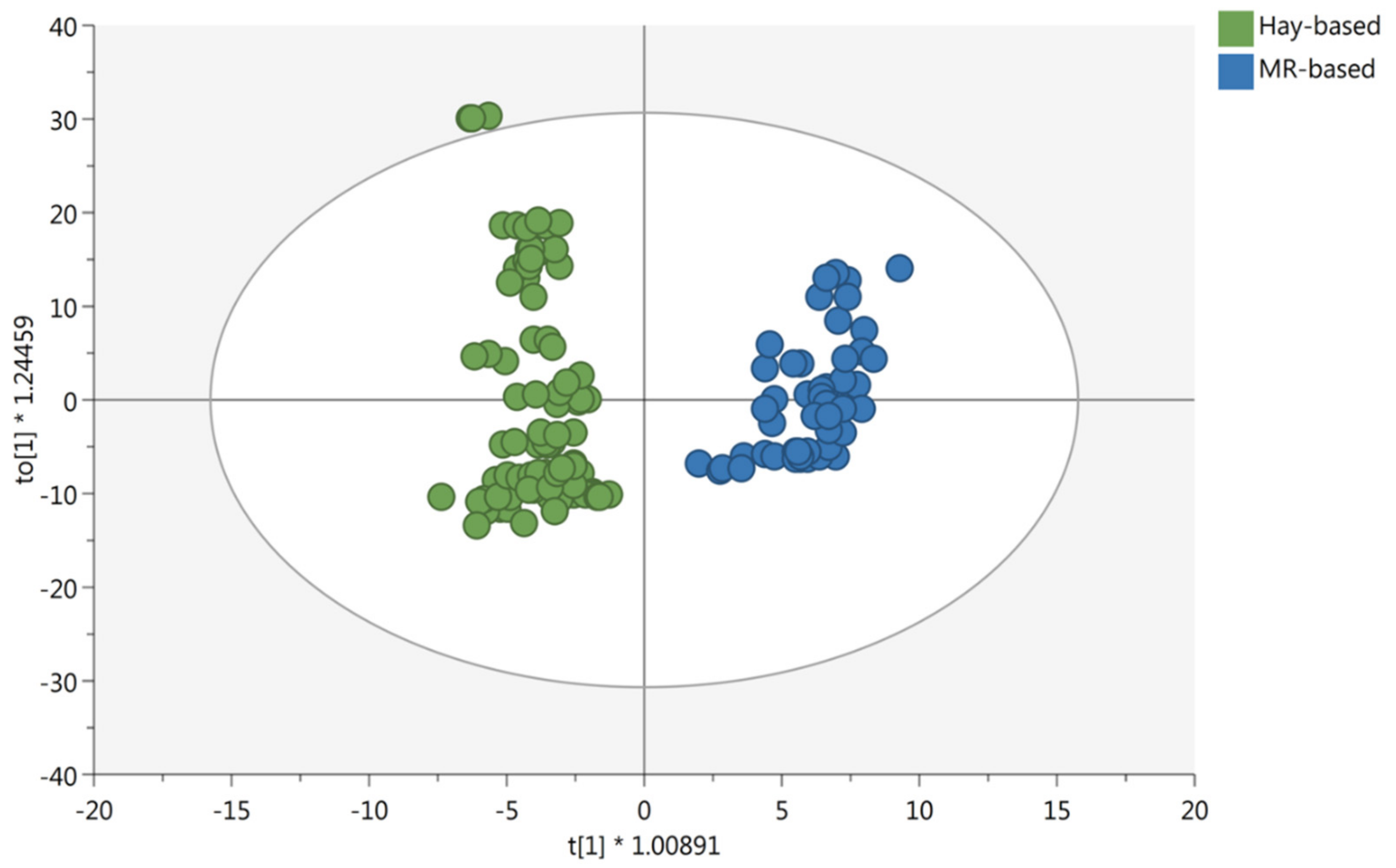
3.1. Multivariate Statistical Discrimination of the Different Milk Samples
3.2. Marker Compounds of the Feeding System
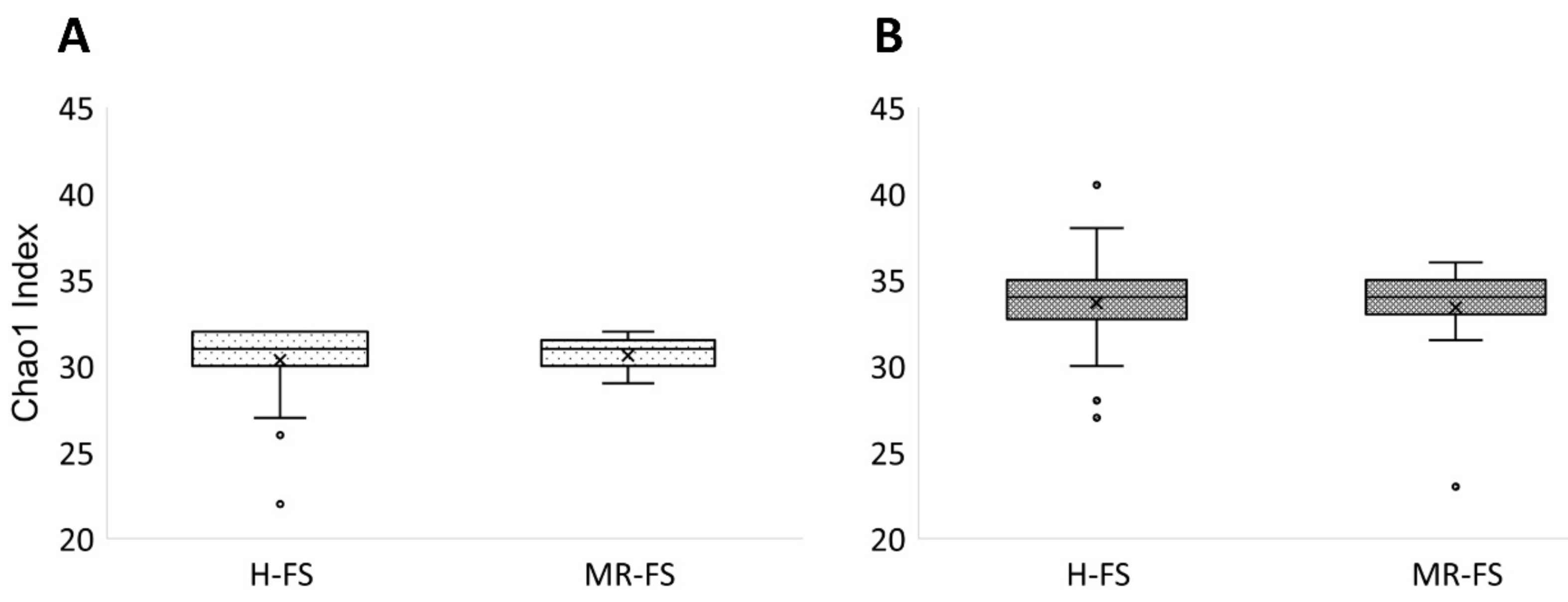
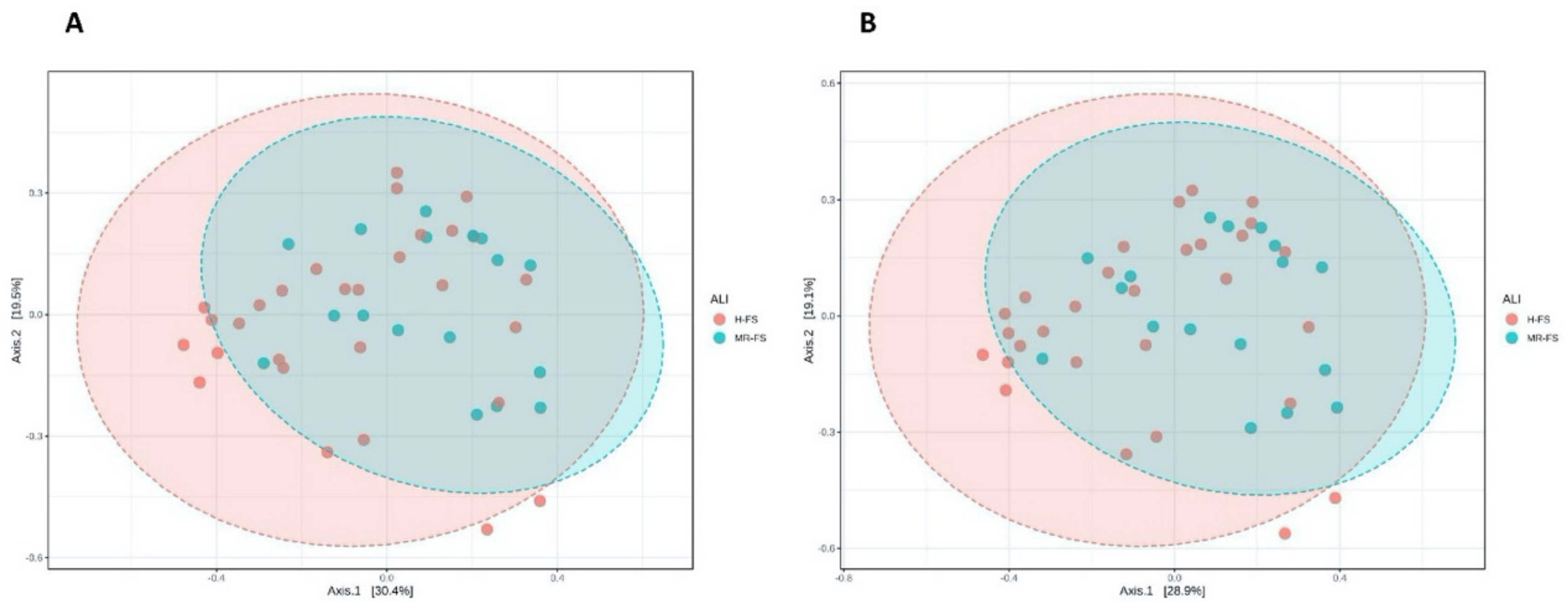
3.3. Microbial Community Diversity
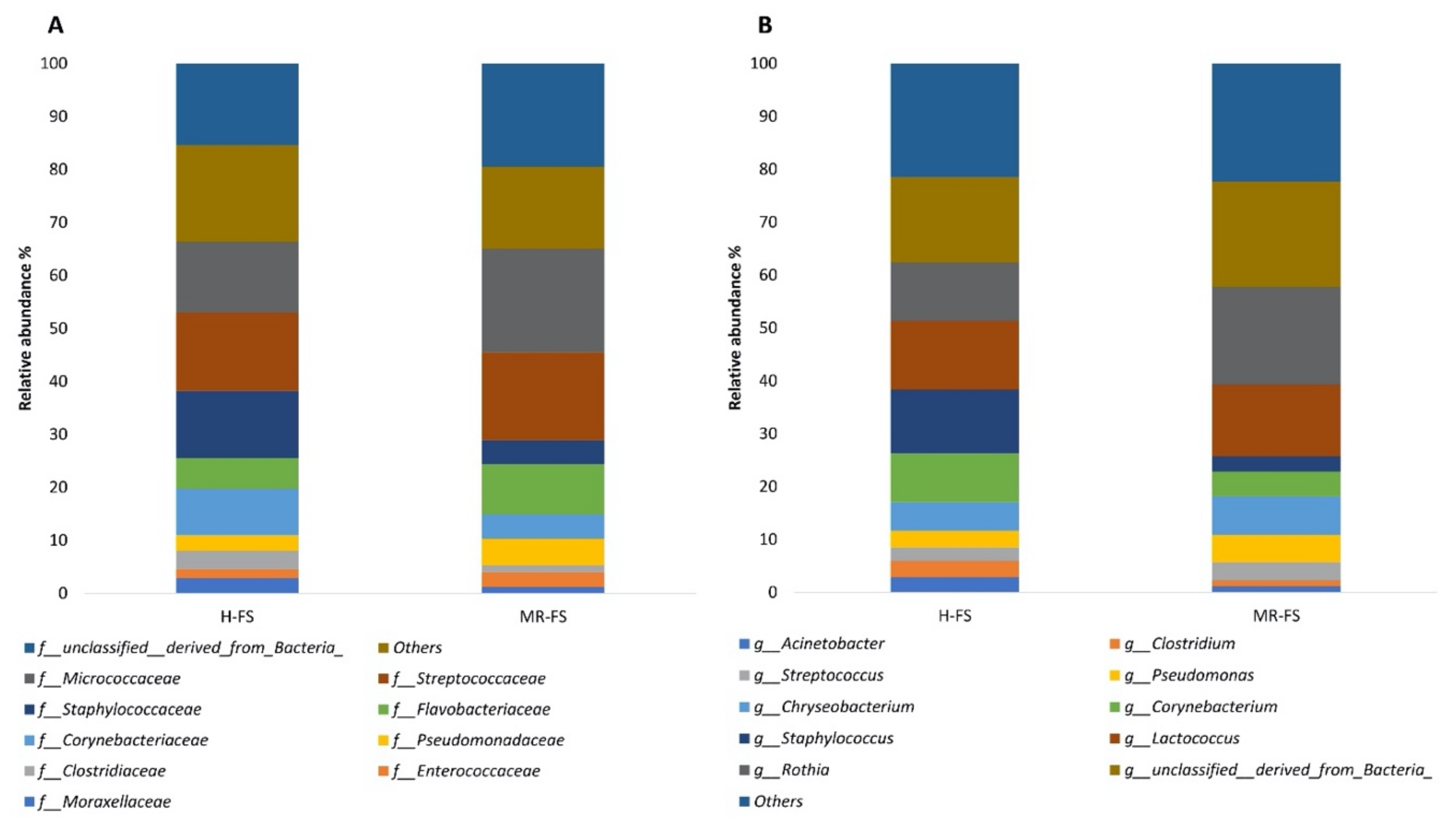
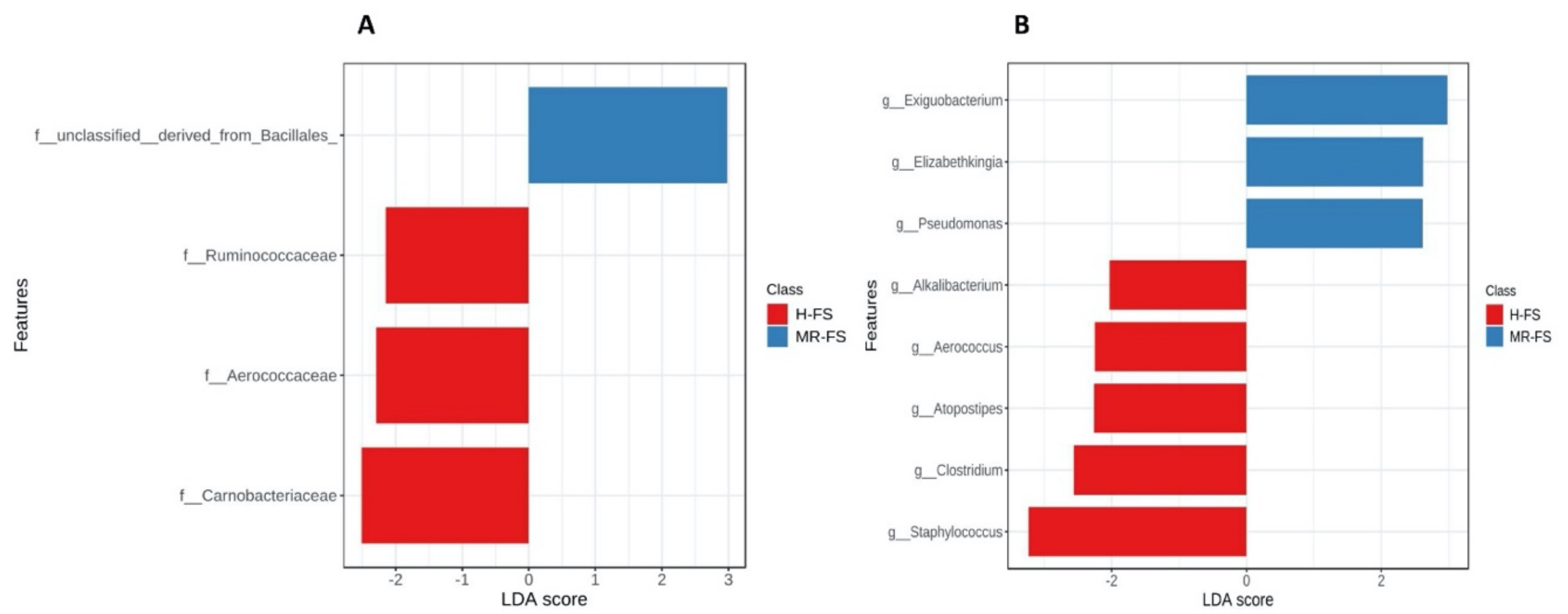
3.4. Microbial Community Taxonomy
3.5. Correlation between Metabolomic and Metagenomic Profiles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corral-Fraga, M.; Carpena, M.; Garcia-Oliveira, P.; Pereira, A.G.; Prieto, M.A.; Simal-Gandara, J. Analytical Metabolomics and Applications in Health, Environmental and Food Science. Crit. Rev. Anal. Chem. 2020. [Google Scholar] [CrossRef]
- Sun, H.-Z.; Wang, D.-M.; Wang, B.; Wang, J.-K.; Liu, H.-Y.; Guan, L.L.; Liu, J.-X. Metabolomics of Four Biofluids from Dairy Cows: Potential Biomarkers for Milk Production and Quality. J. Proteome Res. 2015, 14, 1287–1298. [Google Scholar] [CrossRef]
- Salzano, A.; Manganiello, G.; Neglia, G.; Vinale, F.; De Nicola, D.; D’Occhio, M.; Campanile, G. A Preliminary Study on Metabolome Profiles of Buffalo Milk and Corresponding Mozzarella Cheese: Safeguarding the Authenticity and Traceability of Protected Status Buffalo Dairy Products. Molecules 2020, 25, 304. [Google Scholar] [CrossRef]
- Scano, P.; Carta, P.; Ibba, I.; Manis, C.; Caboni, P. An Untargeted Metabolomic Comparison of Milk Composition from Sheep Kept Under Different Grazing Systems. Dairy 2020, 1, 30–41. [Google Scholar] [CrossRef]
- Saleem, F.; Ametaj, B.N.; Bouatra, S.; Mandal, R.; Zebeli, Q.; Dunn, S.M.; Wishart, D.S. A metabolomics approach to uncover the effects of grain diets on rumen health in dairy cows. J. Dairy Sci. 2012, 95, 6606–6623. [Google Scholar] [CrossRef]
- Xue, M.-Y.; Sun, H.-Z.; Wu, X.-H.; Liu, J.-X.; Guan, L.L. Multi-omics reveals that the rumen microbiome and its metabolome together with the host metabolome contribute to individualized dairy cow performance. Microbiome 2020, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Rocchetti, G.; Gallo, A.; Nocetti, M.; Lucini, L.; Masoero, F. Milk metabolomics based on ultra-high-performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry to discriminate different cows feeding regimens. Food Res. Int. 2020, 134, 109279. [Google Scholar] [CrossRef] [PubMed]
- Popescu, S.; Borda, C.; Diugan, E.A.; Niculae, M.; Stefan, R.; Sandru, C.D. The Effect of the Housing System on the Welfare Quality of Dairy Cows. Ital. J. Anim. Sci. 2014, 13. [Google Scholar] [CrossRef]
- Lambertz, C.; Sanker, C.; Gauly, M. Climatic effects on milk production traits and somatic cell score in lactating Holstein-Friesian cows in different housing systems. J. Dairy Sci. 2014, 97, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Rocchetti, G.; Lucini, L.; Gallo, A.; Masoero, F.; Trevisan, M.; Giuberti, G. Untargeted metabolomics reveals differences in chemical fingerprints between PDO and non-PDO Grana Padano cheeses. Food Res. Int. 2018, 113, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Clausen, M.R.; Dalsgaard, T.K.; Bertram, H.C. Metabolomics to Explore Impact of Dairy Intake. Nutrients 2015, 7, 4875–4896. [Google Scholar] [CrossRef] [PubMed]
- Foroutan, A.; Guo, A.C.; Vazquez-Fresno, R.; Lipfert, M.; Zhang, L.; Zheng, J.; Badran, H.; Budinski, Z.; Mandal, R.; Ametaj, B.N.; et al. Chemical Composition of Commercial Cow’s Milk. J. Agric. Food Chem. 2019, 67, 4897–4914. [Google Scholar] [CrossRef] [PubMed]
- Hou, K.; Tong, J.; Zhang, H.; Gao, S.; Guo, Y.; Niu, H.; Xiong, B.; Jiang, L. Microbiome and metabolic changes in milk in response to artemisinin supplementation in dairy cows. AMB Express 2020, 10, 154. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Zhang, H.; Zhang, Y.; Xiong, B.; Jiang, L. Microbiome and Metabolome Analyses of Milk from Dairy Cows with Subclinical Streptococcus agalactiae Mastitis—Potential Biomarkers. Front. Microbiol. 2019, 10, 2547. [Google Scholar] [CrossRef]
- Martin, N.H.; Kent, D.J.; Evanowski, R.L.; Zuber Hrobuchak, T.J.; Wiedmann, M. Bacterial spore levels in bulk tank raw milk are influenced by environmental and cow hygiene factors. J. Dairy Sci. 2019, 102, 9689–9701. [Google Scholar] [CrossRef]
- Quintana, A.R.; Sesena, S.; Garzón, A.; Arias, R. Factors Affecting Levels of Airborne Bacteria in Dairy Farms: A Review. Animals 2020, 10, 526. [Google Scholar] [CrossRef]
- Gatti, M.; Bottari, B.; Lazzi, C.; Neviani, E.; Mucchetti, G. Invited review: Microbial evolution in raw-milk, long-ripened cheeses produced using undefined natural whey starters. J. Dairy Sci. 2014, 97, 573–591. [Google Scholar] [CrossRef]
- Capodifoglio, E.; Vidal, A.M.C.; Lima, J.A.S.; Bartoletto, F.; D’Abreu, L.F.; Gonçalves, A.C.S.; Vaz, A.C.N.; de Carvalho Balieiro, J.C.; Netto, A.S. Lipolytic and proteolytic activity of Pseudomonas spp. isolated during milking and storage of refrigerated raw milk. J. Dairy. Sci. 2016, 99, 5214–5223. [Google Scholar] [CrossRef]
- Xi, X.; Kwok, L.Y.; Wang, Y.; Ma, C.; Mi, Z.; Zhang, H. Ultra-performance liquid chromatography-quadrupole-time of flight mass spectrometry MSE-based untargeted milk metabolomics in dairy cows with subclinical or clinical mastitis. J. Dairy Sci. 2017, 100, 4884–4896. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef]
- Tsugawa, H.; Kind, T.; Nakabayashi, R.; Yukihira, D.; Tanaka, W.; Cajka, T.; Saito, K.; Fiehn, O.; Arita, M. Hydrogen Rearrangement Rules: Computational MS/MS Fragmentation and Structure Elucidation Using MS-FINDER Software. Anal. Chem. 2016, 88, 7946–7958. [Google Scholar] [CrossRef] [PubMed]
- Foroutan, A.; Fitzsimmons, C.; Mandal, R.; Piri-Moghadam, H.; Zheng, J.; Guo, A.; Li, C.; Guan, L.L.; Wishart, D.S. The Bovine Metabolome. Metabolites 2020, 10, 233. [Google Scholar] [CrossRef] [PubMed]
- Salek, R.M.; Steinbeck, C.; Viant, M.R.; Goodacre, R.; Dunn, W.B. The role of reporting standards for metabolite annotation and identification in metabolomic studies. GigaScience 2013, 2, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41. [Google Scholar] [CrossRef]
- Keegan, K.P.; Glass, E.M.; Meyer, F. MG-RAST, a Metagenomics Service for Analysis of Microbial Community Structure and Function. In Microbial Environmental Genomics (MEG); Martin, F., Uroz, S., Eds.; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016; Volume 1399. [Google Scholar] [CrossRef]
- Vetrovsky, T.; Baldrian, P.; Morais, D. SEED 2: A user-friendly platform for amplicon high-throughput sequencing data analyses. Bioinformatics 2018, 34, 2292–2294. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, 633–642. [Google Scholar] [CrossRef]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68. [Google Scholar] [CrossRef]
- Rombouts, C.; Hemeryck, L.Y.; Van Hecke, T.; De Smet, S.; De Vos, W.J.; Vanhaecke, L. Untargeted metabolomics of colonic digests reveals kynurenine pathway metabolites, dityrosine and 3-dehydroxycarnitine as red versus white meat discriminating metabolites. Sci. Rep. 2017, 7, 42514. [Google Scholar] [CrossRef]
- Xia, J.; Broadhurst, D.I.; Wilson, M.; Wishart, D.S. Translational biomarker discovery in clinical metabolomics: An introductory tutorial. Metabolomics 2013, 9, 280–299. [Google Scholar] [CrossRef]
- Mosele, J.I.; Macià, A.; Motilva, M.-J. Metabolic and Microbial Modulation of the Large Intestine Ecosystem by Non-Absorbed Diet Phenolic Compounds: A Review. Molecules 2015, 20, 17429–17468. [Google Scholar] [CrossRef] [PubMed]
- Mena, P.; Bresciani, L.; Brindani, N.; Ludwig, I.A.; Pereira-Caro, G.; Angelino, D.; Llorach, R.; Calani, L.; Brighenti, F.; Clifford, M.N.; et al. Phenyl-γ-valerolactones and phenylvaleric acids, the main colonic metabolites of flavan-3-ols: Synthesis, analysis, bioavailability, and bioactivity. Nat. Prod. Rep. 2019, 36, 714–752. [Google Scholar] [CrossRef] [PubMed]
- Handa, R.J.; Sharma, D.; Uht, R. A role for the androgen metabolite, 5alpha androstane 3beta, 17beta Diol (3β-Diol) in the regulation of the hypothalamo-pituitary–adrenal axis. Front. Endocrinol. 2011, 2, 65. [Google Scholar] [CrossRef] [PubMed]
- Barbaccia, M.L.; Lello, S.; Sidiropoulou, T.; Cocco, T.; Sorge, R.P.; Cocchiarale, A.; Piermarini, V.; Sabato, A.F.; Trabucchi, M.; Romanini, C. Plasma 5α–androstane–3α,17βdiol, an endogenous steroid that positively modulates GABAA receptor function, and anxiety: A study in menopausal women. Psychoneuroendocrinology 2000, 25, 659–675. [Google Scholar] [CrossRef]
- Burstein, S.H. N-Acyl Amino Acids (Elmiric Acids): Endogenous Signaling Molecules with Therapeutic Potential. Mol. Pharmacol. 2018, 93, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Munsch-Alatossava, P.; Käkelä, R.; Ibarra, D.; Youbi-Idrissi, M.; Alatossava, T. Phospholipolysis Caused by Different Types of Bacterial Phospholipases During Cold Storage of Bovine Raw Milk Is Prevented by N2 Gas Flushing. Front. Microbiol. 2018, 9, 1307. [Google Scholar] [CrossRef]
- Espinosa-Salinas, I.; Rodriguez-Casado, A.; Molina, S.; Rodriguez-Gonzalez, A.; Ordovas, J.M.; de Molina, A.R. Beneficial Effects of Bioactive Phospholipids: Genomic Bases. Curr. Nutr. Food Sci. 2011, 7, 145–154. [Google Scholar] [CrossRef]
- Yuan, L.; Sadiq, F.A.; Liu, T.J.; Li, Y.; Gu, J.S.; Yang, H.Y.; He, G.Q. Spoilage potential of psychrotrophic bacteria isolated from raw milk and the thermo-stability of their enzymes. J. Zhejiang Univ. Sci. B 2018, 19, 630–642. [Google Scholar] [CrossRef]
- Shanks, O.C.; Kelty, C.A.; Archibeque, S.; Jenkins, M.; Newton, R.J.; McLellan, S.L.; Huse, S.M.; Sogin, M.L. Community Structures of Fecal Bacteria in Cattle from Different Animal Feeding Operations. Appl. Environ. Microbiol. 2011, 77, 2992–3001. [Google Scholar] [CrossRef]
- Derakhshani, H.; Fehr, K.B.; Sepehri, S.; Francoz, D.; De Buck, J.; Barkema, H.W.; Plaizier, J.C.; Khafipour, E. Invited review: Microbiota of the bovine udder: Contributing factors and potential implications for udder health and mastitis susceptibility. J. Dairy Sci. 2018, 101, 10605–10625. [Google Scholar] [CrossRef]
- Braem, G.; De Vliegher, S.; Verbist, B.; Heyndrickx, M.; Leroy, F.; De Vuyst, L. Culture-independent exploration of the teat apex microbiota of dairy cows reveals a wide bacterial species diversity. Vet. Microbiol. 2012, 157, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Guan, Q.; Ye, C.; Zhang, C.; Foster, J.A.; Forney, L.J. Network analysis suggests a potentially ‘evil’ alliance of opportunistic pathogens inhibited by a cooperative network in human milk bacterial communities. Sci. Rep. 2015, 5, 8275. [Google Scholar]
- Pang, M.; Xie, X.; Bao, H.; Sun, L.; He, T.; Zhao, H.; Zhou, Y.; Zhang, L.; Zhang, H.; Wei, R.; et al. Insights Into the Bovine Milk Microbiota in Dairy Farms With Different Incidence Rates of Subclinical Mastitis. Front. Microbiol. 2018, 9, 2379. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | VIP Score (OPLS-DA) | LogFC Value (MR-FS vs. H-FS) | p-Value (FDR) | ROC AUC |
|---|---|---|---|---|
| PA(16:0/18:1(11Z)) | 1.13 | 1.11 | 1.54 × 10−2 | 0.65 |
| DG(22:0/0:0/18:2n6) | 1.01 | −0.72 | 5.70 × 10−3 | 0.66 |
| 5alpha-Androstan-3alpha,17beta-diol disulfate | 1.32 | 0.66 | 3.10 × 10−3 | 0.67 |
| DG(24:1(15Z)/15:0/0:0) | 1.10 | −0.94 | 5.00 × 10−3 | 0.68 |
| PC(16:0/14:1(9Z)) | 1.43 | −1.22 | 3.12 × 10−3 | 0.71 |
| PC(P-18:0/14:1(9Z)) | 1.47 | 2.31 | 5.04 × 10−4 | 0.72 |
| PC(P-14:0/18:1(9Z)) | 1.41 | 1.85 | 1.61 × 10−3 | 0.73 |
| SM(d18:1/12:0) | 1.51 | −1.45 | 8.19 × 10−6 | 0.76 |
| DG(22:2n6/0:0/22:4n6) | 1.02 | −1.91 | 1.63 × 10−7 | 0.78 |
| 5-(3′,4′-Dihydroxyphenyl)-gamma-valerolactone-4′-O-glucuronide | 1.41 | 1.20 | 1.49 × 10−3 | 0.78 |
| DG(22:0/18:4(6Z,9Z,12Z,15Z)/0:0) | 1.75 | −2.54 | 2.01 × 10−6 | 0.79 |
| SM(d16:0/17:1(10Z)) | 1.70 | −2.53 | 6.44 × 10−8 | 0.79 |
| DG(20:2n6/0:0/20:2n6) | 1.63 | −1.32 | 6.17 × 10−4 | 0.79 |
| PC(P-18:1(9Z)/14:1(9Z)) | 1.60 | 1.65 | 2.53 × 10−9 | 0.79 |
| LysoPC(10:0) | 1.93 | 0.86 | 9.19 × 10−7 | 0.85 |
| 3-Hexenedioic acid | 2.26 | −1.44 | 6.01 × 10−13 | 0.89 |
| LysoPE(0:0/22:1(13Z)) | 2.45 | 2.62 | 1.02 × 10−6 | 0.89 |
| Benzene-1,2,4-triol | 2.43 | −1.49 | 1.32 × 10−16 | 0.91 |
| N-stearoyl glycine | 2.40 | 1.86 | 1.28 × 10−21 | 0.91 |
| LysoPE(22:1(13Z)/0:0) | 2.53 | 2.61 | 5.83 × 10−9 | 0.93 |
| Family | Number of Significant Correlations (p < 0.05) |
|---|---|
| Staphylococcaceae | 9 |
| Pseudomonadaceae | 9 |
| Dermabacteraceae | 9 |
| Streptococcaceae | 7 |
| Genus | |
| Pseudomonas | 9 |
| Staphylococcus | 8 |
| Brachybacterium | 8 |
| Macrococcus | 7 |
| VIP metabolites | |
| LysoPE(22:1(13Z)/0:0) | 7 (at both family and genus level) |
| LysoPE(0:0/22:1(13Z)) | 6 (at genus level); 5 (at family level) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellassi, P.; Rocchetti, G.; Nocetti, M.; Lucini, L.; Masoero, F.; Morelli, L. A Combined Metabolomic and Metagenomic Approach to Discriminate Raw Milk for the Production of Hard Cheese. Foods 2021, 10, 109. https://doi.org/10.3390/foods10010109
Bellassi P, Rocchetti G, Nocetti M, Lucini L, Masoero F, Morelli L. A Combined Metabolomic and Metagenomic Approach to Discriminate Raw Milk for the Production of Hard Cheese. Foods. 2021; 10(1):109. https://doi.org/10.3390/foods10010109
Chicago/Turabian StyleBellassi, Paolo, Gabriele Rocchetti, Marco Nocetti, Luigi Lucini, Francesco Masoero, and Lorenzo Morelli. 2021. "A Combined Metabolomic and Metagenomic Approach to Discriminate Raw Milk for the Production of Hard Cheese" Foods 10, no. 1: 109. https://doi.org/10.3390/foods10010109
APA StyleBellassi, P., Rocchetti, G., Nocetti, M., Lucini, L., Masoero, F., & Morelli, L. (2021). A Combined Metabolomic and Metagenomic Approach to Discriminate Raw Milk for the Production of Hard Cheese. Foods, 10(1), 109. https://doi.org/10.3390/foods10010109