

MicroRNA Signatures in Dental Pulp Stem Cells Following Nicotine Exposure



Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Wound Healing Assay
2.3. Cell Differentiation
2.4. Quantitative PCR
2.5. MicroRNA Array
2.6. Bioinformatics Analysis
2.7. Data Analysis
3. Results
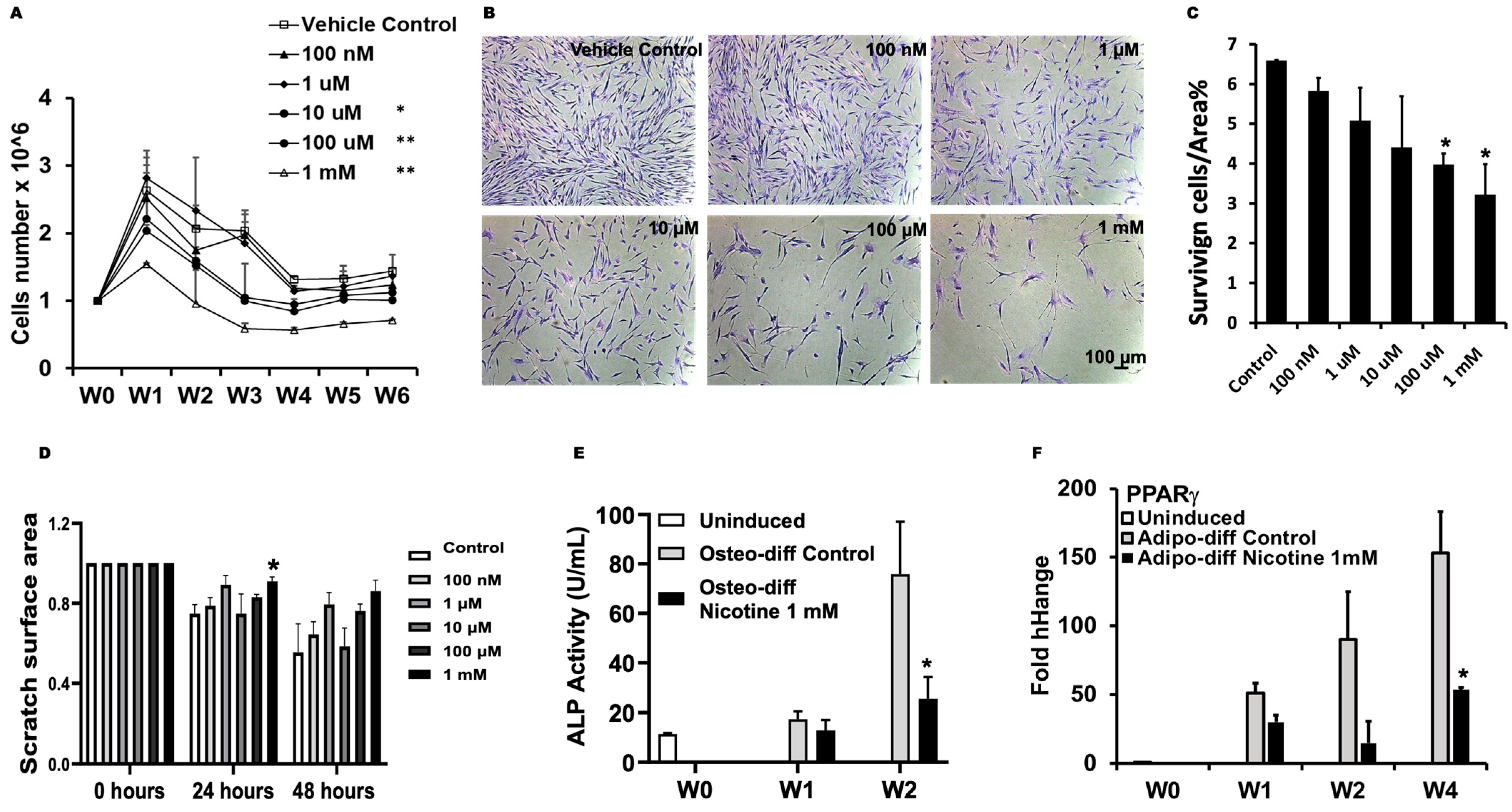
3.1. Nicotine Impaired Regeneration of DPSCs
3.2. Nicotine Induced Distinctive miRNA Expression
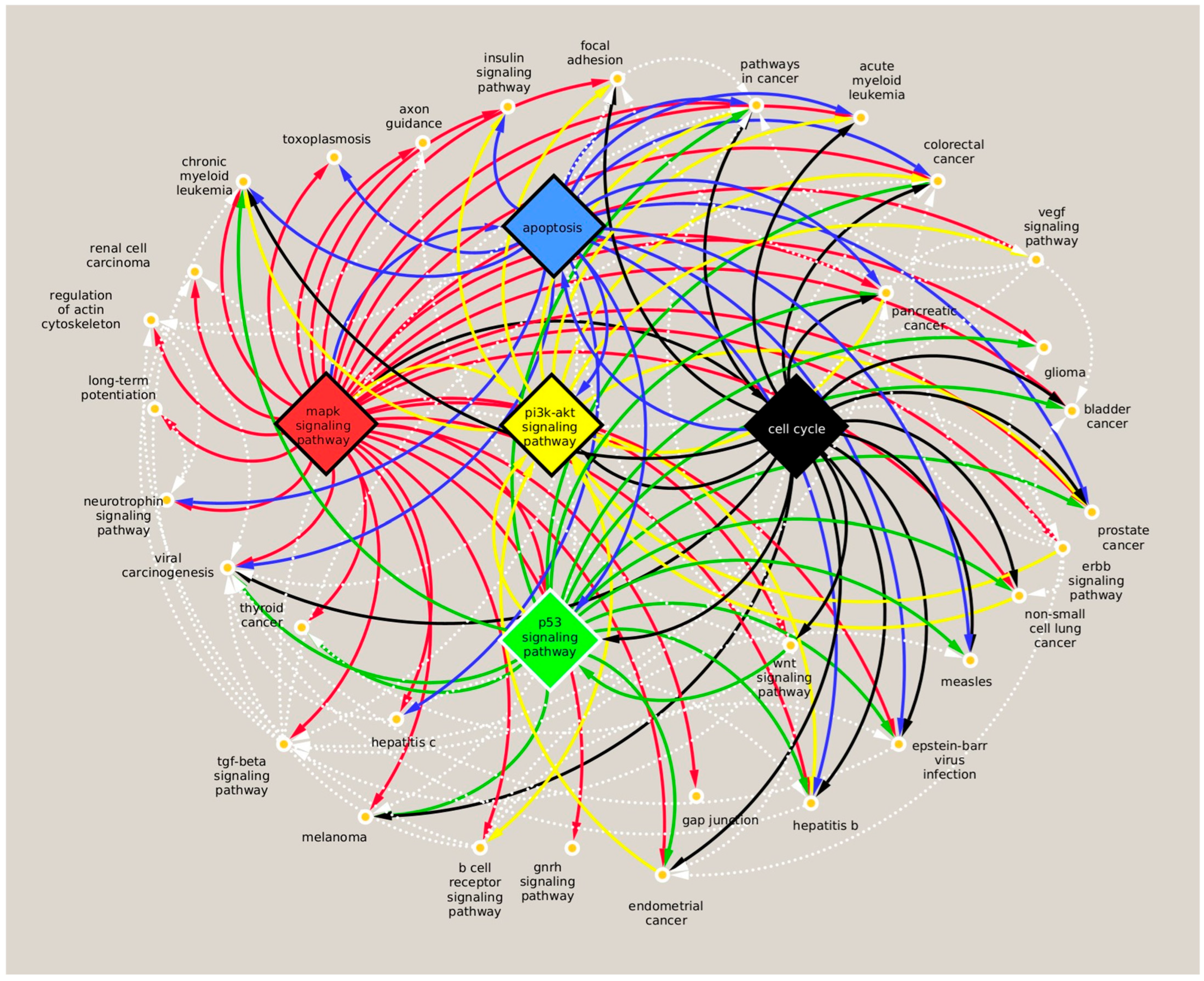
3.3. Analysis of miRNA Targets and Associated Pathway Induced by Nicotine
3.4. Nicotine Induced Unique Molecular Signatures in DPSCs in Comparison to CSC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bilano, V.; Gilmour, S.; Moffiet, T.; D’ESpaignet, E.T.; Stevens, G.A.; Commar, A.; Tuyl, F.; Hudson, I.; Shibuya, K. Global trends and projections for tobacco use, 1990-2025: An analysis of smoking indicators from the WHO Comprehensive Information Systems for Tobacco Control. Lancet 2015, 385, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Taghavi, S.; Khashyarmanesh, Z.; Moalemzadeh-Haghighi, H.; Nassirli, H.; Eshraghi, P.; Jalali, N.; Hassanzadeh-Khayyat, M. Nicotine content of domestic cigarettes, imported cigarettes and pipe tobacco in iran. Addict. Health 2012, 4, 28–35. [Google Scholar] [PubMed]
- Goniewicz, M.L.; Kuma, T.; Gawron, M.; Knysak, J.; Kosmider, L. Nicotine levels in electronic cigarettes. Nicotine Tob. Res. 2013, 15, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-L. FDA summary of adverse events on electronic cigarettes. Nicotine Tob. Res. 2013, 15, 615–616. [Google Scholar] [CrossRef] [PubMed]
- Messner, B.; Frotschnig, S.; Steinacher-Nigisch, A.; Winter, B.; Eichmair, E.; Gebetsberger, J.; Schwaiger, S.; Ploner, C.; Laufer, G.; Bernhard, D. Apoptosis and necrosis: Two different outcomes of cigarette smoke condensate-induced endothelial cell death. Cell Death Dis. 2012, 3, e424. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-H.; An, Y.-J.; Jo, S.; Lee, S.J.; Choi, S.-J.; Lee, K. Comparison of volatile organic compounds between cigarette smoke condensate (CSC) and extract (CSE) samples. Environ. Health Toxicol. 2018, 33, e2018012. [Google Scholar] [CrossRef] [PubMed]
- National Center for Chronic Disease Prevention and Health Promotion (US) Office on Smoking and Health. The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2014. Available online: http://www.ncbi.nlm.nih.gov/books/NBK179276/ (accessed on 28 March 2011).
- Li, T.; Zhang, J.; Zhang, J.; Zhang, N.; Zeng, Y.; Tang, S.; Tao, Z.; Qu, X.; Jia, J.; Zhu, W.; et al. Nicotine-enhanced stemness and epithelial-mesenchymal transition of human umbilical cord mesenchymal stem cells promote tumor formation and growth in nude mice. Oncotarget 2017, 9, 591–606. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-S.; Kim, S.-J.; Kim, H.-J.; Lee, S.-J.; Park, Y.-J.; Lee, J.; You, H.-K. Effects of nicotine on proliferation and osteoblast differentiation in human alveolar bone marrow-derived mesenchymal stem cells. Life Sci. 2012, 90, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Qin, Y.; Chen, H.; Bu, Q.; Li, Y.; Zhong, Q.; Han, X.; Chen, J.; Yu, P.; Liu, G. Effects of nicotine on proliferation and survival in human umbilical cord mesenchymal stem cells. J. Biochem. Mol. Toxicol. 2014, 28, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, B.; Dong, Z.; Liu, F.; Zhang, Y.; Yu, Y.; Shang, F.; Wu, L.; Wang, X.; Jin, Y.; et al. Nicotine deteriorates the osteogenic differentiation of periodontal ligament stem cells through α7 nicotinic acetylcholine receptor regulating Wnt pathway. PLoS ONE 2013, 8, e83102. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, R.; Cerny, L.M.; Torday, J.S.; Rehan, V.K. Mechanism for nicotine-induced up-regulation of Wnt signaling in human alveolar interstitial fibroblasts. Exp. Lung Res. 2011, 37, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Li, T.-Q.; Yan, Y.-E.; Magdalou, J.; Wang, H.; Chen, L.-B. Effect of nicotine on chondrogenic differentiation of rat bone marrow mesenchymal stem cells in alginate bead culture. Bio-Med. Mater. Eng. 2012, 22, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Madhoun, A.A.; Sindhu, S.; Haddad, D.; Atari, M.; Ahmad, R.; Al-Mulla, F. Dental Pulp Stem Cells Derived From Adult Human Third Molar Tooth: A Brief Review. Front. Cell Dev. Biol. 2021, 9, 717624. [Google Scholar] [CrossRef] [PubMed]
- Senthilkumar, S.; Maiya, K.; Jain, N.K.; Mata, S.; Mangaonkar, S.; Prabhu, P.; Rai, K.S.; Kutty, B.M.; Dhanushkodi, A. Reversal of Neuropsychiatric Comorbidities in an Animal Model of Temporal Lobe Epilepsy Following Systemic Administration of Dental Pulp Stem Cells and Bone Marrow Mesenchymal Stem Cells. Curr. Gene Ther. 2023, 23, 198–214. [Google Scholar] [CrossRef] [PubMed]
- Ferrarotti, F.; Romano, F.; Gamba, M.N.; Quirico, A.; Giraudi, M.; Audagna, M.; Aimetti, M. Human Intrabony Defect Regeneration with Micrografts Containing Dental Pulp Stem Cells: A Randomized Controlled Clinical Trial. J. Clin. Periodontol. 2018, 45, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Hardin, L.T.; Vang, D.; Thor, D.; Han, X.; Mashkoor, F.; Alpagot, T.; Ojcius, D.; Xiao, N. Cigarette smoking exposure disrupts the regenerative potential of dental pulp stem cells. Tob. Induc. Dis. 2023, 21, 101. [Google Scholar] [CrossRef] [PubMed]
- Hardin, L.T.; Abid, N.; Vang, D.; Han, X.; Thor, D.; Ojcius, D.M.; Xiao, N. miRNAs mediate impact of smoking on dental pulp stem cells via p53 pathway. Toxicol. Sci. 2024, 200, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Gangaraju, V.K.; Lin, H. MicroRNAs: Key regulators of stem cells. Nat. Rev. Mol. Cell Biol. 2009, 10, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Minadakis, G.; Christodoulou, K.; Tsouloupas, G.; Spyrou, G.M. PathIN: An integrated tool for the visualization of pathway interaction networks. Comput. Struct. Biotechnol. J. 2023, 21, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, Y.; Chen, L.; Liu, W.; Lin, C.; Chen, Y.; Wang, X. HGF Aggravated Periodontitis-Associated Gut Barrier and Microbial Dysfunction: Implications for Oral-Gut Axis Regulation. Biology 2025, 14, 496. [Google Scholar] [CrossRef] [PubMed]
- Yuki, D.; Kikuchi, A.; Miura, N.; Kakehi, A.; Onozawa, M. Good relationship between saliva cotinine kinetics and plasma cotinine kinetics after smoking one cigarette. Regul. Toxicol. Pharmacol. 2013, 67, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.A.; Jarvis, M.; Iyer, R.; Feyerabend, C. Relation of nicotine yield of cigarettes to blood nicotine concentrations in smokers. BMJ 1980, 280, 972–976. [Google Scholar] [CrossRef] [PubMed]
- West, K.A.; Brognard, J.; Clark, A.S.; Linnoila, I.R.; Yang, X.; Swain, S.M.; Harris, C.; Belinsky, S.; Dennis, P.A. Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J. Clin. Investig. 2003, 111, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, H.; Kobune, M.; Kikuchi, S.; Yoshida, M.; Murata, M.; Murase, K.; Iyama, S.; Takada, K.; Sato, T.; Ono, K.; et al. Extracellular vesicle miR-7977 is involved in hematopoietic dysfunction of mesenchymal stromal cells via poly(rC) binding protein 1 reduction in myeloid neoplasms. Haematologica 2016, 101, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Horiguchi, H.; Kikuchi, S.; Iyama, S.; Ikeda, H.; Goto, A.; Kawano, Y.; Murase, K.; Takada, K.; Miyanishi, K.; et al. miR-7977 inhibits the Hippo-YAP signaling pathway in bone marrow mesenchymal stromal cells. PLoS ONE 2019, 14, e0213220. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Chen, L.; Yu, M.; Tao, J.; Liu, J.; Wang, Y.; Pan, H.; Zhou, W.; Wang, S. miR-3178 inhibits cell proliferation and metastasis by targeting Notch1 in triple-negative breast cancer. Cell Death Dis. 2018, 9, 1059. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, K.; Mei, Y.; Huang, X.; Li, Z.; Yang, Q.; Yang, H. Sp1 Suppresses miR-3178 to Promote the Metastasis Invasion Cascade via Upregulation of TRIOBP. Mol. Ther.-Nucleic Acids 2018, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-J.; Cai, Z.-W.; Yang, H.-F.; Tang, X.-D.; Fang, X.; Qiu, L.; Wang, F.; Chen, X.-L. A Next-Generation Sequencing of Plasma Exosome-Derived microRNAs and Target Gene Analysis with a Microarray Database of Thermally Injured Skins: Identification of Blood-to-Tissue Interactions at Early Burn Stage. J. Inflamm. Res. 2021, ume 14, 6783–6798. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, A.; Francescato, G.; Ferrero, G.; Birolo, G.; Tarallo, S.; Francavilla, A.; Piaggeschi, G.; Battista, C.D.; Gallo, G.; Luc, A.R.; et al. The 8q24 region hosts miRNAs altered in biospecimens of colorectal and bladder cancer patients. Cancer Med. 2023, 12, 5859–5873. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, S.; Hu, Y.; Wang, N.; Wu, L.; Ding, M. Role of PI3K/AKT Signaling Pathway in Proliferation, Migration and Odontogenic Differentiation of Human Dental Pulp Stem Cells. J. Hard Tissue Biol. 2020, 29, 99–104. [Google Scholar] [CrossRef]
- Lv, T.; Wu, Y.; Mu, C.; Liu, G.; Yan, M.; Xu, X.; Wu, H.; Du, J.; Yu, J.; Mu, J. Insulin-like growth factor 1 promotes the proliferation and committed differentiation of human dental pulp stem cells through MAPK pathways. Arch. Oral Biol. 2016, 72, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Han, S.; Kwon, C.S.; Lee, D. Biogenesis and regulation of the let-7 miRNAs and their functional implications. Protein Cell 2016, 7, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, J.; Xu, C.; Tang, S.; Ren, H. The insights of Let-7 miRNAs in oncogenesis and stem cell potency. J. Cell. Mol. Med. 2016, 20, 1779–1788. [Google Scholar] [CrossRef] [PubMed]
- Kurniawati, I.; Liu, M.-C.; Hsieh, C.-L.; Do, A.D.; Sung, S.-Y. Targeting Castration-Resistant Prostate Cancer Using Mesenchymal Stem Cell Exosomes for Therapeutic MicroRNA-let-7c Delivery. Front. Biosci. 2022, 27, 256. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Lu, Y.; Wang, Y.; Du, L.; Zhang, Y.; Tao, J. Let-7c regulates proliferation and osteodifferentiation of human adipose-derived mesenchymal stem cells under oxidative stress by targeting SCD-1. Am. J. Physiol. Physiol. 2019, 316, C57–C69. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xu, Y.; Lu, S.; Gao, Y.; Deng, Y. Bone mesenchymal stem cell extracellular vesicles delivered miR let-7-5p alleviate endothelial glycocalyx degradation and leakage via targeting ABL2. Cell Commun. Signal. 2023, 21, 205. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Gao, J.; Chen, M.; Sun, Y.; Qiao, X.; Mao, H.; Guo, L.; Yu, Y.; Yang, D. Let-7a promotes periodontal bone regeneration of bone marrow mesenchymal stem cell aggregates via the Fas/FasL-autophagy pathway. J. Cell. Mol. Med. 2023, 27, 4056–4068. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Han, T.; Guo, R.; Song, P.; Liu, Y.; Wu, Z.; Ai, J.; Shen, C. Micro-RNA let-7a-5p Derived From Mesenchymal Stem Cell-Derived Extracellular Vesicles Promotes the Regrowth of Neurons in Spinal-Cord-Injured Rats by Targeting the HMGA2/SMAD2 Axis. Front. Mol. Neurosci. 2022, 15, 850364. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Lai, D.; Tian, Y.; Lai, F.; Long, M.; Ji, C.; Hao, G. MicroRNA hsa-let-7e-5p in hUC-MSC-EVs alleviates oral mucositis by targeting TAB2. Scand. J. Immunol. 2024, 100, e13399. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Sun, G.; Ye, P.; Li, S.; Shi, Y. MicroRNA let-7d regulates the TLX/microRNA-9 cascade to control neural cell fate and neurogenesis. Sci. Rep. 2013, 3, 1329. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, C.; Fukuda, T.; Kawakami, K.; Toyoda, M.; Nakao, Y.; Watanabe, Y.; Shinjo, T.; Sano, T.; Iwashita, M.; Yotsumoto, K.; et al. miR-1260b inhibits periodontal bone loss by targeting ATF6β mediated regulation of ER stress. Front. Cell Dev. Biol. 2022, 10, 1061216. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shan, B.; Zhao, H.; He, H.; Tian, M.; Cheng, X.; Qin, J.; Jin, G. MiR-130a-3p regulates neural stem cell differentiation in vitro by targeting Acsl4. J. Cell. Mol. Med. 2022, 26, 2717–2727. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Guo, S.; Tong, S.; Sun, X. Exosomal miR-130a-3p regulates osteogenic differentiation of Human Adipose-Derived stem cells through mediating SIRT7/Wnt/β-catenin axis. Cell Prolif. 2020, 53, e12890. [Google Scholar] [CrossRef] [PubMed]
- Camp, E.; Pribadi, C.M.P.; Anderson, P.J.; Zannettino, A.C.W.; Gronthos, S. miRNA-376c-3p Mediates TWIST-1 Inhibition of Bone Marrow-Derived Stromal Cell Osteogenesis and Can Reduce Aberrant Bone Formation of TWIST-1 Haploinsufficient Calvarial Cells. Stem Cells Dev. 2018, 27, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Y.; Yan, R.; Xu, X.; Gao, L.; Mei, J.; Liu, J.; Wang, X.; Zhang, J.; Wu, P.; et al. miR-377-3p regulates adipogenic differentiation of human bone marrow mesenchymal stem cells by regulating LIFR. Mol. Cell. Biochem. 2018, 449, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Wang, J.; Sun, Y.; Zhao, T.; Luo, X.; Lu, J.; Hou, W.; Yu, X.; Xue, L.; Yan, Y.; et al. MiR-222-3p suppresses C2C12 myoblast proliferation and differentiation via the inhibition of IRS-1/PI3K/Akt pathway. J. Cell. Biochem. 2023, 124, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Tan, S.; Wang, H.; Zhu, J.; Li, J.; Zhang, P.; Wang, M.; Zhang, F.; Liu, B. Mesenchymal Stem Cell-Derived Exosomal miRNA-222-3p Increases Th1/Th2 Ratio and Promotes Apoptosis of Acute Myeloid Leukemia Cells. Anal. Cell. Pathol. 2023, 2023, 4024887. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, X.; Wang, J.; Zhang, Y.; Ye, Q.; Wang, Y.; Fei, D.; Wang, Q. Inflammatory Periodontal Ligament Stem Cells Drive M1 Macrophage Polarization via Exosomal miR-143-3p-Mediated Regulation of PI3K/AKT/NF-κB Signaling. Stem Cells 2023, 41, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wang, M.; Ruan, Z.; Zhu, L.; Tang, C. Mesenchymal stem cell-derived exosomal miR-143-3p suppresses myocardial ischemia-reperfusion injury by regulating autophagy. Life Sci. 2021, 280, 119742. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lin, H.; Nie, W.; Wan, J.; Jiang, Q.; Zhang, A.; Zhang, P.-Y. Exosomal miR-221-3p Derived from Bone Marrow Mesenchymal Stem Cells Alleviates Asthma Progression by Targeting FGF2 and Inhibiting the ERK1/2 Signaling Pathway. Evid.-Based Complement. Altern. Med. 2022, 2022, 5910874. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Xu, W.; Liang, Z. Bone marrow mesenchymal stem cell-derived exosomal miR-221-3p promotes angiogenesis and wound healing in diabetes via the downregulation of forkhead box P1. Diabet. Med. 2024, 41, e15386. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Chen, Y.; Guo, Y.; Wang, X.; Chen, D. Hsa-miR-22-3p Inhibits Liver Cancer Cell EMT and Cell Migration/Invasion by Indirectly Regulating SPRY2. PLoS ONE 2023, 18, e0281536. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, C. Exosomes miR-22-3p Derived from Mesenchymal Stem Cells Suppress Colorectal Cancer Cell Proliferation and Invasion by Regulating RAP2B and PI3K/AKT Pathway. J. Oncol. 2021, 2021, 3874478. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Upregulated Nicotine vs. Vehicle | Downregulated Nicotine vs. Vehicle |
|---|---|
| hsa-miR-4497 | hsa-miR-376c-3p |
| hsa-miR-7977 | hsa-miR-377-3p |
| hsa-miR-3178 | hsa-miR-222-3p |
| hsa-miR-1260b | hsa-miR-130a-3p |
| hsa-miR-10400-5p | hsa-miR-143-3p |
| hsa-let-7e-5p | hsa-miR-127-3p |
| hsa-let-7a-5p | hsa-miR-221-3p |
| hsa-let-7d-5p | hsa-miR-12136 |
| hsa-let-7c-5p | hsa-miR-22-3p |
| Upregulated Nicotine vs. CSC | Downregulated Nicotine vs. CSC |
|---|---|
| hsa-miR-199b-5p | hsa-miR-10395-5p |
| hsa-miR-1260b | hsa-miR-222-3p |
| hsa-miR-7977 | hsa-miR-221-3p |
| hsa-miR-30b-5p | hsa-miR-29b-3p |
| hsa-miR-26a-5p | hsa-miR-4321 |
| hsa-miR-26b-5p | |
| hsa-miR-199a-3p | |
| hsa-miR-199a-5p |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vang, D.; Tahrani Hardin, L.; Abid, N.; Thor, D.; Xiao, N. MicroRNA Signatures in Dental Pulp Stem Cells Following Nicotine Exposure. Dent. J. 2025, 13, 338. https://doi.org/10.3390/dj13080338
Vang D, Tahrani Hardin L, Abid N, Thor D, Xiao N. MicroRNA Signatures in Dental Pulp Stem Cells Following Nicotine Exposure. Dentistry Journal. 2025; 13(8):338. https://doi.org/10.3390/dj13080338
Chicago/Turabian StyleVang, David, Leyla Tahrani Hardin, Nabil Abid, Der Thor, and Nan Xiao. 2025. "MicroRNA Signatures in Dental Pulp Stem Cells Following Nicotine Exposure" Dentistry Journal 13, no. 8: 338. https://doi.org/10.3390/dj13080338
APA StyleVang, D., Tahrani Hardin, L., Abid, N., Thor, D., & Xiao, N. (2025). MicroRNA Signatures in Dental Pulp Stem Cells Following Nicotine Exposure. Dentistry Journal, 13(8), 338. https://doi.org/10.3390/dj13080338