
Tris-{Hydridotris(1-pyrazolyl)borato}lanthanide Complexes: Synthesis, Spectroscopy, Crystal Structure and Bonding Properties



Abstract
1. Introduction
2. Results and Discussion
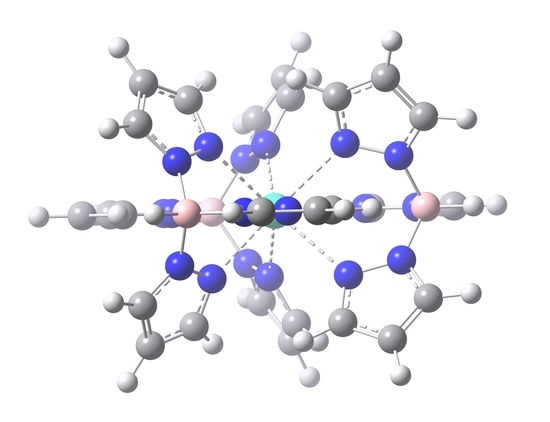
2.1. Crystal and Molecular Structures
2.2. Bonding Analysis
3. Materials and Methods
3.1. Materials
3.2. X-Ray Diffraction
3.3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Trofimenko, S. Boron-Pyrazole Chemistry. J. Am. Chem. Soc. 1966, 88, 1842–1844. [Google Scholar] [CrossRef]
- Trofimenko, S. Recent advances in poly(pyrazolyl)borate (scorpionate) chemistry. Chem. Rev. 1993, 93, 943–980. [Google Scholar] [CrossRef]
- Trofimenko, S. Skorpionates—The Coordination Chemistry of Pyrazolylborate Ligands; World Scientific Publishing: London, UK, 1999. [Google Scholar]
- Marques, N.; Sella, A.; Takats, J. Chemistry of the lanthanides using pyrazolylborate ligands. Chem. Rev. 2002, 102, 2137–2159. [Google Scholar] [CrossRef]
- Pettinari, C. Scorpionates II—Chelating Borate Ligands; Imperial College Press: London, UK, 2008. [Google Scholar]
- Apostolidis, C.; Rebizant, J.; Kanellakopulos, B.; Ammon, R.v.; Dornberger, E.; Müller, J.; Powietzka, B.; Nuber, B. Homoscorpionates (hydridotris(1-pyrazolyl)borato complexes) of the trivalent 4f ions. The crystal and molecular structure of [(HB(N2C3H3)3]3LnIII, (Ln = Pr, Nd). Polyhedron 1997, 16, 1057–1068. [Google Scholar] [CrossRef]
- Apostolidis, C.; Rebizant, J.; Walter, O.; Kanellakopulos, B.; Reddmann, H.; Amberger, H.-D. Electronic structures of highly symmetrical compounds of f elements. 35 [1]—Crystal and molecular structure of tris(hydrotris(1-pyrazolyl)borato)lanthanide(III) (LnTp3; Ln = La, Eu), and electronic structure of EuTp3. Z. Anorg. Allg. Chem. 2002, 628, 2013–2025. [Google Scholar] [CrossRef]
- Reddmann, H.; Apostolidis, C.; Walter, O.; Rebizant, J.; Amberger, H.-D. Electronic structures of highly symmetrical compounds of f elements. 38 [1] Crystal, molecular and electronic structure of tris(hydrotris(1-pyrazolyl) borato)samarium(III). Z. Anorg. Allg. Chem. 2005, 631, 1487–1496. [Google Scholar] [CrossRef]
- Stainer, M.V.R.; Takats, J. X-ray Crystal and Molecular Structure of Tris[hydridotris(pyrazol-1-yl)borato]ytterbium(III), Yb(HBPz3)3. Inorg. Chem. 1982, 21, 4050–4053. [Google Scholar] [CrossRef]
- Seminara, A.; Musumeci, A. Absorption and Emission Spectra of Neodymium(III) and Europium(III) Complexes*. Inorg. Chim. Acta 1984, 95, 291–307. [Google Scholar] [CrossRef]
- Amberger, H.-D.; Reddmann, H.; Apostolidis, C.; Kanellakopulos, B. Electronic structures of highly symmetrical compounds of f elements. 36 [1]: Parametric analysis of the optical spectra of an oriented tris(hydrotris(1-pyrazolyl)borato)praseodymium(III) single crystal. Z. Anorg. Allg. Chem. 2003, 629, 147–160. [Google Scholar] [CrossRef]
- Faltynek, R.A. Lanthanide Coordination Chemistry: Spectroscopic Properties of Terbium and Europium Poly(Pyrazol-1-YL)- and Poly(Imidazol-1-Yl)Borate Complexes. J. Coord. Chem. 1989, 20, 73–80. [Google Scholar] [CrossRef]
- Miranda, P., Jr.; Aricó, E.M.; Máduar, M.F.; Matos, J.R.; De Carvalho, C.A.A. Study of the thermal decomposition of the Nd(III), Eu(III) and Tb(III) scorpionates. J. Alloys Comp. 2002, 344, 105–109. [Google Scholar] [CrossRef]
- Apostolidis, C.; Kovács, A.; Walter, O.; Colineau, E.; Griveau, J.-C.; Morgenstern, A.; Rebizant, J.; Caciuffo, R.; Panak, P.J.; Rabung, T.; et al. Tris-{hydridotris(1-pyrazolyl)borato}actinide Complexes: Synthesis, Spectroscopy, Crystal Structure, Bonding Properties and Magnetic Behaviour. Chem. Eur. J. 2020, 26, 11293–11306. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. On the calculation of bonding energies by the Hartree Fock Slater method. I. The transition state method. Theor. Chim. Acta 1977, 46, 1–10. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Shannon, R.D.; Prewitt, C.T. Revised values of effective ionic radii. Acta Cryst. 1970, B26, 1046–1048. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M.; Baerends, E.J. Kohn-Sham Density Functional Theory: Predicting and Understanding Chemistry. In Reviews in Computational Chemistry; Lipkowitz, K.B., Boyd, D.B., Eds.; Wiley-VCH: New York, NY, USA, 2000; Volume 15, pp. 1–86. [Google Scholar]
- Hopffgarten, M.V.; Frenking, G. Energy decomposition analysis. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 43–62. [Google Scholar] [CrossRef]
- Hayton, T.W.; Kaltsoyannis, N. Organometallic Actinide Complexes with Novel Oxidation States and Ligand Types. In Experimental and Theoretical Approaches to Actinide Chemistry; Gibson, J.K., de Jong, W.A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018; pp. 181–236. [Google Scholar]
- Kaltsoyannis, N. Transuranic Computational Chemistry. Chem. Eur. J. 2018, 24, 2815–2825. [Google Scholar] [CrossRef]
- Kerridge, A. Quantification of f-element covalency through analysis of the electron density: Insights from simulation. Chem. Commun. 2017, 53, 6685–6695. [Google Scholar] [CrossRef]
- Dognon, J.-P. Theoretical insights into the chemical bonding in actinide complexes. Coord. Chem. Rev. 2014, 266–267, 110–122. [Google Scholar] [CrossRef]
- Jones, M.B.; Gaunt, A.J.; Gordon, J.C.; Kaltsoyannis, N.; Neu, M.P.; Scott, B.L. Uncovering f-element bonding differences and electronic structure in a series of 1:3 and 1:4 complexes with a diselenophosphinate ligand. Chem. Sci. 2013, 4, 1189–1203. [Google Scholar] [CrossRef]
- Schnaars, D.D.; Gaunt, A.J.; Hayton, T.W.; Jones, M.B.; Kirker, I.; Kaltsoyannis, N.; May, I.; Reilly, S.D.; Scott, B.L.; Wu, G. Bonding trends traversing the tetravalent actinide series: Synthesis, structural, and computational analysis of AnIV(Aracnac)4 complexes (An = Th, U, Np, Pu; Aracnac = ArNC(Ph)CHC(Ph)O; Ar = 3,5-tBu2C6H3). Inorg. Chem. 2012, 51, 8557–8566. [Google Scholar] [CrossRef]
- Kerridge, A. Oxidation state and covalency in f-element metallocenes (M = Ce, Th, Pu): A combined CASSCF and topological study. Dalton Trans. 2013, 42, 16428–16436. [Google Scholar] [CrossRef]
- Kerridge, A. f-Orbital covalency in the actinocenes (An = Th – Cm): Multiconfigurational studies and topological analysis. RSC Adv. 2014, 4, 12078–12086. [Google Scholar] [CrossRef]
- Huang, Q.R.; Kingham, J.R.; Kaltsoyannis, N. The strength of actinide-element bonds from the quantum theory of atoms-in-molecules. Dalton Trans. 2015, 44, 2554–2566. [Google Scholar] [CrossRef] [PubMed]
- Kaltsoyannis, N. Covalency hinders AnO2(H2O)+ → AnO(OH)2+ isomerisation (An = Pa – Pu). Dalton Trans. 2016, 45, 3158–3162. [Google Scholar] [CrossRef] [PubMed]
- Gupta, T.; Velmurugan, G.; Rajeshkumar, T.; Rajaraman, G. Role of Lanthanide-Ligand bonding in the magnetization relaxation of mononuclear single-ion magnets: A case study on Pyrazole and Carbene ligated LnIII (Ln = Tb, Dy, Ho, Er) complexes. J. Chem. Sci. 2016, 128, 1615–1630. [Google Scholar] [CrossRef]
- Calhorda, M.J.; Costa, P.J. Structure, bonding and reactivity of seven-coordinate allylic Mo(II) and W(II) complexes. Coord. Chem. Rev. 2017, 344, 83–100. [Google Scholar] [CrossRef]
- Wu, Q.Y.; Cheng, Z.P.; Lan, J.H.; Wang, C.Z.; Chai, Z.F.; Gibson, J.K.; Shi, W.Q. Insight into the nature of M-C bonding in the lanthanide/actinide-biscarbene complexes: A theoretical perspective. Dalton Trans. 2018, 47, 12718–12725. [Google Scholar] [CrossRef]
- Carlotto, S.; Sambi, M.; Rancan, M.; Casarin, M. Theoretical Investigation of the Electronic Properties of Three Vanadium Phthalocyaninato (Pc) Based Complexes: PcV, PcVO, and PcVI. Inorg. Chem. 2018, 57, 1859–1869. [Google Scholar] [CrossRef]
- Kovács, A.; Apostolidis, C.; Walter, O. Comparative study of complexes of rare earths and actinides with 2,6-bis(1,2,4-triazin-3-yl)pyridine. Inorganics 2019, 7, 26. [Google Scholar] [CrossRef]
- Kovács, A.; Konings, R.J.M. Thermodynamic Properties of the Lanthanide(III) Halides. In Handbook on the Physics and Chemistry of Rare Earths; Gschneidner, K.A., Jr., Bünzli, J.-C., Pecharsky, V., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; Volume 33. [Google Scholar]
- Konings, R.J.M.; Beneš, O.; Kovács, A.; Manara, D.; Sedmidubský, D.; Gorokhov, L.; Iorish, V.S.; Yungman, V.; Shenyavskaya, E.; Osina, E. The thermodynamic properties of the f-elements and their compounds. Part II. The Lanthanide and Actinide Oxides. J. Phys. Chem. Ref. Data 2014, 43, 013101. [Google Scholar] [CrossRef]
- Kovács, A.; Apostolidis, C.; Walter, O.; Lindqvist-Reis, P. ‘Lanthanide contraction’ in [Ln(BTP)3](CF3SO3)3 complexes. Struct. Chem. 2015, 26, 1287–1295. [Google Scholar] [CrossRef]
- Kovács, A. Structure and bonding of lanthanide dinitrogen complexes, Ln(N2)1-8. Int. J. Quantum. Chem. 2020, 120, e26051. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Fryer-Kanssen, I.; Austin, J.; Kerridge, A. Topological Study of Bonding in Aquo and Bis(triazinyl)pyridine Complexes of Trivalent Lanthanides and Actinides: Does Covalency Imply Stability? Inorg. Chem. 2016, 55, 10034–10042. [Google Scholar] [CrossRef]
- Peters, J.A.; Djanashvili, K.; Geraldes, C.F.G.C.; Platas-Iglesias, C. The chemical consequences of the gradual decrease of the ionic radius along the Ln-series. Coord. Chem. Rev. 2020, 406. [Google Scholar] [CrossRef]
- SMART, SAINT, SADABS; Siemens, Analytical X-ray Instruments Inc.: Karlsruhe, Germany, 1997.
- APEX2, SAINT-Plus, SADABS, Programs for Data Collection, Integration and Absorption Correction; Bruker AXS Inc.: Madison, WI, USA, 2007.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Soltek, R.; Huttner, G. Winray-32; University of Heidelberg: Heidelberg, Germany, 1998. [Google Scholar]
- Enraf-Nonius SDP-Plus Structure Determination Package; Enraf-Nonius: Delft, The Netherlands, 1987.
- Amsterdam Density Functional Package; SCM Theoretical Chemistry, Vrije Universiteit: Amsterdam, The Netherlands, 2020.
- te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J. Optimized Slater-type basis sets for the elements 1-118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.J.; Köster, A.M.; Ayers, P.W.; Santos-Valle, A.; Vela, A.; Merino, G. An efficient grid-based scheme to compute QTAIM atomic properties without explicit calculation of zero-flux surfaces. J. Comput. Chem. 2009, 30, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.J.; Bader, R.F.W.; Ayers, P.W.; Michel, C.; Götz, A.W.; Bo, C. A high performance grid-based algorithm for computing QTAIM properties. Chem. Phys. Lett. 2009, 472, 149–152. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CeTp3 | NdTp3 | SmTp3 | TbTp3 |
|---|---|---|---|---|
| Formula | C4.5H5B0.5N3M0.16 | |||
| Formula weight | 129.87 | 130.56 | 131.58 | 133.01 |
| Temperature | 100(2) K | 100(2) K | 100(2) K | 200(2) K |
| Wavelength | 0.71073 Å | |||
| Crystal system | hexagonal | |||
| Space group | P63/m | |||
| Unit cell dimensions | a = 11.735(2) Å, c = 13.595(3) Å | a = 11.7325(4) Å, c = 13.5550(7) Å | a = 11.692(1) Å, c = 13.569(2) Å | a = 11.699(1) Å, c = 13.628(3) Å |
| Volume | 1621.5(6) Å3 | 1615.9(1) Å3 | 1606.3(4) Å3 | 1615.6(5) Å3 |
| Z | 12 | |||
| Density (calc.) | 1.596 Mg/m3 | 1.610 Mg/m3 | 1.632 Mg/m3 | 1.640 Mg/m3 |
| Abs. coefficient | 1.456 mm−1 | 1.659 mm−1 | 1.880 mm−1 | 2.241 mm−1 |
| F(000) | 782 | 786 | 790 | 796 |
| Crystal size (mm3) | 0.11 × 0.07 × 0.03 | 0.13 × 0.08 × 0.07 | 0.05 × 0.04 × 0.04 | 0.2 × 0.25 × 0.10 |
| θ range | 2.004 to 28.650° | 2.004 to 28.493° | 2.011 to 28.364° | 2.010 to 28.341° |
| Index ranges | −15 ≤ h ≤ 15, −15 ≤ k ≤ 15, −17 ≤ l ≤ 18 | −14 ≤ h ≤ 15, −15 ≤ k ≤ 15, −17 ≤ l ≤ 17 | −15 ≤ h ≤ 15, −11 ≤ k ≤ 15, −17 ≤ l ≤ 16 | −15 ≤ h ≤ 15, −15 ≤ k ≤ 15, −17 ≤ l ≤ 17 |
| Reflections collected: independent: | 29,215 1424 [R(int) = 0.07021] | 29,802 1392 [R(int) = 0.0395] | 20,385 1365 [R(int) = 0.0633] | 16,998 1392 [R(int) = 0.0486] |
| Observed [I > 2σ(I)] | 1323 | 1313 | 1204 | 1249 |
| Coverage (θ = 25°) | 100% | 100% | 100% | 100% |
| Data/restraints/parameters | 1424/0/108 | 1392/0/108 | 1365/0/108 | 1392/0/108 |
| Goof on F2 | 1.119 | 1.080 | 1.068 | 1.080 |
| R indices [I > 2σ(I)] | R1 = 0.0209 | R1 = 0.0179 | R1 = 0.0232 | R1 = 0.0207 |
| R indices (all data) | wR2 = 0.448 | wR2 = 0.0428 | wR2 = 0.0488 | wR2 = 0.0387 |
| Largest peak/hole | 0.412/−0.489 e.Å−3 | 0.766/−0.541 e.Å−3 | 0.566/−0.450 e.Å−3 | 0.329/−0.420 e.Å−3 |
| Ln | M–Napical | M–Nequatorial | ||
|---|---|---|---|---|
| XRD | DFT | XRD | DFT | |
| La | 2.659(6) b | 2.663 | 2.80(1) b | 2.807 |
| Ce | 2.615(1) | 2.637 | 2.777(2) | 2.789 |
| Pr | 2.609(6) c | 2.612 | 2.783(6) c | 2.784 |
| Nd | 2.589(1), 2.599(7) c | 2.594 | 2.763(2), 2.804(7) c | 2.778 |
| Sm | 2.565(2), 2.540(6) d | 2.562 | 2.754(2), 2.753(7) d | 2.773 |
| Eu | 2.533(5) b | 2.544 | 2.76(1) b | 2.784 |
| Gd | 2.518(5) | 2.535 | 2.750(7) | 2.770 |
| Tb | 2.523(2) | 2.520 | 2.757(2) | 2.764 |
| Energy | La(CN9) | Gd(CN9) | Lu(CN9) | Lu(CN8) |
|---|---|---|---|---|
| ΔVelst | −4060.6 | −4175.6 | −4204.3 | −4205.1 |
| ΔEoi | −1723.4 | −1907.7 | −1994.2 | −2035.3 |
| ΔEPauli | 702.2 | 656.4 | 609.3 | 701.3 |
| ΔEint | −5081.7 | −5426.9 | −5589.0 | −5539.2 |
| ΔVelst/(ΔVelst + ΔEoi) (%) | 70.2 | 68.6 | 67.8 | 67.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Apostolidis, C.; Kovács, A.; Morgenstern, A.; Rebizant, J.; Walter, O. Tris-{Hydridotris(1-pyrazolyl)borato}lanthanide Complexes: Synthesis, Spectroscopy, Crystal Structure and Bonding Properties. Inorganics 2021, 9, 44. https://doi.org/10.3390/inorganics9060044
Apostolidis C, Kovács A, Morgenstern A, Rebizant J, Walter O. Tris-{Hydridotris(1-pyrazolyl)borato}lanthanide Complexes: Synthesis, Spectroscopy, Crystal Structure and Bonding Properties. Inorganics. 2021; 9(6):44. https://doi.org/10.3390/inorganics9060044
Chicago/Turabian StyleApostolidis, Christos, Attila Kovács, Alfred Morgenstern, Jean Rebizant, and Olaf Walter. 2021. "Tris-{Hydridotris(1-pyrazolyl)borato}lanthanide Complexes: Synthesis, Spectroscopy, Crystal Structure and Bonding Properties" Inorganics 9, no. 6: 44. https://doi.org/10.3390/inorganics9060044
APA StyleApostolidis, C., Kovács, A., Morgenstern, A., Rebizant, J., & Walter, O. (2021). Tris-{Hydridotris(1-pyrazolyl)borato}lanthanide Complexes: Synthesis, Spectroscopy, Crystal Structure and Bonding Properties. Inorganics, 9(6), 44. https://doi.org/10.3390/inorganics9060044