Metal-Rich Metallaboranes: Synthesis, Structures and Bonding of Bi- and Trimetallic Open-Faced Cobaltaboranes



Abstract
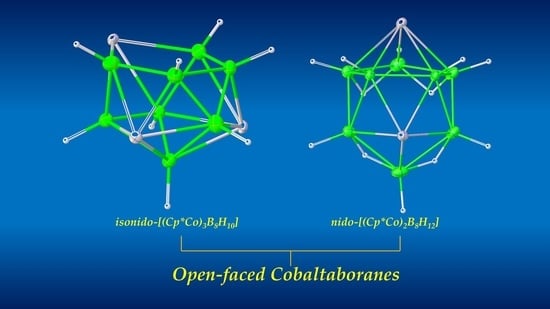
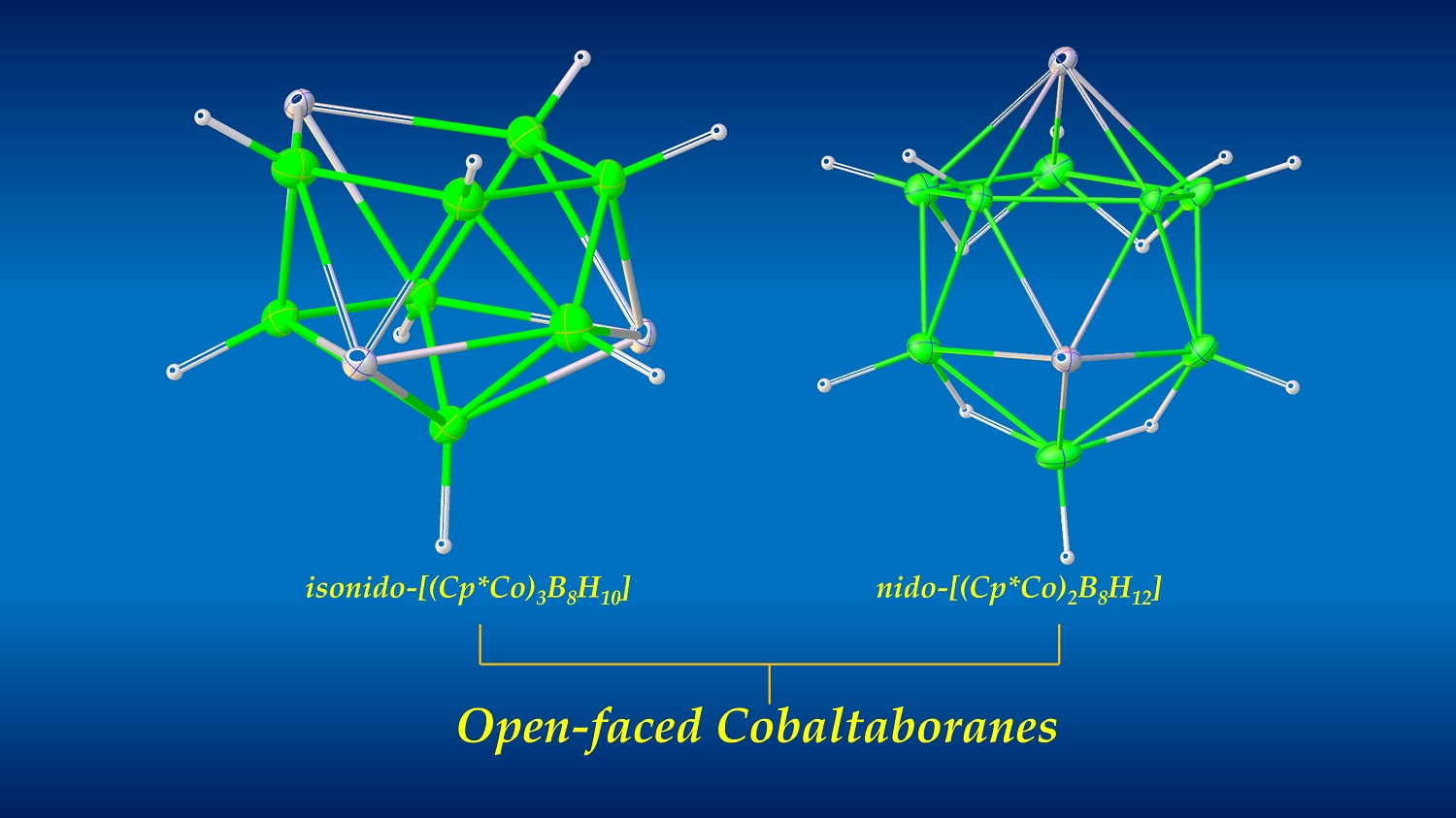
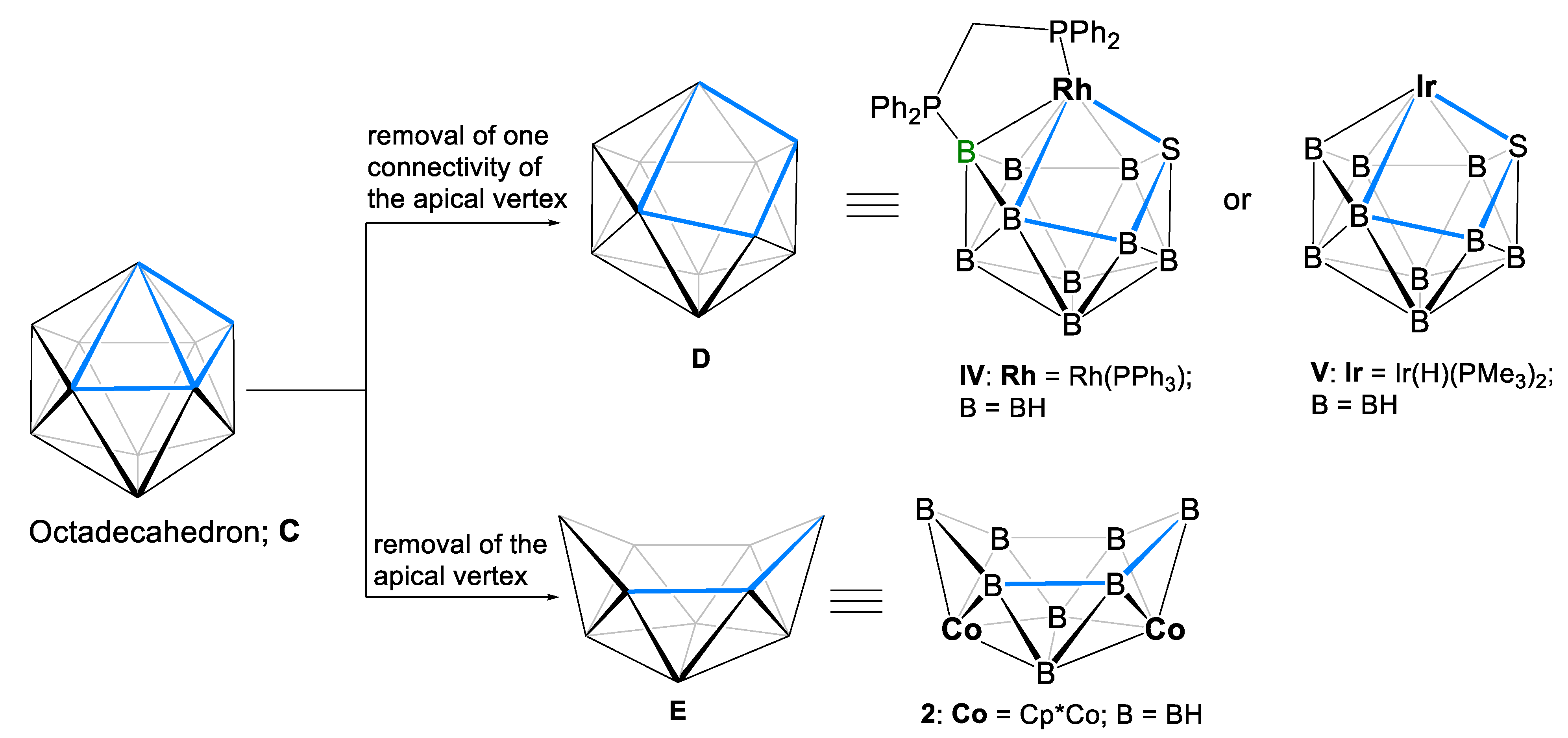
1. Introduction
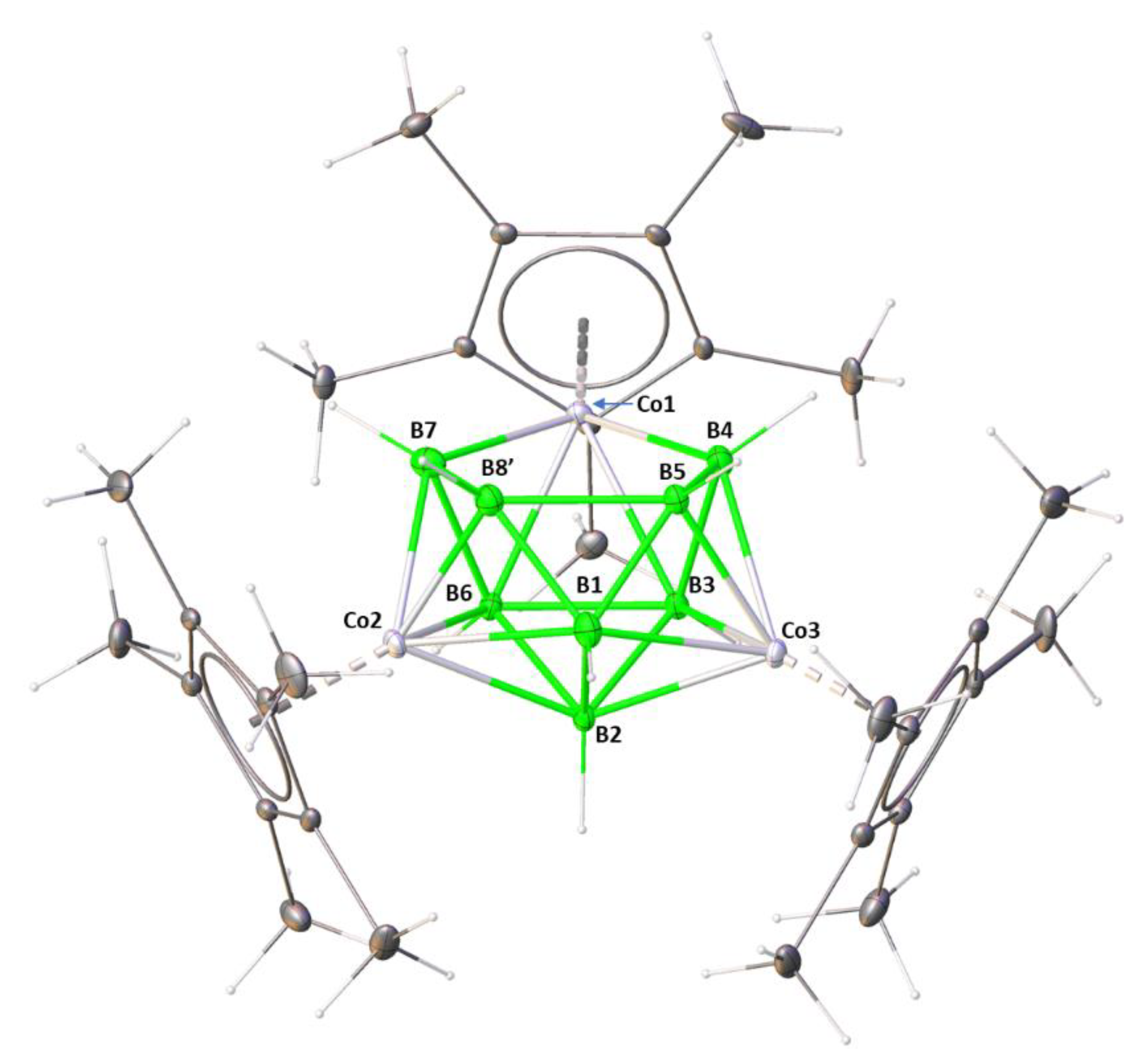
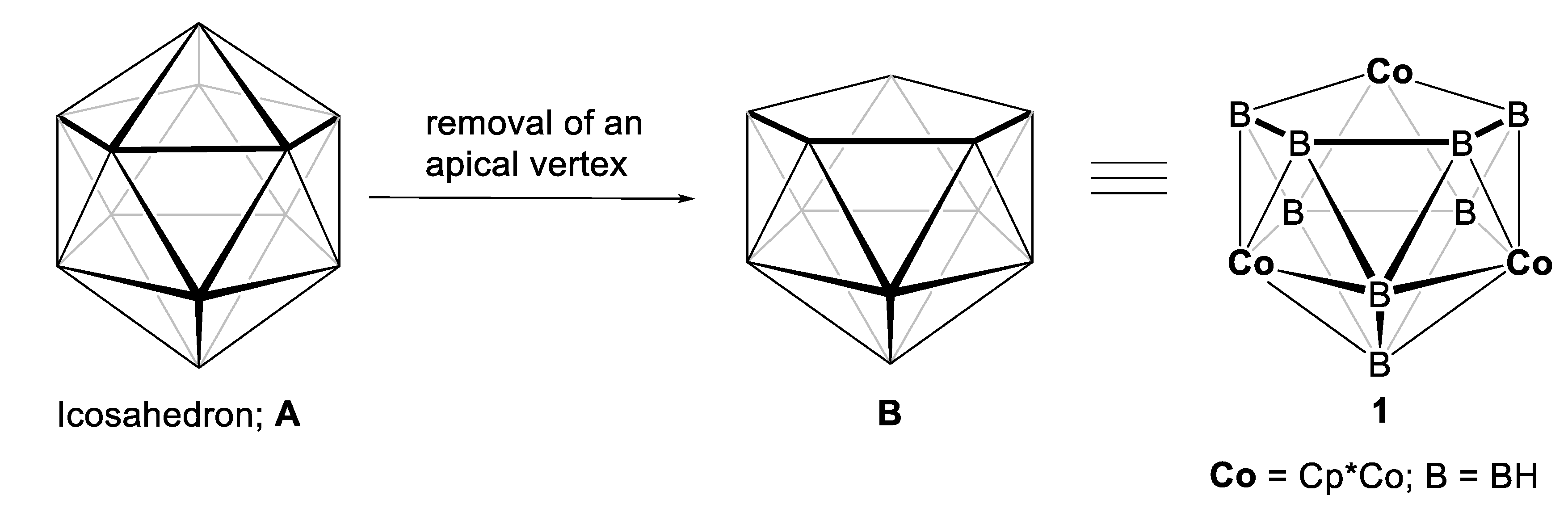
2. Results and Discussion
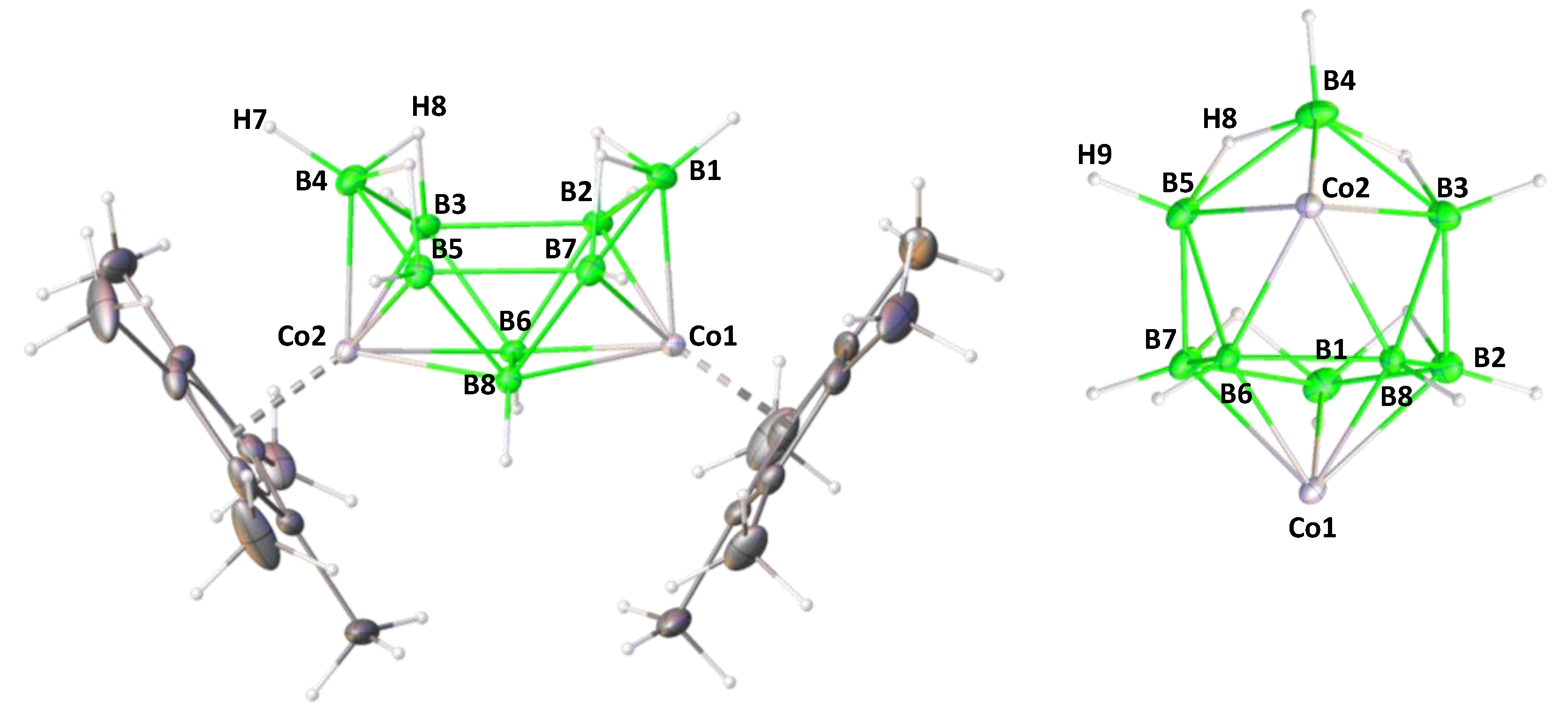
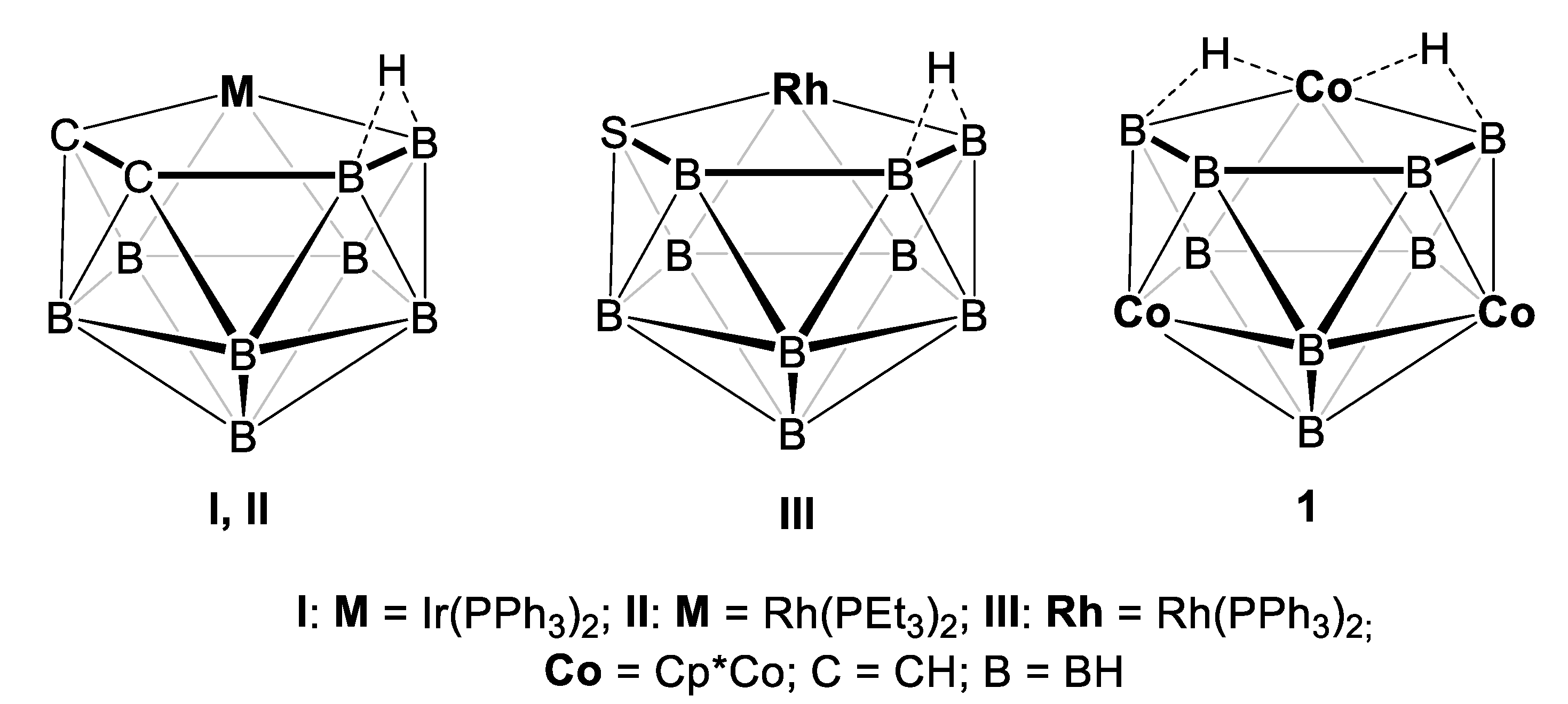
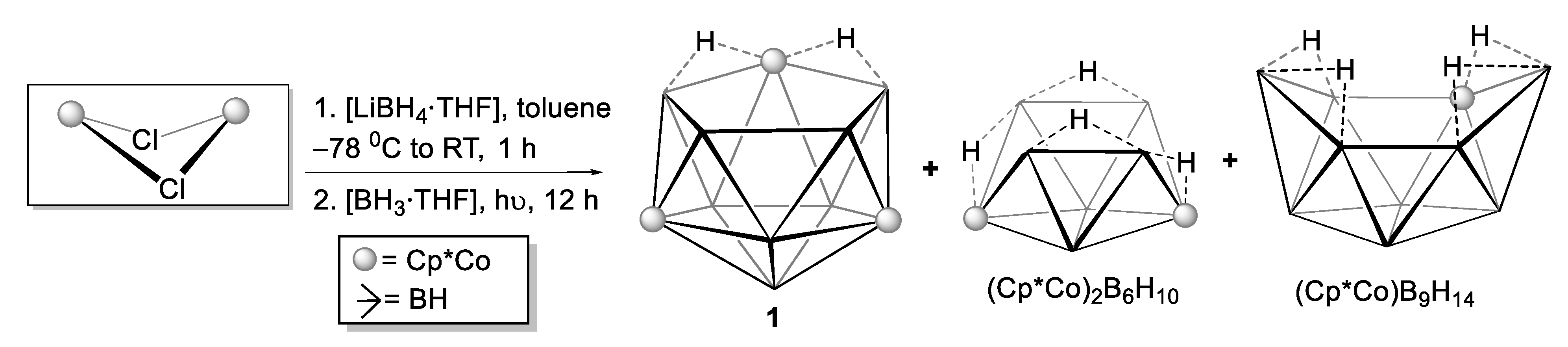
2.1. Reaction of (Cp*CoCl)2 with [LiBH4·THF] and [BH3·THF]
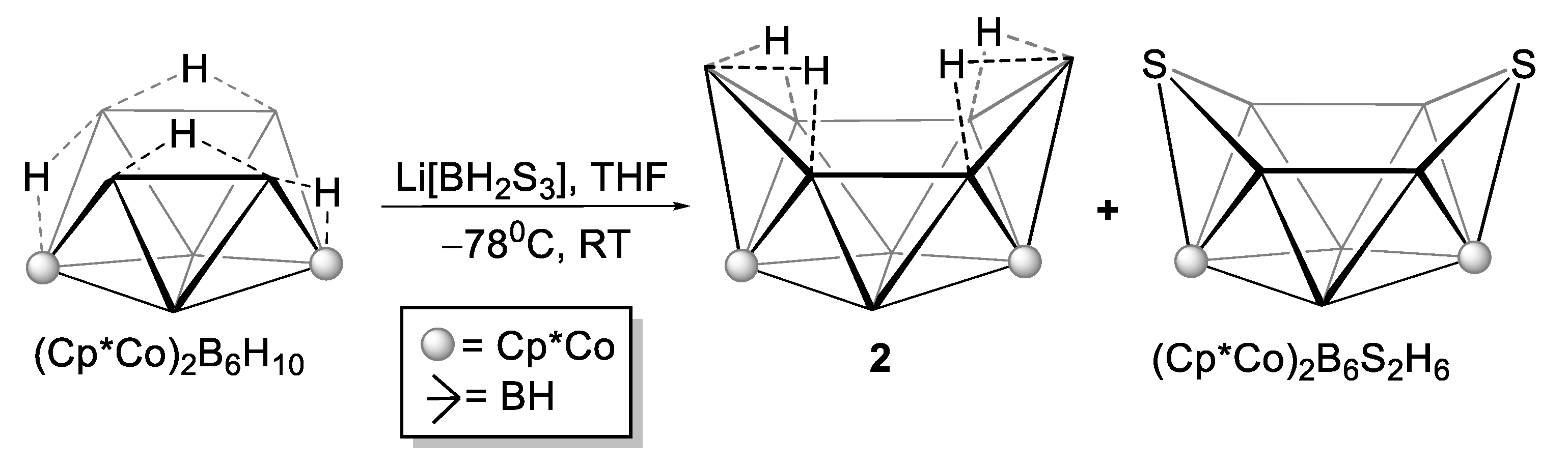
2.2. Reactivity of [(Cp*Co)2B6H12] with Li[BH2S3]
3. Materials and Methods
3.1. General Methods and Instrumentation
3.2. Single Crystal X-ray Diffraction Analysis
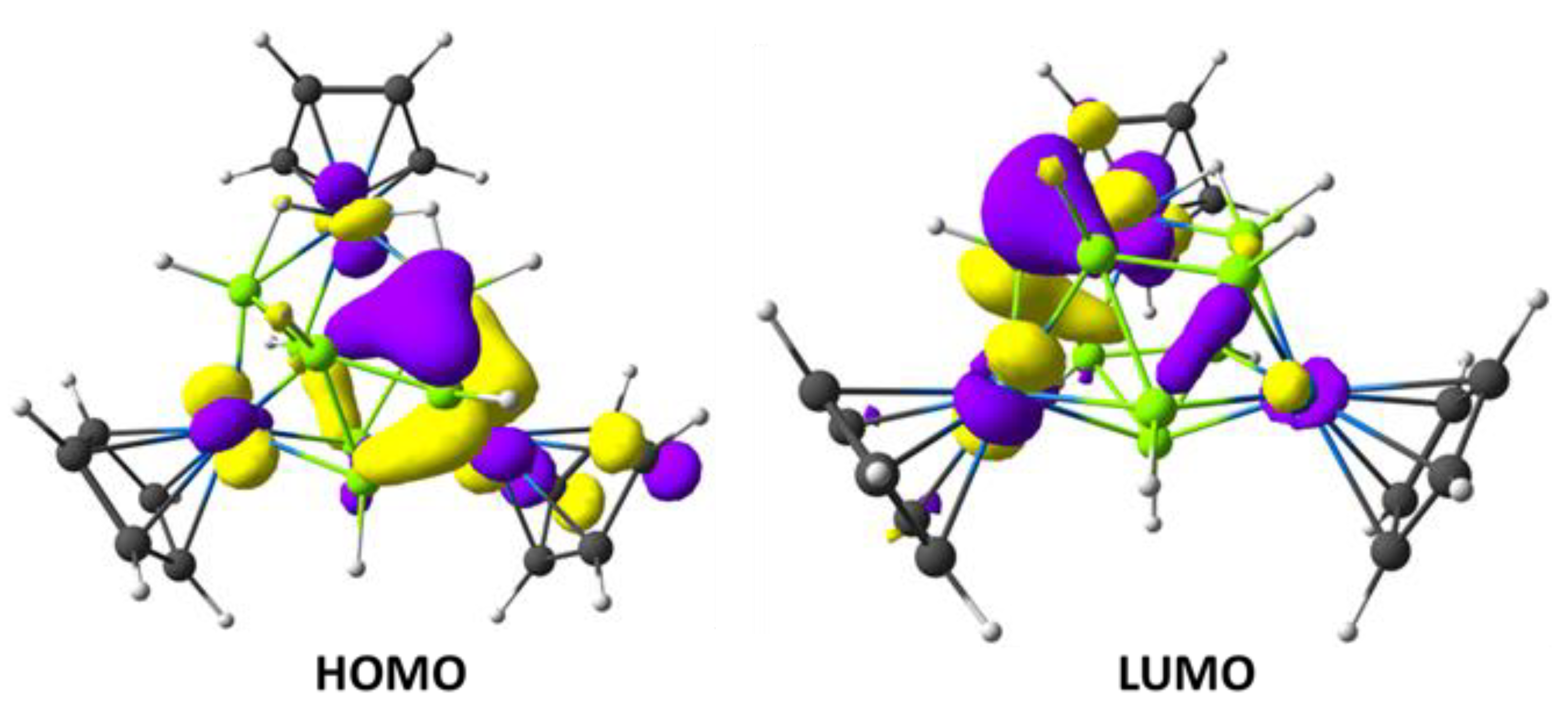
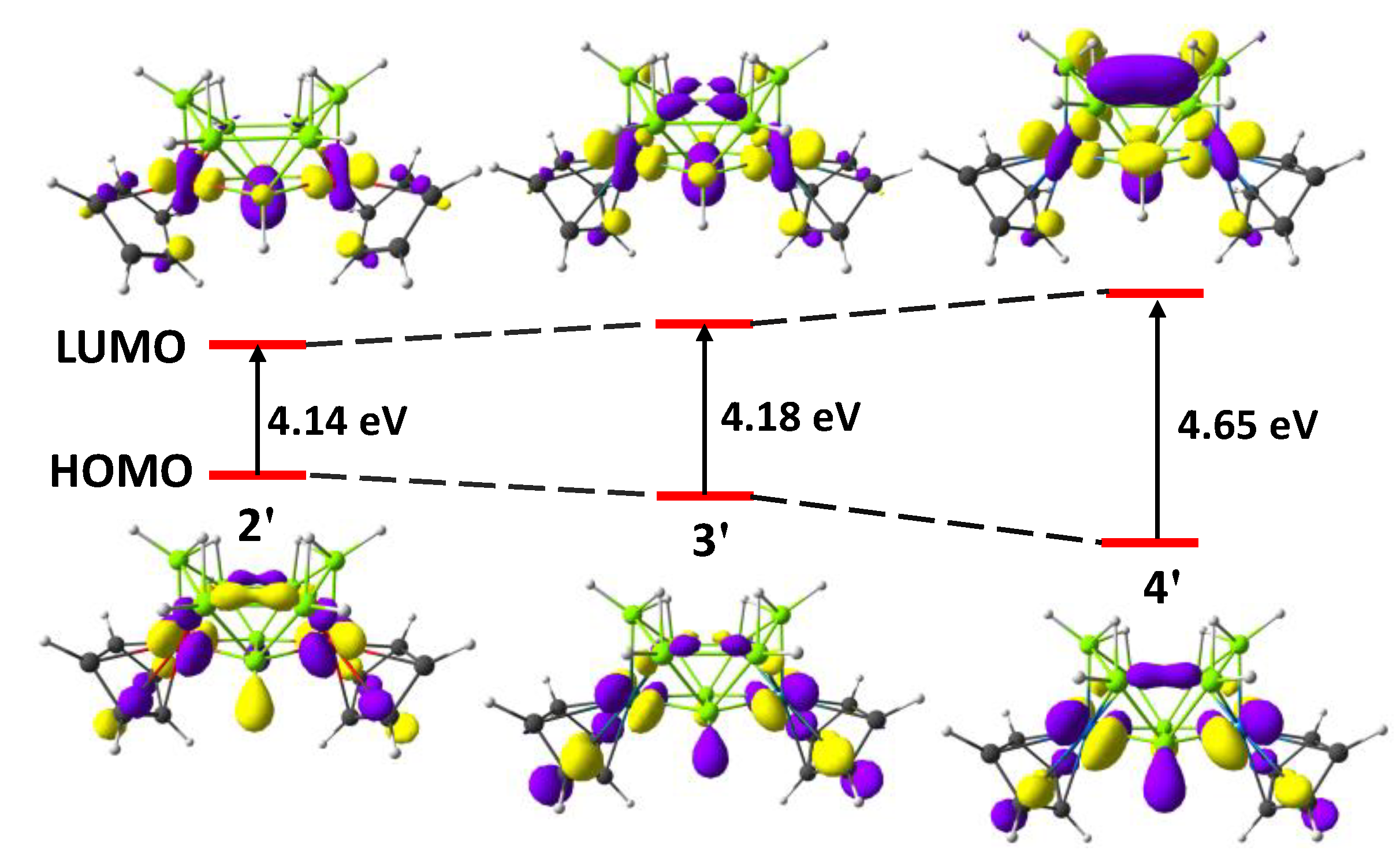
3.3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lipscomb, W.L. Boron Hydrides; Benjamin: New York, NY, USA, 1963. [Google Scholar]
- Muetterties, E.L. Boron Hydride Chemistry; Academic Press: New York, NY, USA, 1975. [Google Scholar]
- Grimes, R.N. Carboranes, 3rd ed.; Elsevier: Oxford, UK, 2016. [Google Scholar]
- King, R.B. Three-Dimensional Aromaticity in Polyhedral Boranes and Related Molecules. Chem. Rev. 2001, 101, 1119–1152. [Google Scholar] [CrossRef]
- Jemmis, E.D. Overlap control and stability of polyhedral molecules. closo-Carboranes. J. Am. Chem. Soc. 1982, 104, 7017–7020. [Google Scholar] [CrossRef]
- Hosmane, N.S.; Maguire, J.A. Metallacarboranes of d- and f-Block Metals. In Comprehensive Organometallic Chemistry III; Mingos, D.M.P., Crabtree, R.H., Eds.; Pergamon: New York, NY, USA, 2007; Chapter 3.05; Volume 3, pp. 175–264. [Google Scholar]
- Hosmane, N.S. Boron Science: New Technologies and Applications; CRC Press, Taylor and Francis Group: Boca Raton, FL, USA, 2011. [Google Scholar]
- Saxena, A.K.; Hosmane, N.S. Recent advances in the chemistry of carborane metal complexes incorporating d- and f-block elements. Chem. Rev. 1993, 93, 1081–1124. [Google Scholar] [CrossRef]
- Grimes, R.N. Transition Metal Metallacarbaboranes. In Comprehensive Organometallic Chemistry II; Abel, E.W., Stone, F.G.A., Wilkinson, G., Eds.; Pergamon Press: Oxford, UK, 1995; Volume 1, pp. 373–430. [Google Scholar]
- Housecroft, C.E. Boron Atoms in Transition Metal Clusters. Adv. Organomet. Chem. 1991, 33, 1–50. [Google Scholar]
- Fehlner, T.P.; Halet, J.-F.; Saillard, J.-Y. Molecular Clusters. A Bridge to Solid-State Chemistry; University Press: Cambridge, UK, 2007. [Google Scholar]
- Zhang, J.; Xie, Z. Synthesis, structure, and reactivity of 13- and 14-Vertex Carboranes. Acc. Chem. Res. 2014, 47, 1623–1633. [Google Scholar] [CrossRef]
- Hawthorne, M.F.; Dunks, G.B.; McKown, M.M. Probable formation of 13-atom polyhedral complexes containing B10C2H122− and cobalt. J. Am. Chem. Soc. 1971, 93, 2541–2543. [Google Scholar] [CrossRef]
- Evans, W.J.; Hawthorne, M.F. Synthesis of fourteen vertex metallocarboranes by polyhedral expansion. J. Chem. Soc. Chem. Commun. 1974, 38–39. [Google Scholar] [CrossRef]
- Burke, A.; Ellis, D.; Giles, B.T.; Hodson, B.E.; Macgregor, S.A.; Rosair, G.M.; Welch, A.J. Beyond the Icosahedron: The First 13-Vertex Carborane. Angew. Chem. Int. Ed. 2003, 42, 225–228. [Google Scholar] [CrossRef]
- Deng, L.; Chan, H.-S.; Xie, Z. Synthesis, Reactivity, and Structural Characterization of a 14-Vertex Carborane. Angew. Chem. Int. Ed. 2005, 44, 2128–2131. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Zhang, J.; Chan, H.-S.; Xie, Z. Synthesis and Structure of 14- and 15-Vertex Ruthenacarboranes. Angew. Chem. Int. Ed. 2006, 45, 4309–4313. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.K.; Bose, S.K.; Anju, R.S.; Mondal, B.; Ramkumar, V.; Ghosh, S. Boron beyond the Icosahedral Barrier: A 16-Vertex Metallaborane. Angew. Chem. Int. Ed. 2013, 52, 3222–3226. [Google Scholar] [CrossRef]
- Roy, D.K.; Mondal, B.; Shankhari, P.; Anju, R.S.; Geetharani, K.; Mobin, S.M.; Ghosh, S. Supraicosahedral Polyhedra in Metallaboranes: Synthesis and Structural Characterization of 12-, 15- and 16-Vertex Rhodaboranes. Inorg. Chem. 2013, 52, 6705–6712. [Google Scholar] [CrossRef]
- Roy, D.K.; Ghosh, S.; Halet, J.-F. Beyond the Icosahedron: The Quest for High-nuclearity Supraicosahedral Metallaboranes. J. Clust. Sci. 2014, 25, 225–237. [Google Scholar] [CrossRef]
- Onak, T. Polyhedral Carboranes. In Comprehensive Organometallic Chemistry II; Abel, E.W., Stone, F.G.A., Wilkinson, G., Eds.; Pergamon Press: Oxford, UK, 1995; Chapter 6; Volume 1, pp. 217–255. [Google Scholar]
- Hosmane, N.S.; Siebert, W. (Eds.) Advances in Boron Chemistry; Royal Society of Chemistry: Cambridge, UK, 1997; p. 349. [Google Scholar]
- Ghosh, S.; Rheingold, A.L.; Fehlner, T.P. Metallaboranes of the Earlier Transition Metals. An Arachno Nine-vertex, Nine-Skeletal Electron Pair Rhenaborane of Novel Shape: Importance of Total Vertex Connectivities in Such Systems. Chem. Commun. 2001, 10, 895–896. [Google Scholar] [CrossRef]
- Ghosh, S.; Lei, X.; Shang, M.; Fehlner, T.P. Role of the Transition Metal in Metallaborane Chemistry. Reactivity of [(Cp*ReH2)2B4H4] with BH3·THF, CO, and [Co2(CO)8]. Inorg. Chem. 2000, 39, 5373–5382. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Noll, B.C.; Fehlner, T.P. Borane Mimics of Classic Organometallic Compounds: [(Cp*Ru)B8H14(RuCp*)]0,+, Isoelectronic Analogues of Dinuclear Pentalene Complexes. Angew. Chem. Int. Ed. 2005, 44, 6568–6571. [Google Scholar] [CrossRef]
- Ghosh, S.; Fehlner, T.P.; Noll, B.C. Condensed metallaborane clusters: Synthesis and structure of Fe2(CO)6(η5-C5Me5RuCO)(η5-C5Me5Ru)B6H10. Chem. Commun. 2005, 3080–3082. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Noll, B.C.; Fehlner, T.P. Expansion of Iridaborane Clusters by Addition of Monoborane. Novel Metallaboranes and Mechanistic Detail. Dalton Trans. 2008, 3, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Geetharani, K.; Bose, S.K.; Pramanik, G.; Saha, T.K.; Ramkumar, V.; Ghosh, S. An Efficient Route to Group 6 and 8 Metallaborane Compounds: Synthesis of arachno-[Cp*Fe(CO)B3H8] and closo-[(Cp*M)2B5H9] (M = Mo, W). Eur. J. Inorg. Chem. 2009, 2009, 1483–1487. [Google Scholar] [CrossRef]
- Dhayal, R.S.; Sahoo, S.; Reddy, K.H.K.; Mobin, S.M.; Jemmis, E.D.; Ghosh, S. Vertex-Fused Metallaborane Clusters: Synthesis, Characterization and Electronic Structure of [(η5-C5Me5Mo)3MoB9H18]. Inorg. Chem. 2010, 49, 900–904. [Google Scholar] [CrossRef]
- Geetharani, K.; Bose, S.K.; Sahoo, S.; Varghese, B.; Mobin, S.M.; Ghosh, S. Cluster Expansion Reactions of Group 6 and 8 Metallaboranes Using Transition Metal Carbonyl Compounds of Groups 7-9. Inorg. Chem. 2011, 50, 5824–5832. [Google Scholar] [CrossRef] [PubMed]
- Chakrahari, K.K.V.; Thakur, A.; Mondal, B.; Dhayal, R.S.; Ramkumar, V.; Ghosh, S. A Close-packed Boron-rich 11-vertex Molybdaborane with Novel Geometry. J. Organomet. Chem. 2012, 710, 75–79. [Google Scholar] [CrossRef]
- Roy, D.K.; Barik, S.K.; Mondal, B.; Varghese, B.; Ghosh, S. A Novel Heterometallic μ9-Boride Cluster: Synthesis and Structural Characterization of [(η5-C5Me5Rh)2{Co6(CO)12}(μ-H)(BH)B]. Inorg. Chem. 2014, 53, 667–669. [Google Scholar] [CrossRef] [PubMed]
- De, A.; Zhang, Q.-F.; Mondal, B.; Cheung, L.F.; Kar, S.; Saha, K.; Varghese, B.; Wang, L.-S.; Ghosh, S. [(Cp2M)2B9H11] (M = Zr or Hf): Early Transition Metal ‘guarded’ Heptaborane with Strong Covalent and Electrostatic Bonding. Chem. Sci. 2018, 9, 1976–1981. [Google Scholar] [CrossRef] [PubMed]
- Bould, J.; Rath, N.P.; Barton, L. Metallaborane heteroatom incorporation reactions: Metallacarboranes, metallathiaboranes, and an iridaazaborane from iridanonaborane precursors. Organometallics 1996, 15, 4916–4929. [Google Scholar] [CrossRef]
- Ferguson, G.; Lough, A.J.; Faridoon; McGrath, M.N.; Spalding, T.R.; Kennedy, J.D.; Fontaine, X.L.R. Metallaheteroborane chemistry. Part 7. Synthesis, crystal structure, and characterisation of two dinuclear rhodatelluraboranes, [{(PPh3)2RhTeB10H10}2] and [(PPh3)(CO)Rh2Te2B20H20]. J. Chem. Soc. Dalton Trans. 1990, 6, 1831–1839. [Google Scholar] [CrossRef]
- Ferguson, G.; Gallagher, J.F.; McGrath, M.; Sheehan, J.P.; Spalding, T.R.; Kennedy, J.D. Metallaheteroborane chemistry. Part 11. Selective syntheses of the palladium heteroborane complexes [2,2-(PR3)2-closo-2,1-PdEB10H10] (R3 = Ph3, MePh2 or Me2Ph; E = Se or Te) and [2-X-2-(PPh3)-closo-2,1-PdTeB10H9(PPh3)] (X = Cl, Br, I, CN, SCN or O2CMe). J. Chem. Soc. Dalton Trans. 1993, 1, 27–34. [Google Scholar] [CrossRef]
- Norman, N.C.; Orpen, A.G.; Quayle, M.J.; Rice, C.R. Diborane(4) compounds incorporating thio- and seleno-carboranyl groups. New J. Chem. 2000, 24, 837–839. [Google Scholar] [CrossRef]
- Hammerschmidt, A.; Doch, M.; Putz, S.; Krebs, B. Na2[B18Se16]: The first 3D polymeric selenoborato-closo-dodecaborate. Z. Anorg. Allg. Chem. 2005, 631, 1125–1128. [Google Scholar] [CrossRef]
- Ferguson, G.; Gallagher, J.F.; Kennedy, J.D.; Kelleher, A.M.; Spalding, T.R. Pentahapto-bonded gold heteroborane clusters [3-(R3P)-closo-2,1-AuTeB10H10]− and [3-(R3P)-closo-3,1,2-AuAs2B9H9]−. Dalton Trans. 2006, 7, 2133–2139. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.K.; Geetharani, K.; Ramkumar, V.; Varghese, B.; Ghosh, S. Chemistry of vanadaboranes: Synthesis, structures and characterization of organ vanadium sulfide clusters with disulfide linkage. Inorg. Chem. 2010, 49, 2881–2888. [Google Scholar] [CrossRef]
- Ghosh, S.; Noll, B.C.; Fehlner, T.P. Synthesis and characterization of [exo-BH2(Cp*M)2B9H14] (M = Ru, Re), and the conversion of the ruthenaborane into [(Cp*Ru)2B10H16] with an open cluster framework based on a capped truncated tetrahedron. Angew. Chem. Int. Ed. 2005, 44, 2916–2918. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Shang, M.; Fehlner, T.P. Comparison of the geometric and molecular orbital structures of (Cp*Cr)2B4H8 and (Cp*Re)2B4H8, Cp* = η5-C5Me5. Structural consequences of delocalized electronic unsaturation in a metallaborane cluster. J. Organomet. Chem. 2000, 614, 92–98. [Google Scholar] [CrossRef]
- Geetharani, K.; Bose, S.K.; Varghese, B.; Ghosh, S. From metallaborane to borylene complexes: Syntheses and structures of triply bridged ruthenium and tantalum borylene complexes. Chem. Eur. J. 2010, 16, 11357–11366. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.K.; Ghosh, S.; Noll, B.C.; Halet, J.-F.; Saillard, J.-Y.; Vega, A. Linked and fused tungstaborane clusters: Synthesis, characterization and electronic structures of bis-{(η5-C5Me5W)2B5H8}2 and (η5-C5Me5W)2{Fe(CO)3}nB6-nH10-n, n = 0,1. Organometallics 2007, 26, 5377–5385. [Google Scholar] [CrossRef]
- Ghosh, S.; Lei, X.; Cahill, C.L.; Fehlner, T.P. Symmetrical scission of the coordinated tetraborane in (Cp*ReH2)B4H4 on CO addition and reassociation of the coordinated diboranes on H2 loss. Angew. Chem. Int. Ed. 2000, 39, 2900–2902. [Google Scholar] [CrossRef]
- Zafar, M.; Kar, S.; Nandi, C.; Ramalakshmi, R.; Ghosh, S. Cluster Fusion: Face-Fused Macropolyhedral Tetracobaltaboranes. Inorg. Chem. 2019, 58, 47–51. [Google Scholar] [CrossRef]
- Venable, T.L.; Sinn, E.; Grimes, R.N. Cobaltaborane analogs of decaborane (B10H14). Crystal and molecular structures of 6-[η5-C5(CH3)5]CoB9H13, 6,9-[η5-C5(CH3)5]2]2Co2B8H12, 5,7-[η5-C5(CH3)5]5]2Co2 B8H12, and 6-Cl-5,7-[η5-C5(CH3)5]2Co2B8H11]. Inorg. Chem. 1982, 21, 895–904. [Google Scholar] [CrossRef]
- Venable, T.L.; Grimes, R.N. (Pentamethylcyclopentadienyl)- cobaltaboranes derived from the octah dropentaborate (B5H8-) and tetradecahydrononabaorate (B9H14−) ions: Studies in synthesis and structure. Inorg. Chem. 1982, 21, 887–892. [Google Scholar] [CrossRef]
- Pipal, J.R.; Grimes, R.N. Crystal structure of a tetracobalt tetraboron cluster, (η5-C5H5)4Co4B4H4. Structural patterns in eight-vertex polyhedra. Inorg. Chem. 1979, 18, 257–263. [Google Scholar] [CrossRef]
- Chakrahari, K.K.; Sharmila, D.; Barik, S.K.; Mondal, B.; Varghese, B.; Ghosh, S. Hypoelectronic metallaboranes: Synthesis, structural characterization and electronic structures of metal-rich cobaltaboranes. J. Organomet. Chem. 2014, 749, 188–196. [Google Scholar] [CrossRef]
- Barik, S.K.; Roy, D.K.; Sharmila, D.; Ramalakshmi, R.; Chakrahari, K.K.; Mobin, S.M.; Ghosh, S. Synthesis, Characterization and Electronic Structures of Rh and Co analogs of Decaborane-14. Proc. Natl. Acad. Sci. India Sect. A 2014, 84, 121–130. [Google Scholar] [CrossRef]
- Barik, S.K.; Roy, D.K.; Ghosh, S. Chemistry of group 9 dimetallaborane analogues of octaborane(12). Dalton Trans. 2015, 44, 669–676. [Google Scholar] [CrossRef]
- Roy, D.K.; Anju, R.S.; Varghese, B.; Ghosh, S. Reactivity of dirhodium analogs of octaborane-12 and decaborane-14 towardstransition-metal moieties. Organometallics 2013, 32, 1964–1970. [Google Scholar] [CrossRef]
- Nandi, C.; Kar, S.; Zafar, M.; Kar, K.; Ghosh, S. Chemistry of Dimetallaoctaborane(12) with Chalcogen-Based Borate Ligands: Obedient versus Disobedient Clusters. Inorg. Chem. 2020, 59, 3537–3541. [Google Scholar] [CrossRef]
- Roy, D.K.; Bose, S.K.; Anju, R.S.; Ramkumar, V.; Ghosh, S. Synthesis and Structure of Dirhodium Analogue of Octaborane-12 and Decaborane-14. Inorg. Chem. 2012, 51, 10715–10722. [Google Scholar] [CrossRef]
- Stibr, B.; Holub, J.; Bakardjiev, M.; Hnyk, D.; Tok, O.L.; Milius, W.; Wrackmeyer, B. Phosphacarborane Chemistry: The Synthesis of the Parent Phosphadicarbaboranes nido-7,8,9-PC2B8H11 and [nido-7,8,9-PC2B8H10]−, and Their 10-Cl Derivatives-Analogs of the Cyclopentadienide Anion. Eur. J. Inorg. Chem. 2002, 2002, 2320–2326. [Google Scholar] [CrossRef]
- Volkov, O.; Macias, R.; Rath, N.P.; Barton, L. Phosphine−Boranes as Bidentate Ligands: Formation of [8,8-η2-{η2-(BH3)-dppm}-nido-8,7-RhSB9H10] and [9,9-η2-{η2-(BH3)-dppm}-nido-9,7,8-RhC2B8H11] from [8,8-(η2-dppm)-8-(η1-dppm)-nido-8,7-RhSB9H10] and [9,9-(η2-dppm)-9-(η1-dppm)-nido-9,7,8-RhC2B8H11], Respectively. Inorg. Chem. 2002, 41, 5837–5843. [Google Scholar] [PubMed]
- Dou, J.; Wu, L.; Guo, Q.; Li, D.; Wang, D. A New Strategy for the Preparation of Metallaboranes-Solvothermal Synthesis and Structural Characterisation of Two Nido 11-vertex Diplatinaundecaborane Clusters. Eur. J. Inorg. Chem. 2005, 2005, 63–65. [Google Scholar] [CrossRef]
- Mingos, D.M.P. Polyhedral skeletal electron pair approach. Acc. Chem. Res. 1984, 17, 311–319. [Google Scholar] [CrossRef]
- Jung, C.W.; Hawthorne, M.F. Eleven-Vertex Rhodium, Iridium, and Ruthenium Phosphinometallocarborane Complexes Formed from Sodium Undecahydro-5,6-dicarba-nido-decaborate (1-). J. Am. Chem. Soc. 1980, 102, 3024–3032. [Google Scholar] [CrossRef]
- Ferguson, G.; Jennings, M.C.; Lough, A.J.; Coughlan, S.; Spalding, T.R.; Kennedy, J.D.; Fontaine, X.L.R.; Stibr, B. Novel Rhodathiaborane Complexes Derived from [(PPh3)2RhSB9H10]. J. Chem. Soc. Chem. Commun. 1990, 12, 891–894. [Google Scholar] [CrossRef]
- Coughlan, S.; Spalding, T.R.; Ferguson, G.; Gallagher, J.F.; Lough, A.J.; Fontaine, X.L.R.; Kennedy, J.D.; Stıbr, B. Metallaheteroborane chemistry. Part 10. Synthesis and characterisation of closo-structured rhodathiaborane complexes [1-(CO)-1-L-3-L′-1,2-RhSB9H8](L = L′ = PPh3; L = PMe2Ph, L′ = PMe2Ph or PPh3). J. Chem. Soc. Dalton Trans. 1992, 19, 2865–2871. [Google Scholar] [CrossRef]
- Volkov, O.; Macias, R.; Rath, N.P.; Barton, L. Chemistry on a metallathiaborane cluster Part 4: Reactions of 11-vertex rhodathiaboranes with bidentate phosphines and their subsequent rearrangements. J. Organomet. Chem. 2002, 657, 40–47. [Google Scholar] [CrossRef]
- Volkov, O.; Rath, N.P.; Barton, L. Chemistry of a Metallathiaborane Cluster. 5. Reaction of [8,8-(η2-dppm)-8-(η1-dppm)-nido-8,7-RhSB9H10] and Its Derivatives with Organotransition-Metal Reagents. Organometallics 2002, 21, 5505–5514. [Google Scholar] [CrossRef]
- Bould, J.; Cunchillos, C.; Lahoz, F.J.; Oro, L.A.; Kennedy, J.D.; Macias, R. New Iridathiaboranes with Reversible Isonido–Nido Cluster Flexibility. Inorg. Chem. 2010, 49, 7353–7361. [Google Scholar] [CrossRef]
- Kasper, J.S.; Lucht, C.M.; Harker, D. The Crystal Structure of Decaborane, B10H14. Acta Cryst. 1950, 3, 436–455. [Google Scholar] [CrossRef]
- Kölle, U.; Khouzami, F.; Fuss, B. Bridged C5Me5CoII Complexes—Reactive Intermediates in the Cyclopentadienylation of Cobalt(II) Halides. Angew. Chem. Int. Ed. 1982, 21, 131–132. [Google Scholar] [CrossRef]
- Yoshino, T.; Ikemoto, H.; Matsunaga, S.; Kanai, M.A. Cationic High-Valent Cp*CoIII Complex for the Catalytic Generation of Nucleophilic Organometallic Species: Directed C–H Bond Activation. Angew. Chem. Int. Ed. 2013, 52, 2207–2211. [Google Scholar] [CrossRef] [PubMed]
- Lalancette, J.M.; Frêche, A.; Monteux, R. Reductions with sulfurated borohydrides. I. Preparation of sulfurated borohydrides. Can. J. Chem. 1968, 46, 2754–2757. [Google Scholar] [CrossRef]
- Lalancette, J.M.; Arnac, M. Reductions with sulfurated borohydrides. III. Borohydrides incorporating selenium and tellurium. Can. J. Chem. 1969, 47, 3695–3697. [Google Scholar] [CrossRef]
- Ryschkewitsch, G.E.; Nainan, K.C. Octahydrotriborate(1-) ([B3H8]-) salts. Method A. Inorg. Synth. 1974, 15, 113–114. [Google Scholar]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M.C.; Polidori, G.; Camalli, M. SIRPOW. 92—A program for automatic solution of crystal structures by direct methods optimized for powder data. J. Appl. Cryst. 1994, 27, 435–436. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar]
- Sheldrick, G.M. SHELXS97 and SHELXL97: Program for Crystal Structure Solution and Refinement; University of Gottingen: Gottingen, Germany, 1997. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.G.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; Caricato, M.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Wadt, W.R.; Hay, P.J. Ab Initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. A New Basis Set Exchange: An Open, Up-to-date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- London, F.J. Quantum theory of interatomic currents in aromatic combinations. J. Phys. Radium 1937, 8, 397–409. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Chemcraft-Graphical Software for Visualization of Quantum Chemistry Computations. Available online: https://www.chemcraftprog.com (accessed on 21 March 2021).
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO Program 6.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2013. [Google Scholar]
- Wiberg, K. Application of the Pople-Santry-Segal CNDO Method to the Cyclopropylcarbinyl and Cyclobutyl Cation and to Bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Geometry | d(B-B)av [Å] | 11B NMR (δ,ppm) | Ref. | Geometry | d(B-B)av [Å] | 11B NMR (δ,ppm) | Ref. |
|---|---|---|---|---|---|---|---|
 6-[(Cp*Co)B9H13] | 1.78 | 20.5, 15.4, 5.2, −1.2, −12.4, −29.8 | [47] |  3,5-[(Cp*Co)2B8H12] | 1.80 | 42.4, 30.1, 25.3, 20.1, 9.1, 6.3, −1.2, −12.9 | [52] |
 5-[(Cp*Co)B9H13] | 1.78 | 29.4, 27.1, 12.5, 4.1, 3.5, 1.7, −1.1, −14.5, −36.9 | [52] |  6-Cl-5,7-[(Cp*Co)2B8H12] | - | 49.9, 23.1, 19.4, 7.1, −7.1, −40.7 | [47] |
 2-[(CpCo)B9H13] | - | 17.0, 14.6, 9.0, 1.7, −38.2 | [49] |  5,7-[(Cp*Co)2B8H12] | 1.80 | 57.3, 24.4, 6.0, −7.4, 40.3 | [47] |
 [B10H14] | 1.78 | 11.7, 9.7, 0.28, −36.5 | [66] |  [(Cp*Co)2B8H11] [(Cp*Co)2B8H11] | 1.76 | 54.0, 29.7, 22.2, 8.7, 3.3, −40.1 | [51] |
 6,9-[(Cp*Co)2B8H12] | 2.02 | 20.8, 2.3 | [47] |  2,4-[(Cp*Co)2B8H12] | 1.81 | 26.6, 11.4, 5.6 | This wok |

| Cluster | 1H NMR δ (ppm) | 11B NMR δ (ppm) | Ref. | ||
|---|---|---|---|---|---|
| Expt. | Calc. | Expt. | Calc. | ||
| 2/2′ | −4.25 | −6.22 | 26.6, 11.4, 5.6 | 20.0, 11.0, 3.0 | This work |
| 3/3′ | −3.52 | −5.43 | 21.1, 9.4, 1.9 | 16.2, 6.5, 0.8 | 56 (expt.) |
| 4/4″ | −3.73 | −5.45 | 1.8, −0.2, −12.9 | 7.3, −1.7, −11.0 | 27 (expt.) |
| Compound | 1 | 2 |
|---|---|---|
| CCCDC No. | 2043679 | 2060786 |
| Empirical formula | C30H53B8Co3 | C20H42B8Co2 |
| Formula weight | 676.99 | 486.87 |
| Crystal system | Triclinic | Monoclinic |
| Space group | P | P21/n |
| a (Å) | 8.785 (4) | 8.3902 (2) |
| b (Å) | 11.638 (6) | 40.6669 (12) |
| c (Å) | 16.993 (9) | 8.5097 (2) |
| α (°) | 96.244 (18) | 90 |
| β (°) | 96.56 (2) | 119.0361 (11) |
| γ (°) | 101.532 (18) | 90 |
| Volume (Å3) | 1675.6 (15) | 2538.60 (12) |
| Z | 2 | 4 |
| ρcalc(g/cm−3) | 1.342 | 1.274 |
| μ (mm−1) | 1.487 | 1.312 |
| F (000) | 708 | 1024 |
| 2θ range for data collection (°) | 3.6−44.9 | 2−50 |
| Independent reflections | 3990 | 4472 |
| Final R indices [I >= 2σ (I)] | R1 = 0.0889, wR2 = 0.2412 | R1 = 0.0439, wR2 = 0.0958 |
| Parameters | 452 | 329 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pathak, K.; Nandi, C.; Halet, J.-F.; Ghosh, S. Metal-Rich Metallaboranes: Synthesis, Structures and Bonding of Bi- and Trimetallic Open-Faced Cobaltaboranes. Inorganics 2021, 9, 28. https://doi.org/10.3390/inorganics9040028
Pathak K, Nandi C, Halet J-F, Ghosh S. Metal-Rich Metallaboranes: Synthesis, Structures and Bonding of Bi- and Trimetallic Open-Faced Cobaltaboranes. Inorganics. 2021; 9(4):28. https://doi.org/10.3390/inorganics9040028
Chicago/Turabian StylePathak, Kriti, Chandan Nandi, Jean-François Halet, and Sundargopal Ghosh. 2021. "Metal-Rich Metallaboranes: Synthesis, Structures and Bonding of Bi- and Trimetallic Open-Faced Cobaltaboranes" Inorganics 9, no. 4: 28. https://doi.org/10.3390/inorganics9040028
APA StylePathak, K., Nandi, C., Halet, J.-F., & Ghosh, S. (2021). Metal-Rich Metallaboranes: Synthesis, Structures and Bonding of Bi- and Trimetallic Open-Faced Cobaltaboranes. Inorganics, 9(4), 28. https://doi.org/10.3390/inorganics9040028