
Metal–Organic Frameworks as Versatile Platforms for Organometallic Chemistry

 , , and
, , and 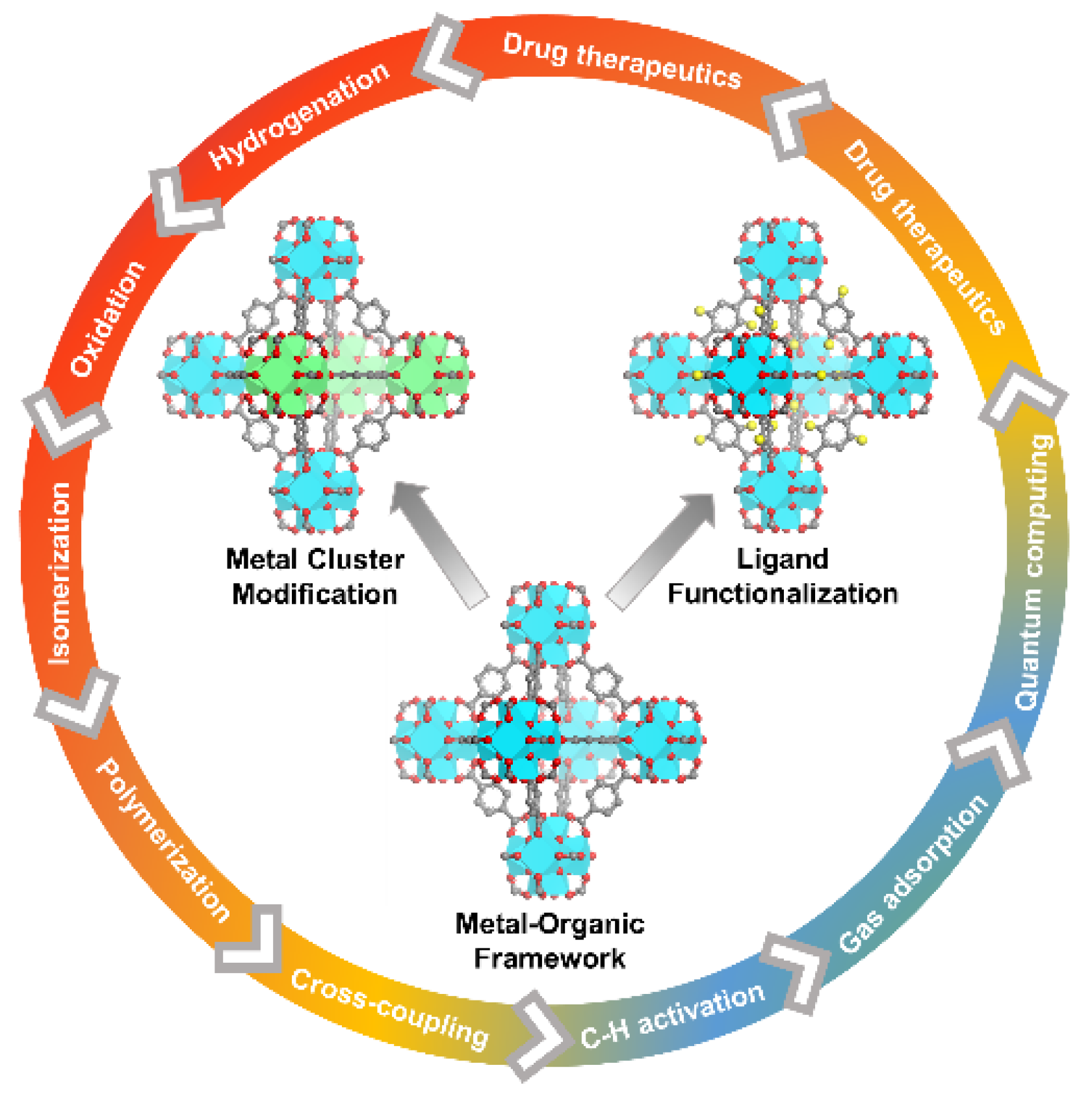{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
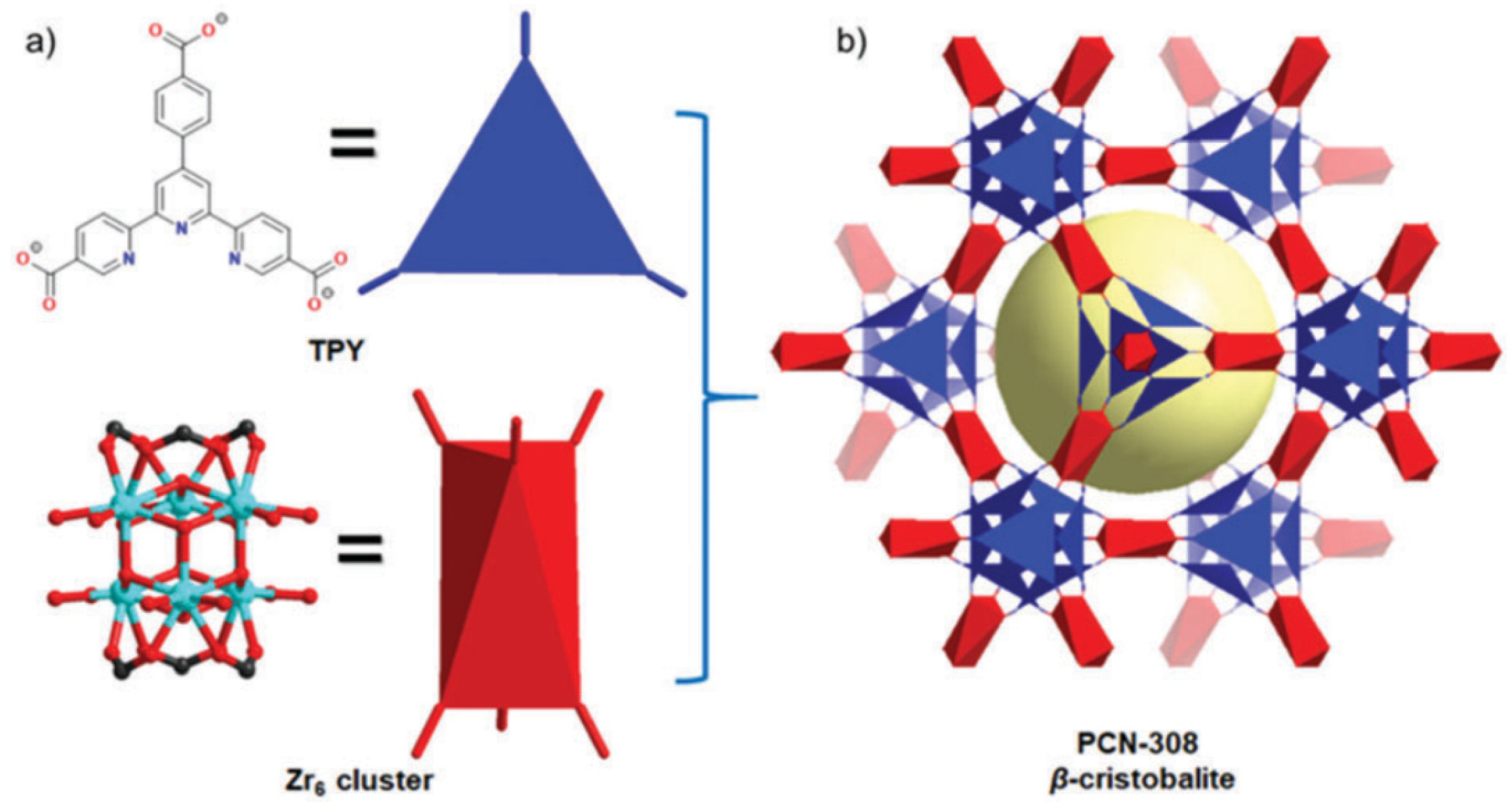{kind=link}
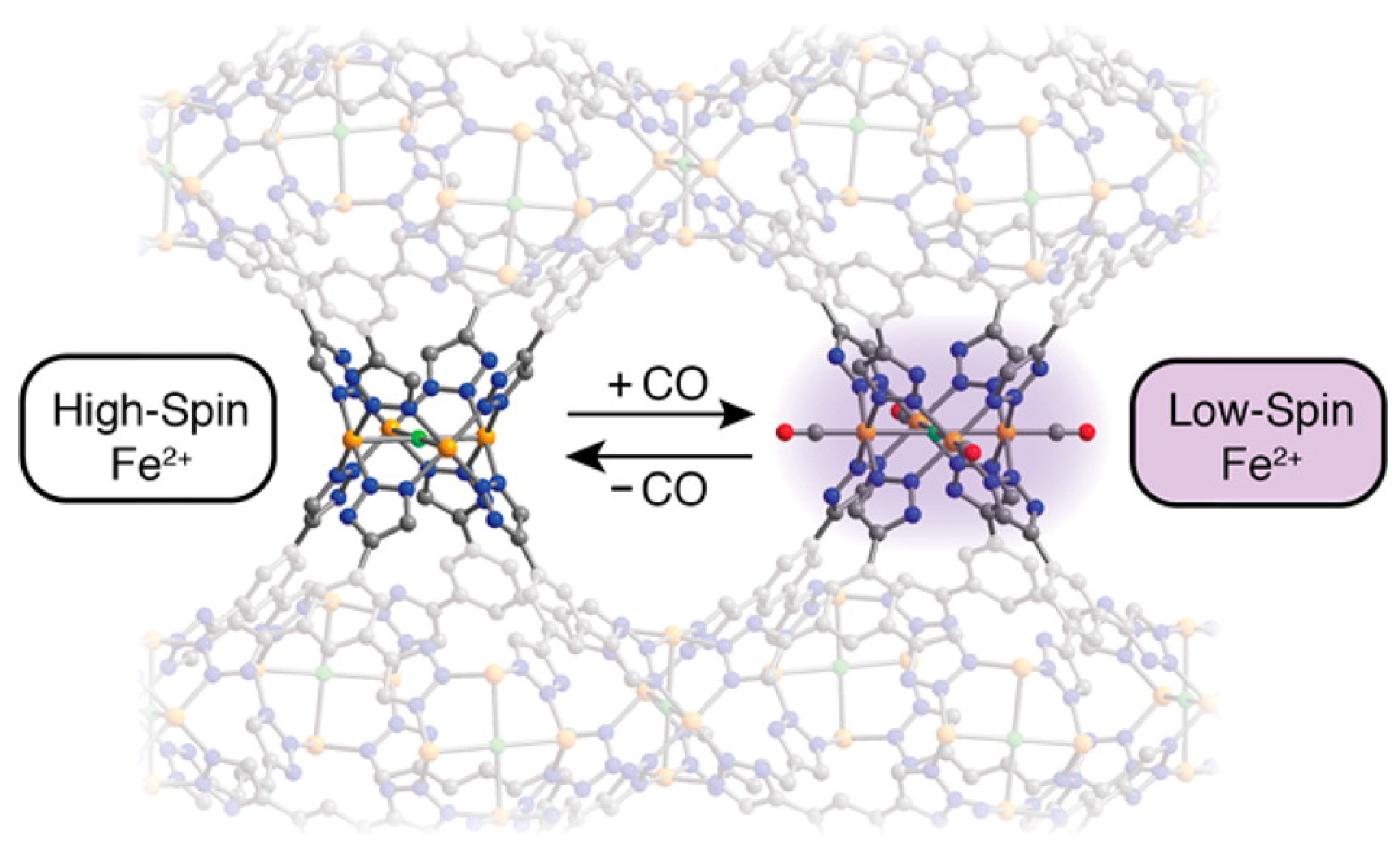{kind=link}
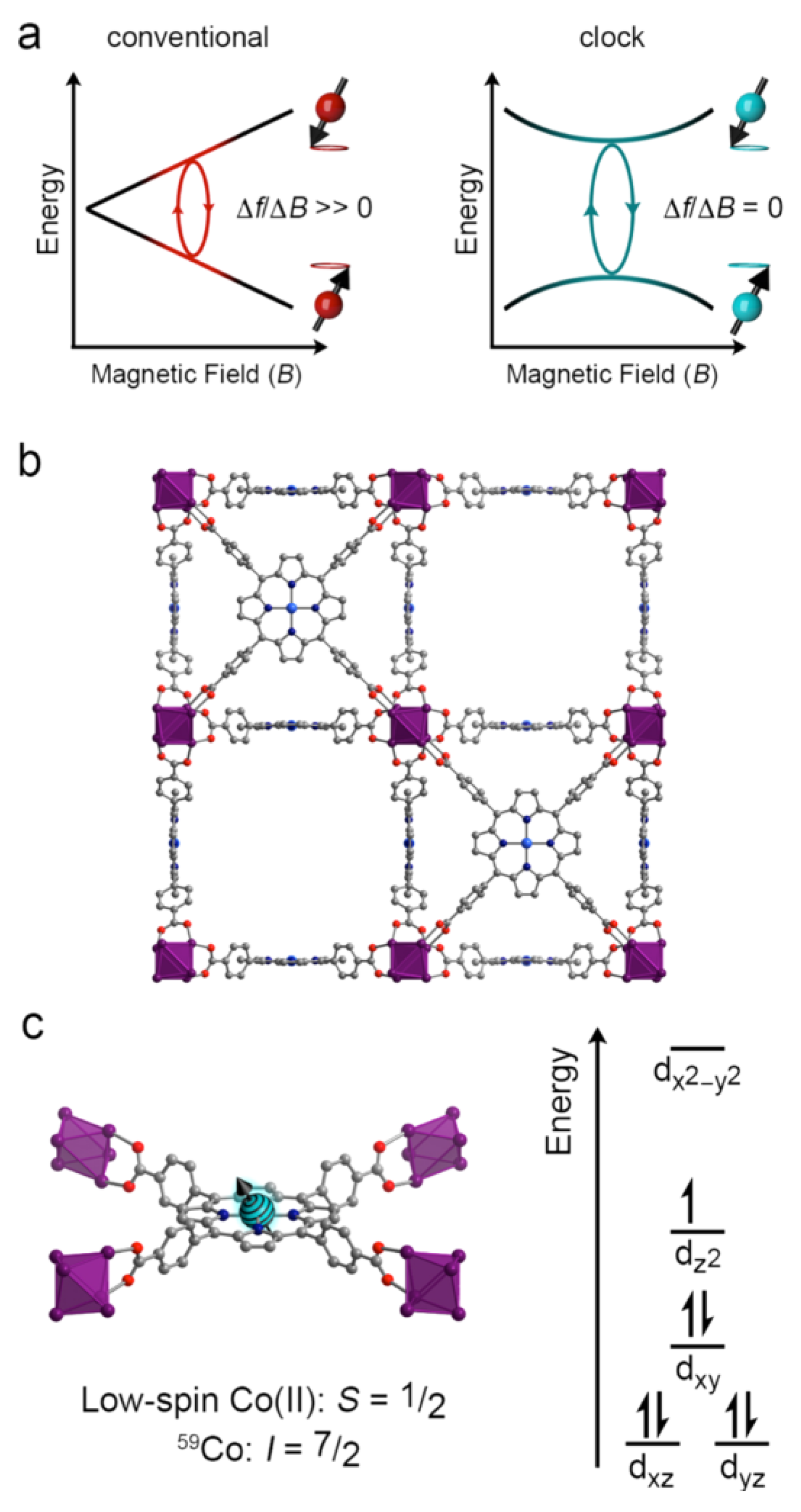{kind=link}
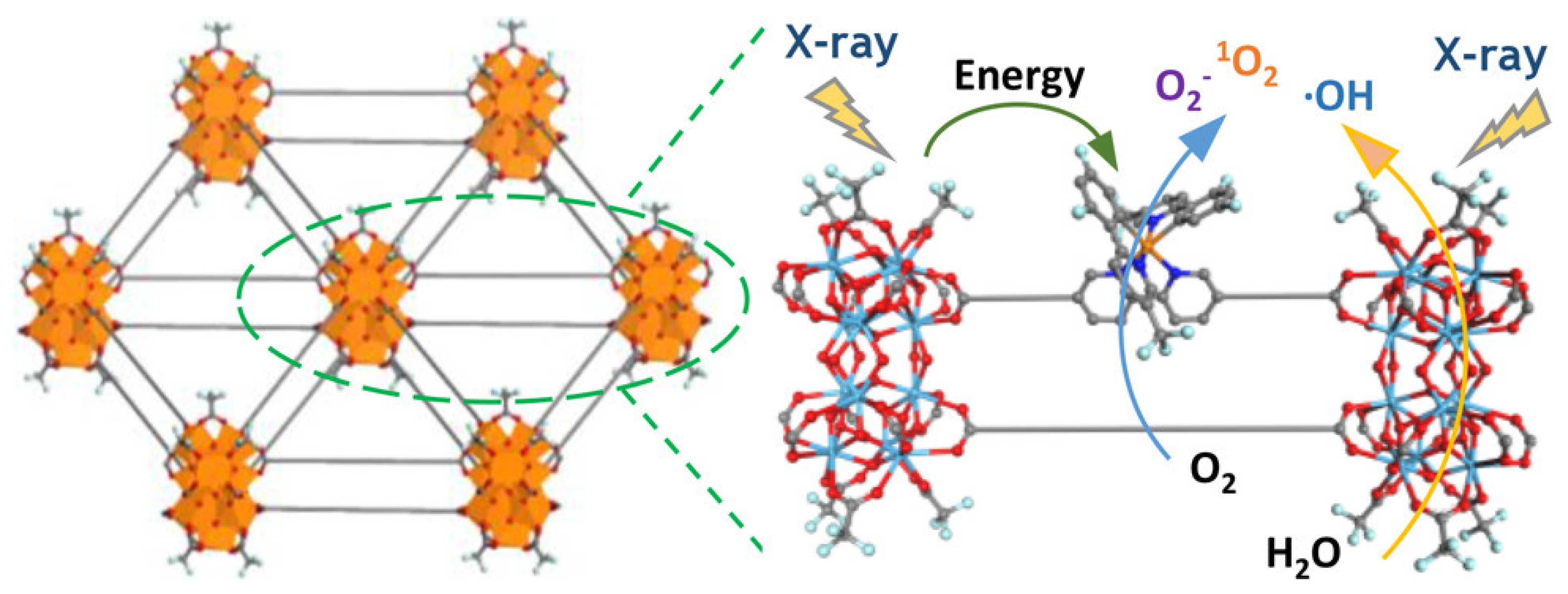{kind=link}
Abstract
1. Introduction
2. Linker-Based Functional Organometallic MOFs
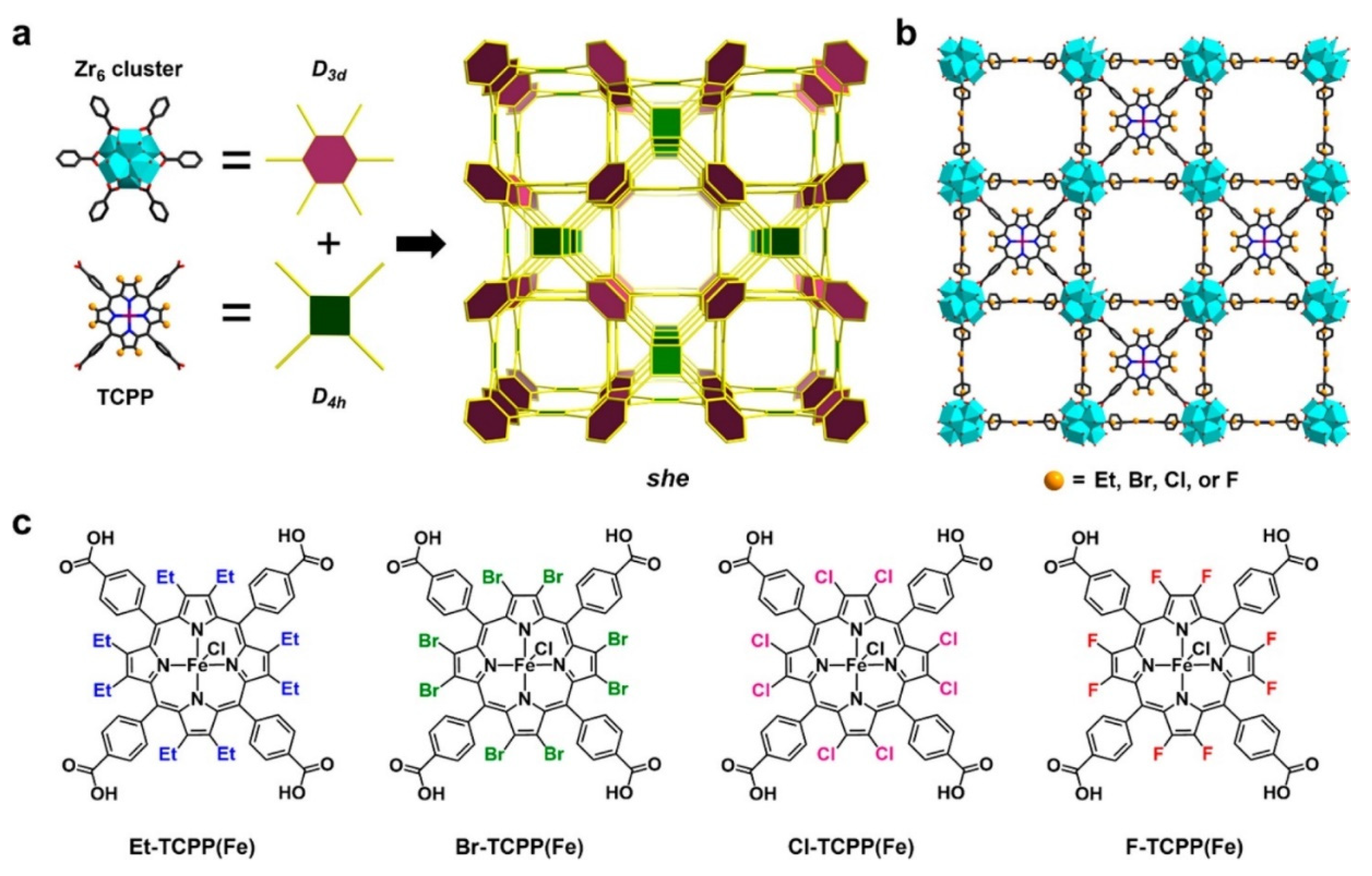
2.1. Porphyrin-Based Linkers
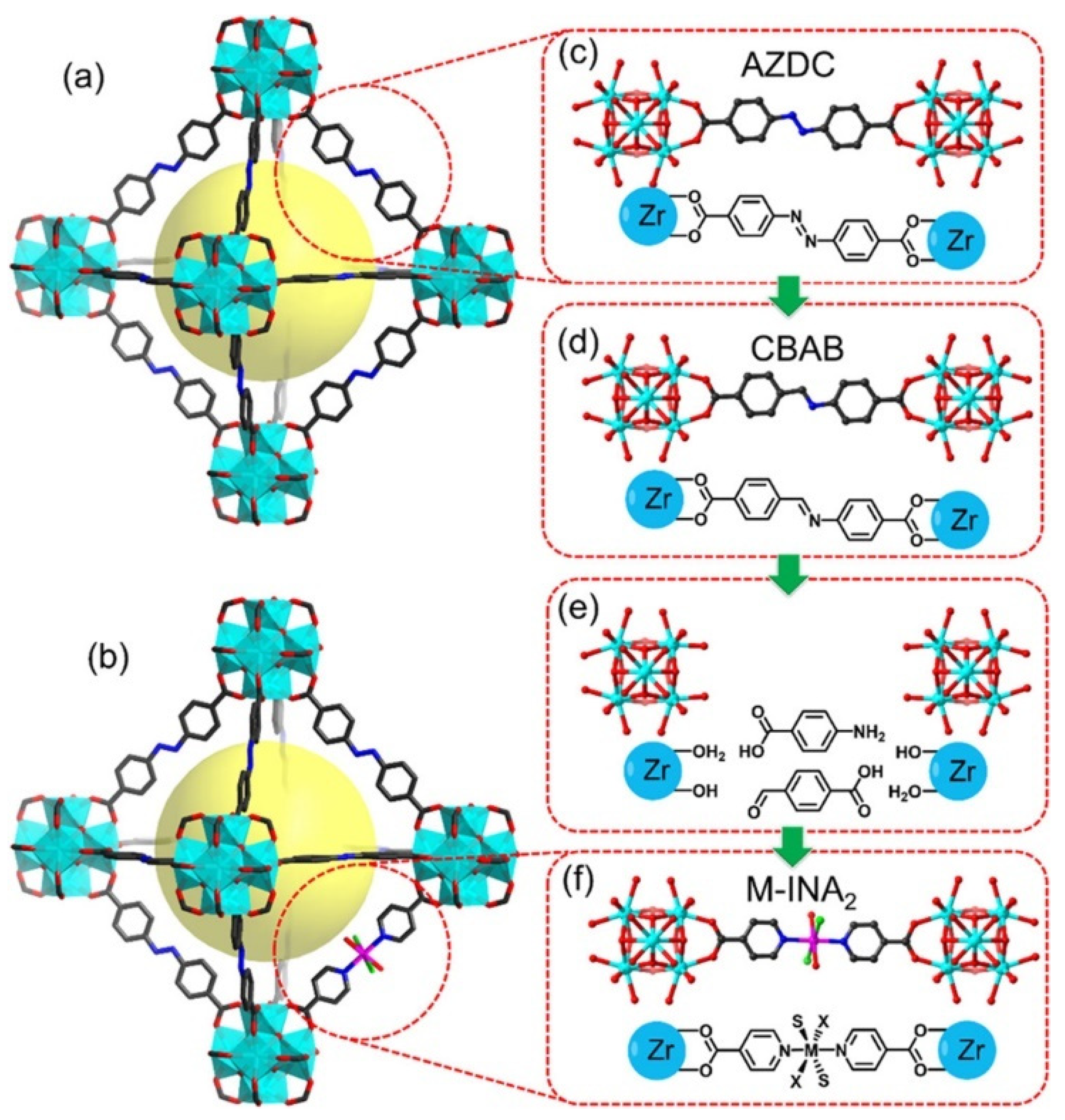
2.2. Polypyridyl-Based Linkers
2.3. Pyridyl-Based Linkers
2.4. Pincer-Based Linkers
2.5. Other Linkers in MOFs
3. Cluster-Based Functional Organometallic MOFs
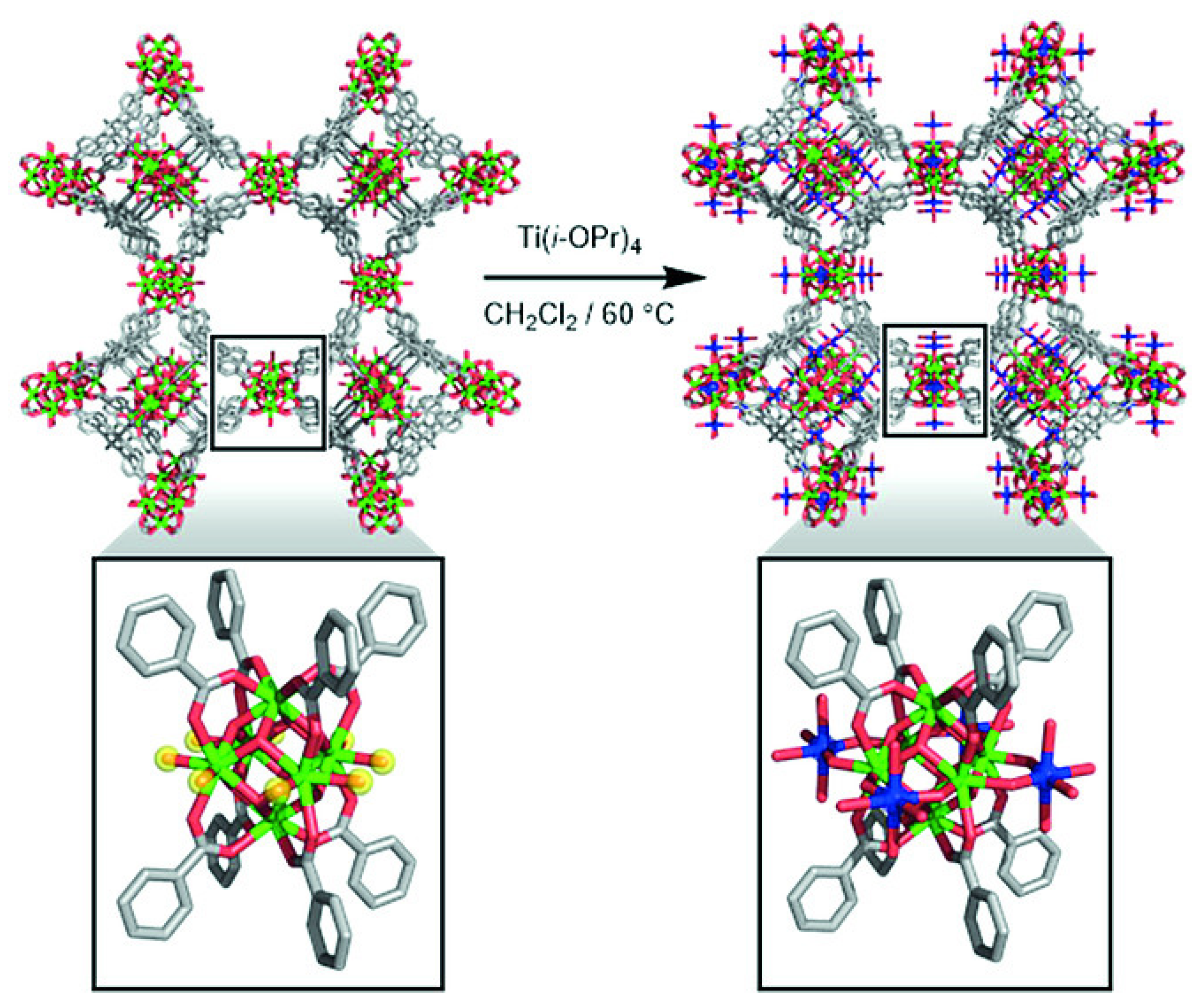
3.1. Post-Synthetic Metalation
3.1.1. Platforms Suitable for Post-Synthetic Metalation
3.1.2. Post-Synthetic Metalation Techniques
3.2. Post-Synthetic Metal Exchange
3.3. In Situ Cation Doping
4. Organometallic Catalysis in MOFs
4.1. Reactions of Alkenes
4.2. Pd-Catalyzed Cross-Coupling Reactions
4.3. C–H Activation Reactions
5. New Horizons to the Use of MOFs for Organometallic Transformations
5.1. Gas Adsorption
5.2. Magnetism
5.3. Quantum Computation
5.4. Therapeutic
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whitesides, G.M. Reinventing chemistry. Angew. Chem. Int. Ed. 2015, 54, 3196–3209. [Google Scholar] [CrossRef] [PubMed]
- Barnaby, R.; Liefeld, A.; Jackson, B.P.; Hampton, T.H.; Stanton, B.A. Effectiveness of table top water pitcher filters to remove arsenic from drinking water. Environ. Res. 2017, 158, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Benavides, P.T.; Sun, P.; Han, J.; Dunn, J.B.; Wang, M. Life-cycle analysis of fuels from post-use non-recycled plastics. Fuel 2017, 203, 11–22. [Google Scholar] [CrossRef]
- Phalke, V.S.; Arote, A.P.; Sonawane, R.R. Design and development of machine which generates fuel using pyrolysis of waste plastic. Int. J. Sci. Res. Dev. 2017, 5, 1–6. [Google Scholar]
- Li, H.; Eddaoudi, M.; Groy, T.L.; Yaghi, O.M. Establishing microporosity in open metal-organic frameworks: Gas sorption isotherms for Zn(BDC) (BDC = 1,4-benzenedicarboxylate). J. Am. Chem. Soc. 1998, 120, 8571–8572. [Google Scholar] [CrossRef]
- Yaghi, O.M.; Li, G.; Li, H. Selective binding and removal of guests in a microporous metal-organic framework. Nature 1995, 378, 703–706. [Google Scholar] [CrossRef]
- Kirchon, A.; Feng, L.; Drake, H.F.; Joseph, E.A.; Zhou, H.-C. From fundamentals to applications: A toolbox for robust and multifunctional MOF materials. Chem. Soc. Rev. 2018, 47, 8611–8638. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Garcia, H. Metal–organic frameworks as solid catalysts for the synthesis of nitrogen-containing heterocycles. Chem. Soc. Rev. 2014, 43, 5750–5765. [Google Scholar] [CrossRef] [PubMed]
- Dhakshinamoorthy, A.; Santiago-Portillo, A.; Asiri, A.M.; Garcia, H. Engineering UiO-66 metal organic framework for heterogeneous catalysis. ChemCatChem 2019, 11, 899–923. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Asiri, A.M.; García, H. Metal organic frameworks as multifunctional solid catalysts. Trends Chem. 2020, 2, 454–466. [Google Scholar] [CrossRef]
- Dincă, M.; Gabbaï, F.P.; Long, J.R. Organometallic chemistry within metal–organic frameworks. Organometallics 2019, 38, 3389–3391. [Google Scholar] [CrossRef]
- An, B.; Zeng, L.Z.; Jia, M.; Li, Z.; Lin, Z.K.; Song, Y.; Zhou, Y.; Cheng, J.; Wang, C.; Lin, W.B. Molecular iridium complexes in metal-organic frameworks catalyze CO2 hydrogenation via concerted proton and hydride transfer. J. Am. Chem. Soc. 2017, 139, 17747–17750. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Dou, Y.; Xie, L.-H.; Rutledge, W.; Li, J.-R.; Zhou, H.-C. Zr-based metal-organic frameworks: Design, synthesis, structure, and applications. Chem. Soc. Rev. 2016, 45, 2327–2367. [Google Scholar] [CrossRef] [PubMed]
- Bosch, M.; Yuan, S.; Rutledge, W.; Zhou, H.-C. Stepwise synthesis of metal-organic frameworks. Acc. Chem. Res. 2017, 50, 857–865. [Google Scholar] [CrossRef]
- Burgess, S.A.; Kassie, A.; Baranowski, S.A.; Fritzsching, K.J.; Schmidt-Rohr, K.; Brown, C.M.; Wade, C.R. Improved catalytic activity and stability of a palladium pincer complex by incorporation into a metal-organic framework. J. Am. Chem. Soc. 2016, 138, 1780–1783. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Li, B.; He, H.; Zhou, W.; Chen, B.; Qian, G. Metal-organic frameworks as platforms for functional materials. Acc. Chem. Res. 2016, 49, 483–493. [Google Scholar] [CrossRef]
- Zhou, H.C.; Long, J.R.; Yaghi, O.M. Introduction to metal-organic frameworks. Chem. Rev. 2012, 112, 673–674. [Google Scholar] [CrossRef]
- Ji, Z.; Wang, H.; Canossa, S.; Wuttke, S.; Yaghi, O.M. Pore chemistry of metal–organic frameworks. Adv. Funct. Mater. 2020, 30, 2000238. [Google Scholar] [CrossRef]
- Kreno, L.E.; Leong, K.; Farha, O.K.; Allendorf, M.; Van Duyne, R.P.; Hupp, J.T. Metal-organic framework materials as chemical sensors. Chem. Rev. 2012, 112, 1105–1125. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Farha, O.K.; Roberts, J.; Scheidt, K.A.; Nguyen, S.T.; Hupp, J.T. Metal-organic framework materials as catalysts. Chem. Soc. Rev. 2009, 38, 1450–1459. [Google Scholar] [CrossRef]
- Li, J.R.; Sculley, J.; Zhou, H.C. Metal-organic frameworks for separations. Chem. Rev. 2012, 112, 869–932. [Google Scholar] [CrossRef] [PubMed]
- Nath, I.; Chakraborty, J.; Verpoort, F. Metal organic frameworks mimicking natural enzymes: A structural and functional analogy. Chem. Soc. Rev. 2016, 45, 4127–4170. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Yuan, S.; Day, G.; Wang, X.; Yang, X.Y.; Zhou, H.C. Luminescent sensors based on metal-organic frameworks. Coord. Chem. Rev. 2018, 354, 28–45. [Google Scholar] [CrossRef]
- Furukawa, H.; Cordova, K.E.; O’Keeffe, M.; Yaghi, O.M. The chemistry and applications of metal-organic frameworks. Science 2013, 341, 974. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.M.; Yang, X.Y.; Zhou, H.C. Synthesis of MOFs for heterogeneous catalysis via linker design. Polyhedron 2018, 154, 189–201. [Google Scholar] [CrossRef]
- Zhao, M.; Ou, S.; Wu, C.D. Porous Metal-organic frameworks for heterogeneous biomimetic catalysis. Acc. Chem. Res. 2014, 47, 1199–1207. [Google Scholar] [CrossRef]
- Gao, W.Y.; Chrzanowski, M.; Ma, S.Q. Metal-metalloporphyrin frameworks: A resurging class of functional materials. Chem. Soc. Rev. 2014, 43, 5841–5866. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.W.; Chung, W.C.; Wei, Z.W.; Gu, Z.Y.; Jiang, H.L.; Chen, Y.P.; Darensbourg, D.J.; Zhou, H.C. Construction of ultrastable porphyrin zr metal-organic frameworks through linker elimination. J. Am. Chem. Soc. 2013, 135, 17105–17110. [Google Scholar] [CrossRef]
- Huang, N.; Yuan, S.; Drake, H.; Yang, X.Y.; Pang, J.D.; Qin, J.S.; Li, J.L.; Zhang, Y.M.; Wang, Q.; Jiang, D.L.; et al. Systematic engineering of single substitution in zirconium metal-organic frameworks toward high-performance catalysis. J. Am. Chem. Soc. 2017, 139, 18590–18597. [Google Scholar] [CrossRef] [PubMed]
- Fateeva, A.; Chater, P.A.; Ireland, C.P.; Tahir, A.A.; Khimyak, Y.Z.; Wiper, P.V.; Darwent, J.R.; Rosseinsky, M.J. A water-stable porphyrin-based metal-organic framework active for visible-light photocatalysis. Angew. Chem. Int. Ed. 2012, 51, 7440–7444. [Google Scholar] [CrossRef]
- Feng, D.W.; Gu, Z.Y.; Chen, Y.P.; Park, J.; Wei, Z.W.; Sun, Y.J.; Bosch, M.; Yuan, S.; Zhou, H.C. A highly stable porphyrinic zirconium metal-organic framework with shp-a topology. J. Am. Chem. Soc. 2014, 136, 17714–17717. [Google Scholar] [CrossRef]
- Huang, N.; Wang, K.C.; Drake, H.; Cai, P.Y.; Pang, J.D.; Li, J.L.; Che, S.; Huang, L.; Wang, Q.; Zhou, H.C. Tailor-made pyrazolide-based metal-organic frameworks for selective catalysis. J. Am. Chem. Soc. 2018, 140, 6383–6390. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xie, Z.G.; deKrafft, K.E.; Lin, W.L. Doping Metal-organic frameworks for water oxidation, carbon dioxide reduction, and organic photocatalysis. J. Am. Chem. Soc. 2011, 133, 13445–13454. [Google Scholar] [CrossRef]
- Zhu, Y.Y.; Lan, G.X.; Fan, Y.J.; Veroneau, S.S.; Song, Y.; Micheroni, D.; Lin, W.B. Merging photoredox and organometallic catalysts in a metal-organic framework significantly boosts photocatalytic activities. Angew. Chem. Int. Ed. 2018, 57, 14090–14094. [Google Scholar] [CrossRef] [PubMed]
- Lan, G.X.; Li, Z.; Veroneau, S.S.; Zhu, Y.Y.; Xu, Z.W.; Wang, C.; Lin, W.B. Photosensitizing metal-organic layers for efficient sunlight-driven carbon dioxide reduction. J. Am. Chem. Soc. 2018, 140, 12369–12373. [Google Scholar] [CrossRef] [PubMed]
- Elumalai, P.; Mamlouk, H.; Yiming, W.; Feng, L.; Yuan, S.; Zhou, H.C.; Madrahimov, S.T. Recyclable and reusable heteroleptic nickel catalyst immobilized on metal-organic framework for suzuki-miyaura coupling. ACS Appl. Mater. Interfaces 2018, 10, 41431–41438. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.-L.; Wang, K.; Wang, B.; Su, J.; Zou, X.; Xie, Y.; Li, J.-R.; Zhou, H.-C. A base-resistant metalloporphyrin metal-organic framework for C-H bond halogenation. J. Am. Chem. Soc. 2017, 139, 211–217. [Google Scholar] [CrossRef]
- Sikma, R.E.; Kunal, P.; Dunning, S.G.; Reynolds, J.E.; Lee, J.S.; Chang, J.S.; Humphrey, S.M. Organoarsine metal-organic framework with cis-diarsine pockets for the installation of uniquely confined metal complexes. J. Am. Chem. Soc. 2018, 140, 9806–9809. [Google Scholar] [CrossRef]
- Yuan, S.; Qin, J.S.; Su, J.; Li, B.; Li, J.L.; Chen, W.M.; Drake, H.F.; Zhang, P.; Yuan, D.Q.; Zuo, J.L.; et al. Sequential transformation of zirconium(IV)-MOFs into heterobimetallic MOFs bearing magnetic anisotropic cobalt(II) centers. Angew. Chem. Int. Ed. 2018, 57, 12578–12583. [Google Scholar] [CrossRef]
- Yuan, S.; Zhang, P.; Zhang, L.L.; Garcia-Esparza, A.T.; Sokaras, D.; Qin, J.S.; Feng, L.; Day, G.S.; Chen, W.M.; Drake, H.F.; et al. Exposed equatorial positions of metal centers via sequential ligand elimination and installation in MOFs. J. Am. Chem. Soc. 2018, 140, 10814–10819. [Google Scholar] [CrossRef]
- Kosanovich, A.J.; Komatsu, C.H.; Bhuvanesh, N.; Ozerov, O.V.; Perez, L.M. Dearomatization of the PCP pincer ligand in a Re(V) oxo complex. Chem. Eur. J. 2018, 24, 13754–13757. [Google Scholar] [CrossRef] [PubMed]
- He, J.P.; Waggoner, N.W.; Dunning, S.G.; Steiner, A.; Lynch, V.M.; Humphrey, S.M. A PCP Pincer Ligand for coordination polymers with versatile chemical reactivity: Selective activation of CO2 gas over CO Gas in the solid state. Angew. Chem. Int. Ed. 2016, 55, 12351–12355. [Google Scholar] [CrossRef] [PubMed]
- Carson, F.; Martinez-Castro, E.; Marcos, R.; Miera, G.G.; Jansson, K.; Zou, X.D.; Martin-Matute, B. Effect of the functionalisation route on a Zr-MOF with an Ir-NHC complex for catalysis. Chem. Commun. 2015, 51, 10864–10867. [Google Scholar] [CrossRef] [PubMed]
- Manna, K.; Ji, P.; Lin, Z.; Greene, F.X.; Urban, A.; Thacker, N.C.; Lin, W. Chemoselective single-site Earth-abundant metal catalysts at metal–organic framework nodes. Nat. Commun. 2016, 7, 12610. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Qin, J.S.; Lollar, C.T.; Zhou, H.C. Stable Metal-organic frameworks with group 4 metals: Current status and trends. ACS Cent. Sci. 2018, 4, 440–450. [Google Scholar] [CrossRef]
- Cavka, J.H.; Jakobsen, S.; Olsbye, U.; Guillou, N.; Lamberti, C.; Bordiga, S.; Lillerud, K.P. A new zirconium inorganic building brick forming metal organic frameworks with exceptional stability. J. Am. Chem. Soc. 2008, 130, 13850–13851. [Google Scholar] [CrossRef]
- Schaate, A.; Roy, P.; Godt, A.; Lippke, J.; Waltz, F.; Wiebcke, M.; Behrens, P. Modulated synthesis of Zr-based metal-organic frameworks: From nano to single crystals. Chem. Eur. J. 2011, 17, 6643–6651. [Google Scholar] [CrossRef]
- Yang, D.; Odoh, S.O.; Borycz, J.; Wang, T.C.; Farha, O.K.; Hupp, J.T.; Cramer, C.J.; Gagliardi, L.; Gates, B.C. Tuning Zr6 metal–organic framework (MOF) nodes as catalyst supports: Site densities and electron-donor properties influence molecular iridium complexes as ethylene conversion catalysts. ACS Catal. 2016, 6, 235–247. [Google Scholar] [CrossRef]
- Denny, M.S.; Parent, L.R.; Patterson, J.P.; Meena, S.K.; Pham, H.; Abellan, P.; Ramasse, Q.M.; Paesani, F.; Gianneschi, N.C.; Cohen, S.M. Transmission electron microscopy reveals deposition of metal oxide coatings onto metal–organic frameworks. J. Am. Chem. Soc. 2018, 140, 1348–1357. [Google Scholar] [CrossRef]
- Liu, L.; Chen, Z.; Wang, J.; Zhang, D.; Zhu, Y.; Ling, S.; Huang, K.-W.; Belmabkhout, Y.; Adil, K.; Zhang, Y.; et al. Imaging defects and their evolution in a metal-organic framework at sub-unit-cell resolution. Nat. Chem. 2019, 11, 622–628. [Google Scholar] [CrossRef]
- Yuan, S.; Zou, L.; Li, H.; Chen, Y.-P.; Qin, J.; Zhang, Q.; Lu, W.; Hall, M.B.; Zhou, H.-C. Flexible zirconium metal-organic frameworks as bioinspired switchable catalysts. Angew. Chem. Int. Ed. 2016, 55, 10776–10780. [Google Scholar] [CrossRef]
- Wang, T.C.; Vermeulen, N.A.; Kim, I.S.; Martinson, A.B.F.; Stoddart, J.F.; Hupp, J.T.; Farha, O.K. Scalable synthesis and post-modification of a mesoporous metal-organic framework called NU-1000. Nat. Protoc. 2015, 11, 149. [Google Scholar] [CrossRef]
- Morris, W.; Volosskiy, B.; Demir, S.; Gandara, F.; McGrier, P.L.; Furukawa, H.; Cascio, D.; Stoddart, J.F.; Yaghi, O.M. Synthesis, structure, and metalation of two new highly porous zirconium metal-organic frameworks. Inorg. Chem. 2012, 51, 6443–6445. [Google Scholar] [CrossRef]
- Liu, T.-F.; Vermeulen, N.A.; Howarth, A.J.; Li, P.; Sarjeant, A.A.; Hupp, J.T.; Farha, O.K. Adding to the arsenal of zirconium-based metal–organic frameworks: The topology as a platform for solvent-assisted metal incorporation. Eur. J. Inorg. Chem. 2016, 2016, 4349–4352. [Google Scholar] [CrossRef]
- Clegg, W.; Harbron, D.R.; Homan, C.D.; Hunt, P.A.; Little, I.R.; Straughan, B.P. Crystal structures of three basic zinc carboxylates together with infrared and FAB mass spectrometry studies in solution. Inorg. Chim. Acta 1991, 186, 51–60. [Google Scholar] [CrossRef]
- Köberl, M.; Cokoja, M.; Herrmann, W.A.; Kühn, F.E. From molecules to materials: Molecular paddle-wheel synthons of macromolecules, cage compounds and metal–organic frameworks. Dalton Trans. 2011, 40, 6834–6859. [Google Scholar] [CrossRef] [PubMed]
- Corma, A.; García, H.; Llabrés i Xamena, F.X. Engineering Metal organic frameworks for heterogeneous catalysis. Chem. Rev. 2010, 110, 4606–4655. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Lu, W.; Chen, Y.-P.; Zhang, Q.; Liu, T.-F.; Feng, D.; Wang, X.; Qin, J.; Zhou, H.-C. Sequential linker installation: Precise placement of functional groups in multivariate metal–organic frameworks. J. Am. Chem. Soc. 2015, 137, 3177–3180. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Lyu, J.; Otake, K.I.; Wang, X.; Redfern, L.R.; Malliakas, C.D.; Li, Z.; Islamoglu, T.; Wang, B.; et al. A flexible metal-organic framework with 4-connected Zr6 nodes. J. Am. Chem. Soc. 2018, 140, 11179–11183. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.W.; Li, Z.; Farha, O.K.; Hupp, J.T. Toward inexpensive photocatalytic hydrogen evolution: A nickel sulfide catalyst supported on a high-stability metal–organic framework. ACS Appl. Mater. Interfaces 2016, 8, 20675–20681. [Google Scholar] [CrossRef] [PubMed]
- Platero-Prats, A.E.; League, A.B.; Bernales, V.; Ye, J.; Gallington, L.C.; Vjunov, A.; Schweitzer, N.M.; Li, Z.; Zheng, J.; Mehdi, B.L.; et al. Bridging zirconia nodes within a metal–organic framework via catalytic ni-hydroxo clusters to form heterobimetallic nanowires. J. Am. Chem. Soc. 2017, 139, 10410–10418. [Google Scholar] [CrossRef]
- Noh, H.; Cui, Y.; Peters, A.W.; Pahls, D.R.; Ortuño, M.A.; Vermeulen, N.A.; Cramer, C.J.; Gagliardi, L.; Hupp, J.T.; Farha, O.K. An exceptionally stable metal–organic framework supported molybdenum(vi) oxide catalyst for cyclohexene epoxidation. J. Am. Chem. Soc. 2016, 138, 14720–14726. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Thornburg, N.E.; Li, Z.; Wang, T.C.; Gallington, L.C.; Chapman, K.W.; Notestein, J.M.; Hupp, J.T.; Farha, O.K. Stable Metal–organic framework-supported niobium catalysts. Inorg. Chem. 2016, 55, 11954–11961. [Google Scholar] [CrossRef]
- Li, Z.; Peters, A.W.; Bernales, V.; Ortuño, M.A.; Schweitzer, N.M.; DeStefano, M.R.; Gallington, L.C.; Platero-Prats, A.E.; Chapman, K.W.; Cramer, C.J.; et al. Metal–organic framework supported cobalt catalysts for the oxidative dehydrogenation of propane at low temperature. ACS Cent. Sci. 2017, 3, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Kung, C.-W.; Otake, K.-I.; Peters, A.W.; Li, Z.; Liao, Y.; Gong, X.; Farha, O.K.; Hupp, J.T. Redox-mediator-assisted electrocatalytic hydrogen evolution from water by a molybdenum sulfide-functionalized metal–organic framework. ACS Catal. 2018, 8, 9848–9858. [Google Scholar] [CrossRef]
- Kung, C.-W.; Audu, C.O.; Peters, A.W.; Noh, H.; Farha, O.K.; Hupp, J.T. Copper nanoparticles installed in metal–organic framework thin films are electrocatalytically competent for CO2 reduction. ACS Energy Lett. 2017, 2, 2394–2401. [Google Scholar] [CrossRef]
- Nguyen, H.G.T.; Schweitzer, N.M.; Chang, C.-Y.; Drake, T.L.; So, M.C.; Stair, P.C.; Farha, O.K.; Hupp, J.T.; Nguyen, S.T. Vanadium-Node-Functionalized UiO-66: A thermally stable MOF-supported catalyst for the gas-phase oxidative dehydrogenation of cyclohexene. ACS Catal. 2014, 4, 2496–2500. [Google Scholar] [CrossRef]
- Yang, D.; Odoh, S.O.; Wang, T.C.; Farha, O.K.; Hupp, J.T.; Cramer, C.J.; Gagliardi, L.; Gates, B.C. Metal–organic framework nodes as nearly ideal supports for molecular catalysts: NU-1000- and UiO-66-supported iridium complexes. J. Am. Chem. Soc. 2015, 137, 7391–7396. [Google Scholar] [CrossRef]
- Pi, Y.; Feng, X.; Song, Y.; Xu, Z.; Li, Z.; Lin, W.B. Metal–organic frameworks integrate cu photosensitizers and secondary building unit-supported fe catalysts for photocatalytic hydrogen evolution. J. Am. Chem. Soc. 2020, 142, 10302–10307. [Google Scholar] [CrossRef]
- An, B.; Li, Z.; Song, Y.; Zhang, J.; Zeng, L.; Wang, C.; Lin, W. Cooperative copper centres in a metal–organic framework for selective conversion of CO2 to ethanol. Nat. Chem. 2019, 2, 709–717. [Google Scholar] [CrossRef]
- Ji, P.; Manna, K.; Lin, Z.; Feng, X.; Urban, A.; Song, Y.; Lin, W. Single-Site Cobalt Catalysts at New Zr12(μ3-O)8(μ3-OH)8(μ2-OH)6 metal–organic framework nodes for highly active hydrogenation of nitroarenes, nitriles, and isocyanides. J. Am. Chem. Soc. 2017, 139, 7004–7011. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Song, Y.; Drake, T.; Veroneau, S.S.; Lin, Z.; Pan, X.; Lin, W. Titanium(III)-oxo clusters in a metal–organic framework support single-site Co(II)-hydride catalysts for arene hydrogenation. J. Am. Chem. Soc. 2018, 140, 433–440. [Google Scholar] [CrossRef]
- Comito, R.J.; Fritzsching, K.J.; Sundell, B.J.; Schmidt-Rohr, K.; Dincă, M. Single-site heterogeneous catalysts for olefin polymerization enabled by cation exchange in a metal-organic framework. J. Am. Chem. Soc. 2016, 138, 10232–10237. [Google Scholar] [CrossRef]
- Lollar, C.T.; Qin, J.-S.; Pang, J.; Yuan, S.; Becker, B.; Zhou, H.-C. Interior decoration of stable metal–organic frameworks. Langmuir 2018, 34, 13795–13807. [Google Scholar] [CrossRef] [PubMed]
- Brozek, C.K.; Bellarosa, L.; Soejima, T.; Clark, T.V.; López, N.; Dincă, M. Solvent-dependent cation exchange in metal–organic frameworks. Chem. Eur. J. 2014, 20, 6871–6874. [Google Scholar] [CrossRef]
- Kim, M.; Cahill, J.F.; Fei, H.; Prather, K.A.; Cohen, S.M. Postsynthetic ligand and cation exchange in robust metal–organic frameworks. J. Am. Chem. Soc. 2012, 134, 18082–18088. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Kim, H.; Kim, K. Metathesis in single crystal: Complete and reversible exchange of metal ions constituting the frameworks of metal−organic frameworks. J. Am. Chem. Soc. 2009, 131, 3814–3815. [Google Scholar] [CrossRef] [PubMed]
- Denysenko, D.; Werner, T.; Grzywa, M.; Puls, A.; Hagen, V.; Eickerling, G.; Jelic, J.; Reuter, K.; Volkmer, D. Reversible gas-phase redox processes catalyzed by Co-exchanged MFU-4l(arge). Chem. Commun. 2012, 48, 1236–1238. [Google Scholar] [CrossRef]
- Sun, D.; Sun, F.; Deng, X.; Li, Z. Mixed-Metal Strategy on Metal–organic frameworks (MOFs) for functionalities expansion: Co substitution induces aerobic oxidation of cyclohexene over inactive Ni-MOF-74. Inorg. Chem. 2015, 54, 8639–8643. [Google Scholar] [CrossRef]
- Zou, L.; Feng, D.; Liu, T.-F.; Chen, Y.-P.; Yuan, S.; Wang, K.; Wang, X.; Fordham, S.; Zhou, H.-C. A versatile synthetic route for the preparation of titanium metal–organic frameworks. Chem. Sci. 2016, 7, 1063–1069. [Google Scholar] [CrossRef]
- Huxley, M.; Coghlan, C.J.; Burgun, A.; Tarzia, A.; Sumida, K.; Sumby, C.J.; Doonan, C.J. Site-specific metal and ligand substitutions in a microporous Mn2+-based metal–organic framework. Dalton Trans. 2016, 45, 4431–4438. [Google Scholar] [CrossRef]
- Banerjee, P.C.; Lobo, D.E.; Middag, R.; Ng, W.K.; Shaibani, M.E.; Majumder, M. Electrochemical capacitance of Ni-doped metal organic framework and reduced graphene oxide composites: More than the sum of its parts. ACS Appl. Mater. Interfaces 2015, 7, 3655–3664. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.J.; Bloch, E.D.; Mason, J.A.; Queen, W.L.; Hudson, M.R.; Planas, N.; Borycz, J.; Dzubak, A.L.; Verma, P.; Lee, K.; et al. Oxidation of ethane to ethanol by N2O in a metal–organic framework with coordinatively unsaturated iron(II) sites. Nat. Chem. 2014, 6, 590. [Google Scholar] [CrossRef]
- Li, H.; Shi, W.; Zhao, K.; Li, H.; Bing, Y.; Cheng, P. Enhanced hydrostability in Ni-doped MOF-5. Inorg. Chem. 2012, 51, 9200–9207. [Google Scholar] [CrossRef]
- Botas, J.A.; Calleja, G.; Sánchez-Sánchez, M.; Orcajo, M.G. Cobalt doping of the MOF-5 framework and its effect on gas-adsorption properties. Langmuir 2010, 26, 5300–5303. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Telfer, S.G. Systematic ligand modulation enhances the moisture stability and gas sorption characteristics of quaternary metal–organic frameworks. J. Am. Chem. Soc. 2015, 137, 3901–3909. [Google Scholar] [CrossRef] [PubMed]
- Kaye, S.S.; Dailly, A.; Yaghi, O.M.; Long, J.R. Impact of preparation and handling on the hydrogen storage properties of Zn4O(1,4-benzenedicarboxylate)3 (MOF-5). J. Am. Chem. Soc. 2007, 129, 14176–14177. [Google Scholar] [CrossRef] [PubMed]
- Cychosz, K.A.; Matzger, A.J. Water stability of microporous coordination polymers and the adsorption of pharmaceuticals from water. Langmuir 2010, 26, 17198–17202. [Google Scholar] [CrossRef]
- Drake, T.; Ji, P.; Lin, W. Site isolation in metal-organic frameworks enables novel transition metal catalysis. Acc. Chem. Res. 2018, 51, 2129–2138. [Google Scholar] [CrossRef]
- Pascanu, V.; Miera, G.G.; Inge, A.K.; Martin-Matute, B. Metal–organic frameworks as catalysts for organic synthesis: A critical perspective. J. Am. Chem. Soc. 2019, 141, 7223–7234. [Google Scholar] [CrossRef]
- Paciello, R.A. High Throughput screening of homogeneous catalysts: Selected trends and applications in process development. In Applied Homogeneous Catalysis with Organometallic Compounds, 3rd ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2019; pp. 1085–1096. [Google Scholar]
- Schaub, T.; Paciello, R.A.; Limbach, M. Homogeneous catalysis with CO2 as a building block: An industrial perspective. In Applied Homogeneous Catalysis with Organometallic Compounds, 3rd ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2019; pp. 1601–1614. [Google Scholar]
- Manna, K.; Ji, P.; Greene, F.X.; Lin, W. Metal-organic framework nodes support single-site magnesium-alkyl catalysts for hydroboration and hydroamination reactions. J. Am. Chem. Soc. 2016, 138, 7488–7491. [Google Scholar] [CrossRef]
- Yang, X.; Yuan, S.; Zou, L.; Drake, H.; Zhang, Y.; Qin, J.; Alsalme, A.; Zhou, H.-C. One-step synthesis of hybrid core-shell metal-organic frameworks. Angew. Chem. Int. Ed. 2018, 57, 3927–3932. [Google Scholar] [CrossRef] [PubMed]
- Thiam, Z.; Abou-Hamad, E.; Dereli, B.; Liu, L.; Emwas, A.-H.; Ahmad, R.; Jiang, H.; Isah, A.A.; Ndiaye, P.B.; Taoufik, M.; et al. Extension of surface organometallic chemistry to metal–organic frameworks: Development of a well-defined single site [(≡Zr–O−)W(═O)(CH2tBu)3] olefin metathesis catalyst. J. Am. Chem. Soc. 2020, 142, 16690–16703. [Google Scholar] [CrossRef]
- Yuan, S.; Zou, L.; Qin, J.-S.; Li, J.; Huang, L.; Feng, L.; Wang, X.; Bosch, M.; Alsalme, A.; Cagin, T.; et al. Construction of hierarchically porous metal–organic frameworks through linker labilization. Nat. Commun. 2017, 8, 15356. [Google Scholar] [CrossRef] [PubMed]
- Molnar, A. (Ed.) Palladium-Catalyzed Coupling Reactions: Practical Aspects and Future Developments; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; p. 570. [Google Scholar] [CrossRef]
- De Vries, A.H.M.; Parlevliet, F.J.; Schmieder-Van De Vondervoort, L.; Mommers, J.H.M.; Henderickx, H.J.W.; Walet, M.A.M.; De Vries, J.G. A practical recycle of a ligand-free palladium catalyst for Heck reactions. Adv. Synth. Catal. 2002, 344, 996–1002. [Google Scholar] [CrossRef]
- Wei, Y.-L.; Li, Y.; Chen, Y.-Q.; Dong, Y.; Yao, J.-J.; Han, X.-Y.; Dong, Y.-B. Pd(II)-NHDC-functionalized UiO-67 type MOF for catalyzing Heck cross-coupling and intermolecular benzyne-benzyne-alkene insertion reactions. Inorg. Chem. 2018, 57, 4379–4386. [Google Scholar] [CrossRef]
- Bahadori, M.; Tangestaninejad, S.; Moghadam, M.; Mirkhani, V.; Mechler, A.; Mohammadpoor-Baltork, I.; Zadehahmadi, F. Metal organic framework-supported N-heterocyclic carbene palladium complex: A highly efficient and reusable heterogeneous catalyst for Suzuki-Miyaura C-C coupling reaction. Microporous Mesoporous Mater. 2017, 253, 102–111. [Google Scholar] [CrossRef]
- Chen, L.; Rangan, S.; Li, J.; Jiang, H.; Li, Y. A molecular Pd(II) complex incorporated into a MOF as a highly active single-site heterogeneous catalyst for C-Cl bond activation. Green Chem. 2014, 16, 3978–3985. [Google Scholar] [CrossRef]
- Fei, H.; Cohen, S.M. A robust, catalytic metal-organic framework with open 2,2′-bipyridine sites. Chem. Commun. 2014, 50, 4810–4812. [Google Scholar] [CrossRef]
- Chen, Y.-Z.; Jiang, H.-L. Porphyrinic metal-organic framework catalyzed Heck-reaction: Fluorescence “turn-on” sensing of Cu(II) ion. Chem. Mater. 2016, 28, 6698–6704. [Google Scholar] [CrossRef]
- Godula, K.; Sames, D. C-H bond functionalization in complex organic synthesis. Science 2006, 312, 67–72. [Google Scholar] [CrossRef]
- Schwach, P.; Pan, X.; Bao, X. Direct conversion of methane to value-added chemicals over heterogeneous catalysts: Challenges and prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef]
- Liao, P.; Getman, R.B.; Snurr, R.Q. Optimizing open iron sites in metal-organic frameworks for ethane oxidation: A first-principles study. ACS Appl. Mater. Interfaces 2017, 9, 33484–33492. [Google Scholar] [CrossRef]
- Bergman, R.G. Organometallic chemistry: C-H activation. Nature 2007, 446, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Santoro, S.; Kozhushkov, S.I.; Ackermann, L.; Vaccaro, L. Heterogeneous catalytic approaches in C-H activation reactions. Green Chem. 2016, 18, 3471–3493. [Google Scholar] [CrossRef]
- Qin, J.-S.; Yuan, S.; Lollar, C.; Pang, J.; Alsalme, A.; Zhou, H.-C. Stable metal-organic frameworks as a host platform for catalysis and biomimetics. Chem. Commun. 2018, 54, 4231–4249. [Google Scholar] [CrossRef]
- Sawano, T.; Lin, Z.; Boures, D.; An, B.; Wang, C.; Lin, W. Metal-organic frameworks stabilize mono(phosphine)-metal complexes for broad-scope catalytic reactions. J. Am. Chem. Soc. 2016, 138, 9783–9786. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Yang, X.; Zhang, P.; Pang, J.; Li, B.; Zhou, H.-C. A mesoporous NNN-pincer-based metal-organic framework scaffold for the preparation of noble-metal-free catalysts. Chem. Commun. 2019, 55, 2023–2026. [Google Scholar] [CrossRef] [PubMed]
- Kalaj, M.; Cohen, S.M. Postsynthetic modification: An enabling technology for the advancement of metal–organic frameworks. ACS Cent. Sci. 2020, 6, 1046–1057. [Google Scholar] [CrossRef]
- Anderson, J.S.; Gallagher, A.T.; Mason, J.A.; Harris, T.D. A five-coordinate heme dioxygen adduct isolated within a metal-organic framework. J. Am. Chem. Soc. 2014, 136, 16489–16492. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, A.T.; Lee, J.Y.; Kathiresan, V.; Anderson, J.S.; Hoffman, B.M.; Harris, T.D. A structurally-characterized peroxomanganese(IV) porphyrin from reversible O-2 binding within a metal-organic framework. Chem. Sci. 2018, 9, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Brozek, C.K.; Miller, J.T.; Stoian, S.A.; Dincă, M. NO disproportionation at a mononuclear site-isolated Fe2+ center in Fe2+-MOF-5. J. Am. Chem. Soc. 2015, 137, 7495–7501. [Google Scholar] [CrossRef]
- Bloch, E.D.; Queen, W.L.; Chavan, S.; Wheatley, P.S.; Zadrozny, J.M.; Morris, R.; Brown, C.M.; Lamberti, C.; Bordiga, S.; Long, J.R. Gradual release of strongly bound nitric oxide from Fe2(NO)2(dobdc). J. Am. Chem. Soc. 2015, 137, 3466–3469. [Google Scholar] [CrossRef]
- Rieth, A.J.; Tulchinsky, Y.; Dinca, M. High and reversible ammonia uptake in mesoporous azolate metal organic frameworks with open Mn, Co, and Ni sites. J. Am. Chem. Soc. 2016, 138, 9401–9404. [Google Scholar] [CrossRef]
- Reed, D.A.; Keitz, B.K.; Oktawiec, J.; Mason, J.A.; Runcevski, T.; Xiao, D.J.; Darago, L.E.; Crocella, V.; Bordiga, S.; Long, J.R. A spin transition mechanism for cooperative adsorption in metal-organic frameworks. Nature 2017, 550, 96–100. [Google Scholar] [CrossRef]
- Reed, D.A.; Xiao, D.J.; Gonzalez, M.I.; Darago, L.E.; Herm, Z.R.; Grandjean, F.; Long, J.R. Reversible CO scavenging via adsorbate-dependent spin state transitions in an iron(II)-triazolate metal-organic framework. J. Am. Chem. Soc. 2016, 138, 5594–5602. [Google Scholar] [CrossRef]
- Bloch, E.D.; Queen, W.L.; Krishna, R.; Zadrozny, J.M.; Brown, C.M.; Long, J.R. Hydrocarbon separations in a metal-organic framework with open iron(II) coordination sites. Science 2012, 335, 1606. [Google Scholar] [CrossRef]
- Tulchinsky, Y.; Hendon, C.H.; Lomachenko, K.A.; Borfecchia, E.; Melot, B.C.; Hudson, M.R.; Tarver, J.D.; Korzynski, M.D.; Stubbs, A.W.; Kagan, J.J.; et al. Reversible capture and release of Cl2 and Br2 with a redox-active metal-organic framework. J. Am. Chem. Soc. 2017, 139, 5992–5997. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.H.; Yin, Z.; Tan, Y.X.; Zhang, W.X.; He, Y.P.; Kurmoo, M. Nanoporous cobalt(II) MOF exhibiting four magnetic ground states and changes in gas sorption upon post-synthetic modification. J. Am. Chem. Soc. 2014, 136, 4680–4688. [Google Scholar] [CrossRef] [PubMed]
- Motokawa, N.; Miyasaka, H.; Yamashita, M.; Dunbar, K.R. An electron-transfer ferromagnet with T-c=107 K based on a three-dimensional [Ru-2](2)/TCNQ system. Angew. Chem. Int. Ed. 2008, 47, 7760–7763. [Google Scholar] [CrossRef]
- Stone, K.H.; Stephens, P.W.; McConnell, A.C.; Shurdha, E.; Pokhodnya, K.I.; Miller, J.S. Mn-II(TCNE)(3/2)(I-3)(1/2)-A 3D network-structured organic-based magnet and comparison to a 2D analog. Adv. Mater. 2010, 22, 2514–2519. [Google Scholar] [CrossRef]
- Lapidus, S.H.; McConnell, A.C.; Stephens, P.W.; Miller, J.S. Structure and magnetic ordering of a 2-D Mn-II (TCNE)I(OH2) (TCNE = tetracyanoethylene) organic-based magnet (T-c = 171 K). Chem. Commun. 2011, 47, 7602–7604. [Google Scholar] [CrossRef] [PubMed]
- Jeon, I.R.; Negru, B.; Van Duyne, R.P.; Harris, T.D. A 2D Semiquinone Radical-Containing Microporous Magnet with Solvent-Induced Switching from T-c=26 to 80 K. J. Am. Chem. Soc. 2015, 137, 15699–15702. [Google Scholar] [CrossRef]
- DeGayner, J.A.; Jeon, I.R.; Sun, L.; Dinca, M.; Harris, T.D. 2D Conductive Iron-Quinoid Magnets Ordering up to T-c=105 K via Heterogenous Redox Chemistry. J. Am. Chem. Soc. 2017, 139, 4175–4184. [Google Scholar] [CrossRef]
- Zadrozny, J.M.; Gallagher, A.T.; Harris, T.D.; Freedman, D.E. A Porous Array of Clock Qubits. J. Am. Chem. Soc. 2017, 139, 7089–7094. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-J.; Krzyaniak, M.D.; Fataftah, M.S.; Wasielewski, M.R.; Freedman, D.E. A concentrated array of copper porphyrin candidate qubits. Chem. Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Pashow, K.M.L.; Della Rocca, J.; Xie, Z.G.; Tran, S.; Lin, W.B. Postsynthetic modifications of iron-carboxylate nanoscale metal-organic frameworks for imaging and drug delivery. J. Am. Chem. Soc. 2009, 131, 14261–14263. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Lu, K.; Liu, D.; Lin, W. Nanoscale metal–organic frameworks for the Co-delivery of cisplatin and pooled siRNAs to enhance therapeutic efficacy in drug-resistant ovarian cancer cells. J. Am. Chem. Soc. 2014, 136, 5181–5184. [Google Scholar] [CrossRef]
- Ni, K.Y.; Lan, G.X.; Veroneau, S.S.; Duan, X.P.; Song, Y.; Lin, W.B. Nanoscale metal-organic frameworks for mitochondria-targeted radiotherapy-radiodynamic therapy. Nat. Commun. 2018, 9, 4321. [Google Scholar] [CrossRef]
- Lan, G.X.; Ni, K.Y.; Veroneau, S.S.; Song, Y.; Lin, W.B. Nanoscale metal-organic layers for radiotherapy-radiodynamic therapy. J. Am. Chem. Soc. 2018, 140, 16971–16975. [Google Scholar] [CrossRef] [PubMed]
- Diring, S.; Carne-Sanchez, A.; Zhang, J.C.; Ikemura, S.; Kim, C.; Inaba, H.; Kitagawa, S.; Furukawa, S. Light responsive metal-organic frameworks as controllable CO-releasing cell culture substrates. Chem. Sci. 2017, 8, 2381–2386. [Google Scholar] [CrossRef] [PubMed]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, F.; Drake, H.F.; Feng, L.; Powell, J.A.; Wang, K.-Y.; Yan, T.-H.; Zhou, H.-C. Metal–Organic Frameworks as Versatile Platforms for Organometallic Chemistry. Inorganics 2021, 9, 27. https://doi.org/10.3390/inorganics9040027
Chen F, Drake HF, Feng L, Powell JA, Wang K-Y, Yan T-H, Zhou H-C. Metal–Organic Frameworks as Versatile Platforms for Organometallic Chemistry. Inorganics. 2021; 9(4):27. https://doi.org/10.3390/inorganics9040027
Chicago/Turabian StyleChen, Fan, Hannah F. Drake, Liang Feng, Joshua A. Powell, Kun-Yu Wang, Tian-Hao Yan, and Hong-Cai Zhou. 2021. "Metal–Organic Frameworks as Versatile Platforms for Organometallic Chemistry" Inorganics 9, no. 4: 27. https://doi.org/10.3390/inorganics9040027
APA StyleChen, F., Drake, H. F., Feng, L., Powell, J. A., Wang, K.-Y., Yan, T.-H., & Zhou, H.-C. (2021). Metal–Organic Frameworks as Versatile Platforms for Organometallic Chemistry. Inorganics, 9(4), 27. https://doi.org/10.3390/inorganics9040027