
Functionalized Tris(anilido)triazacyclononanes as Hexadentate Ligands for the Encapsulation of U(III), U(IV) and La(III) Cations

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
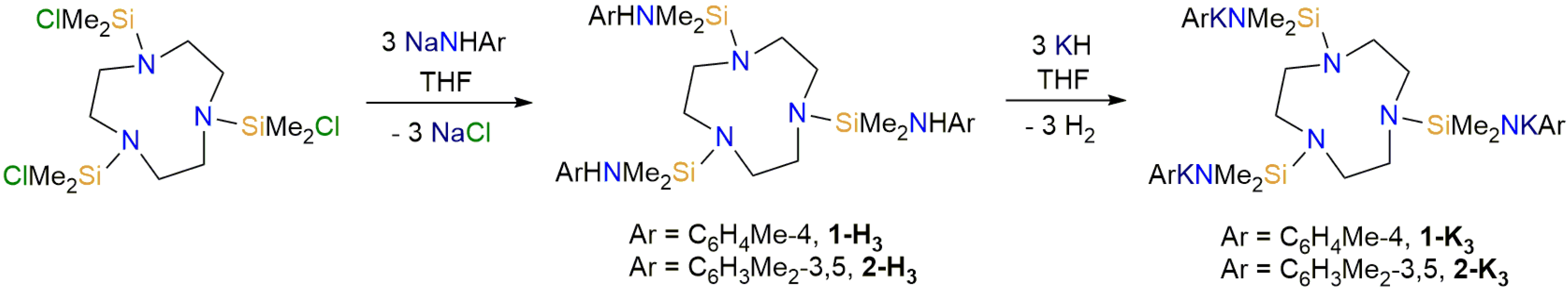
2.1. Synthesis
2.2. NMR and IR Spectroscopy
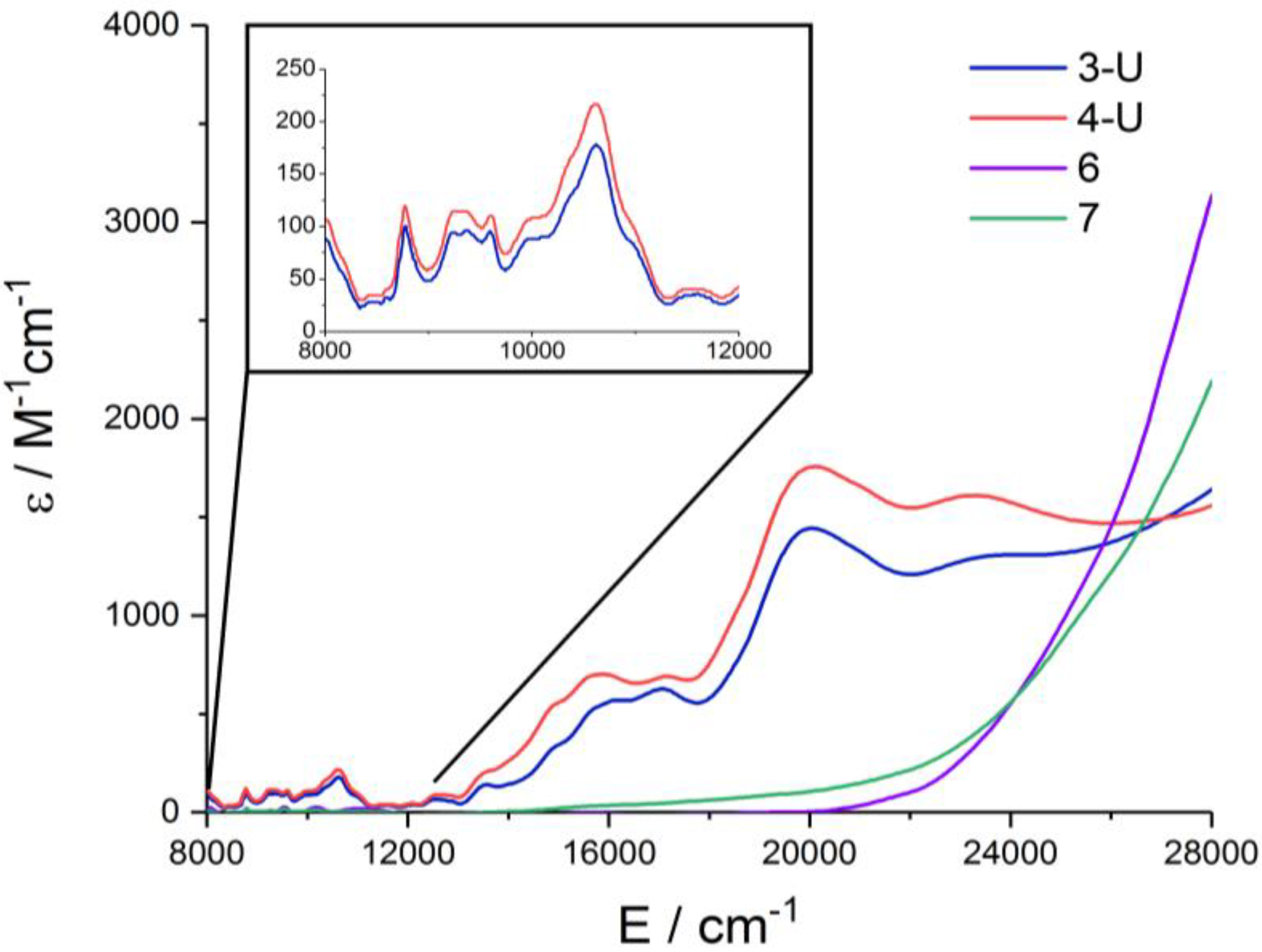
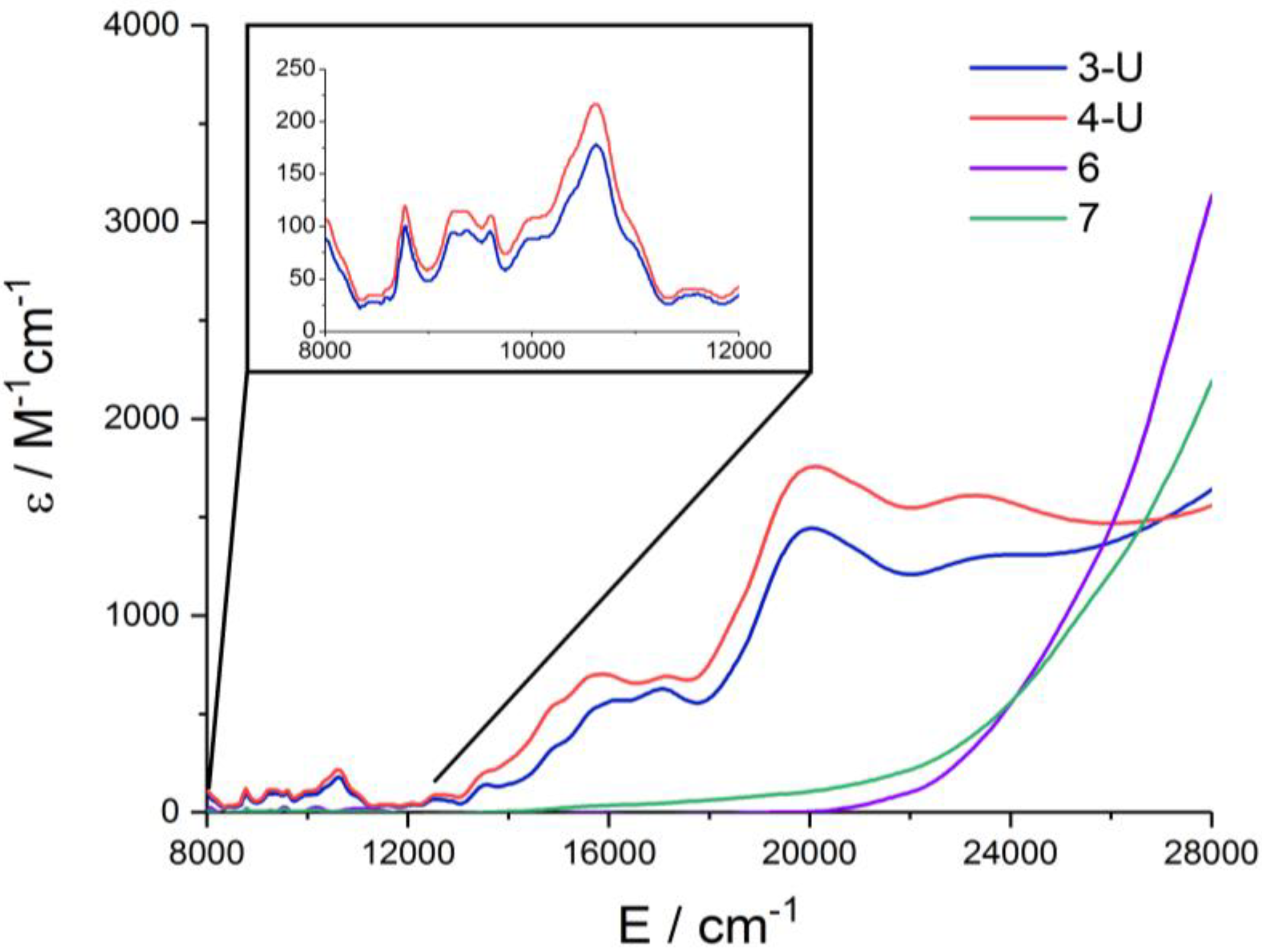
2.3. NIR/Vis/UV Spectroscopy
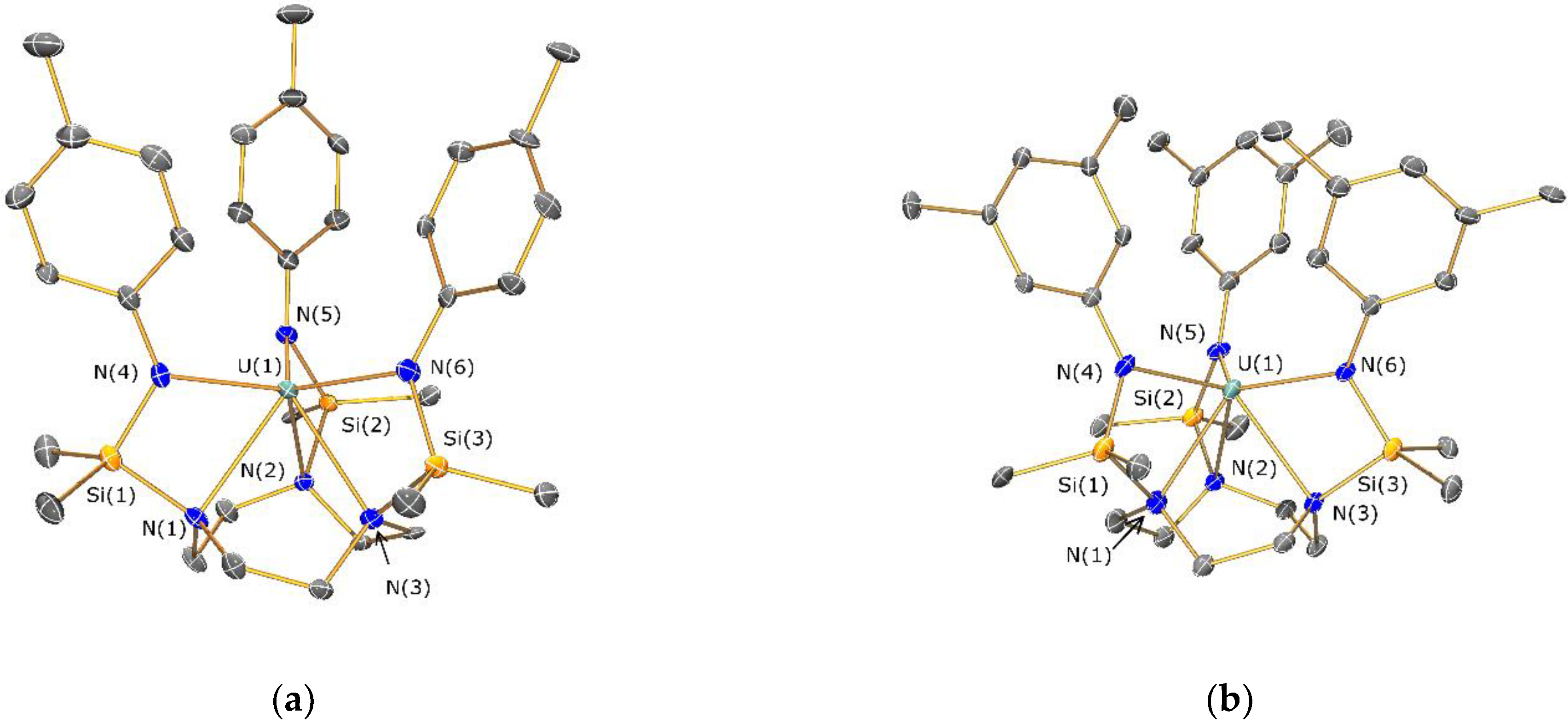
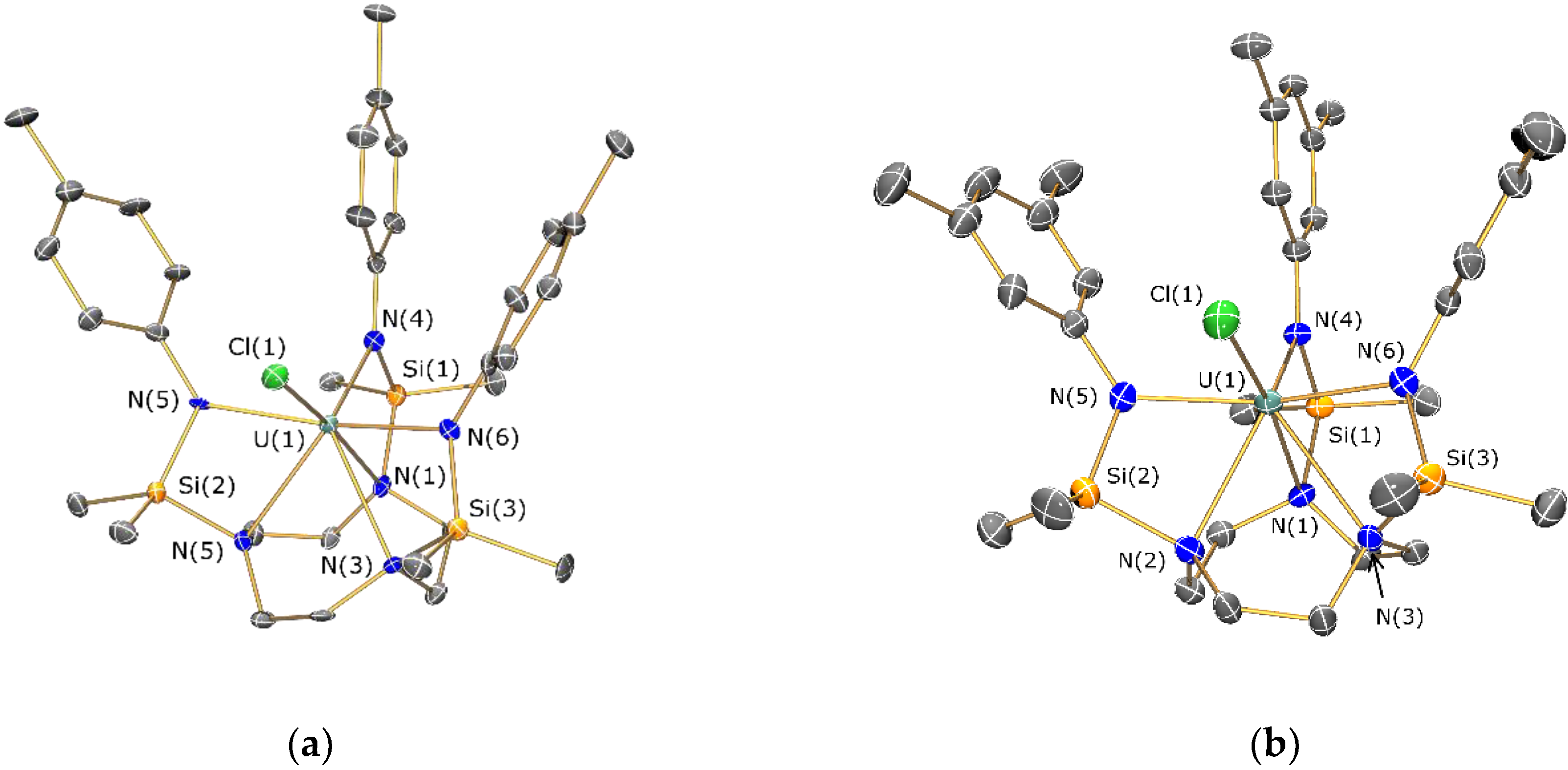
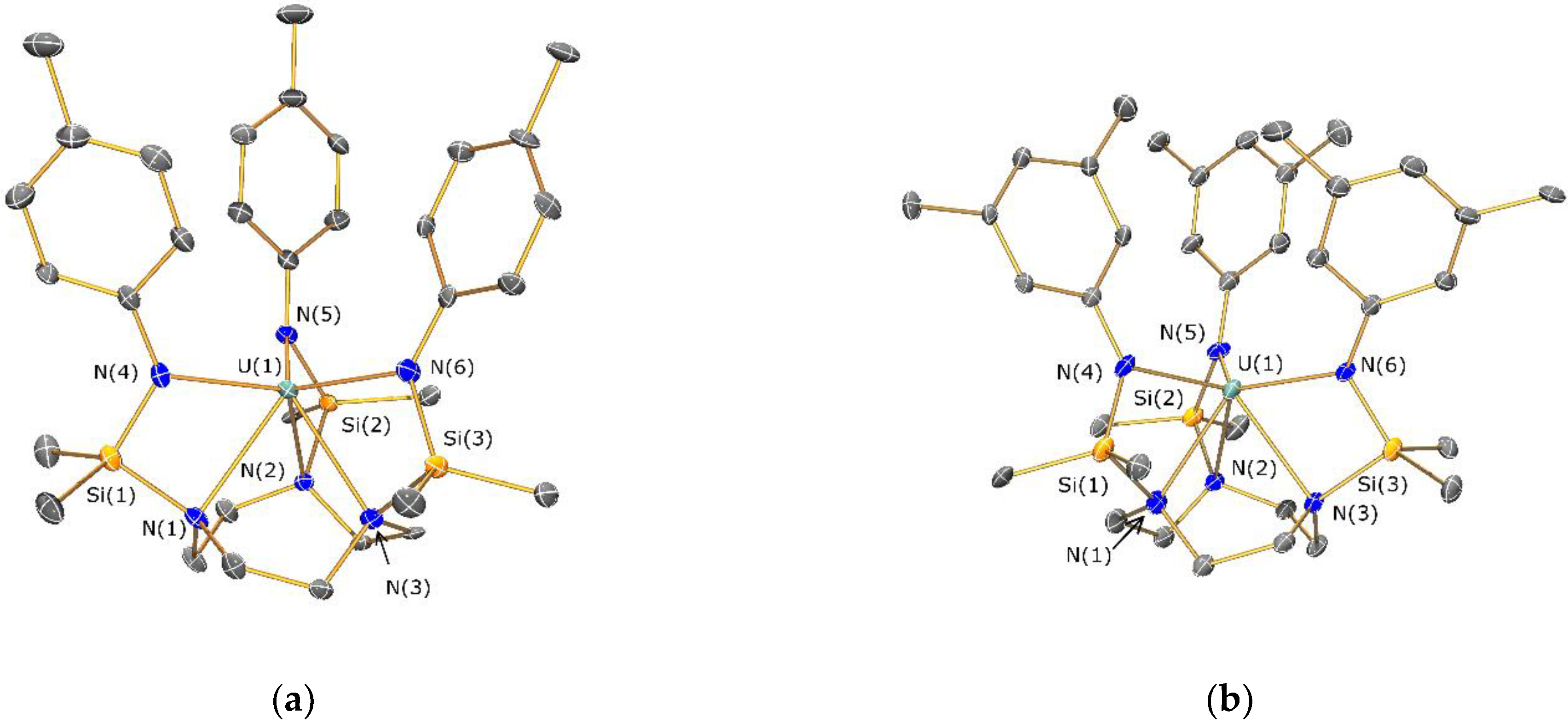
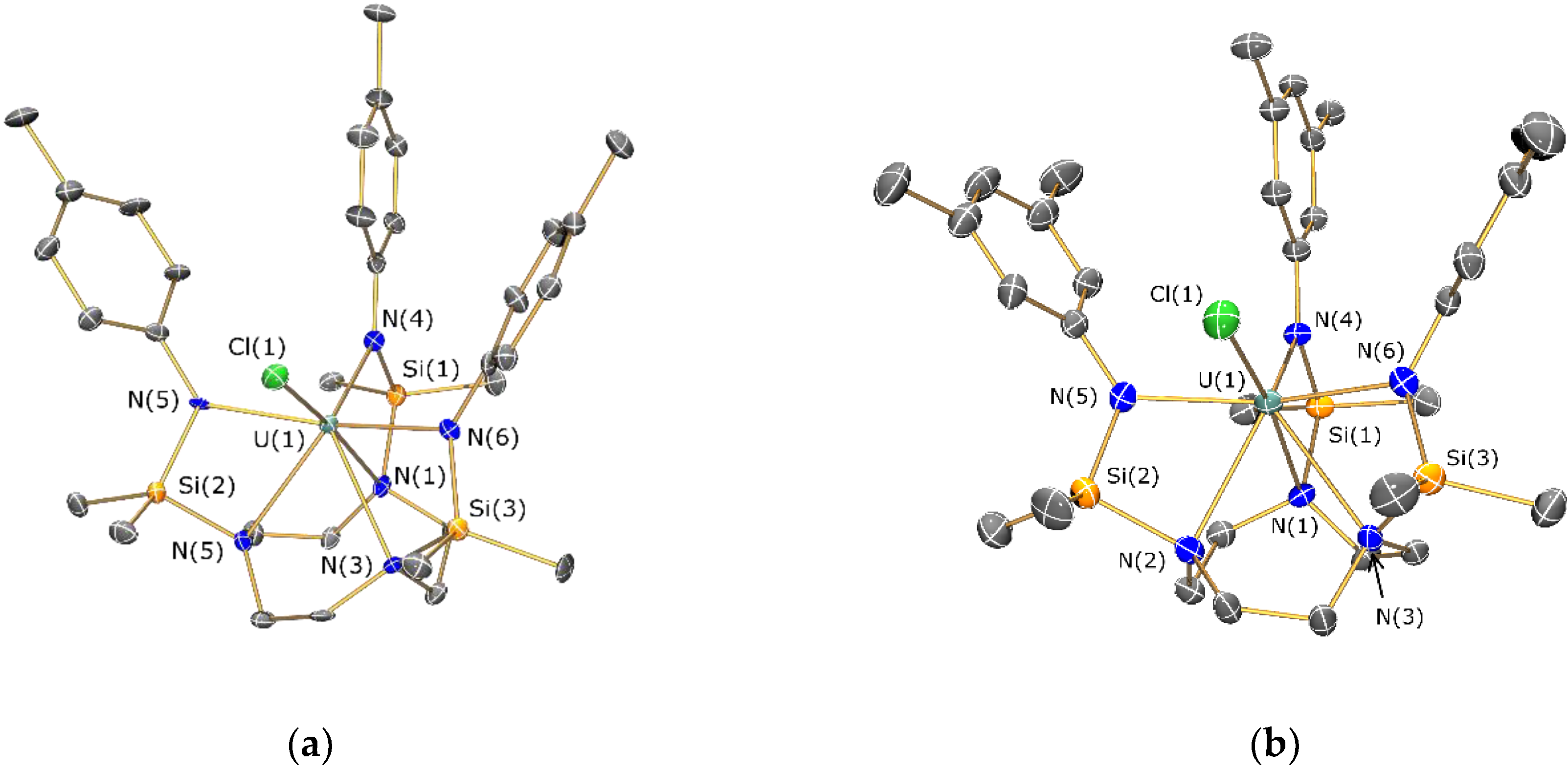
2.4. Structural Characterization
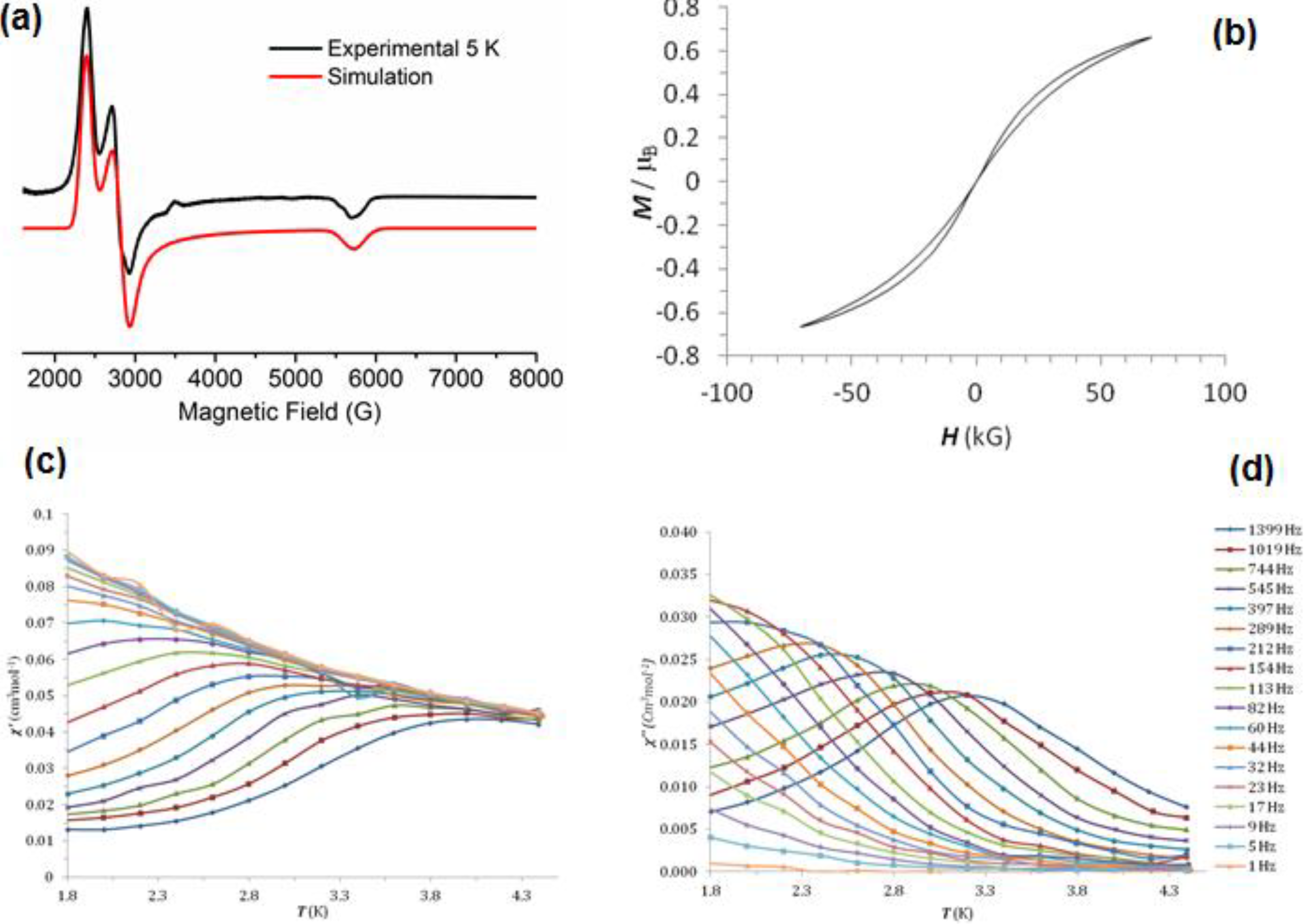
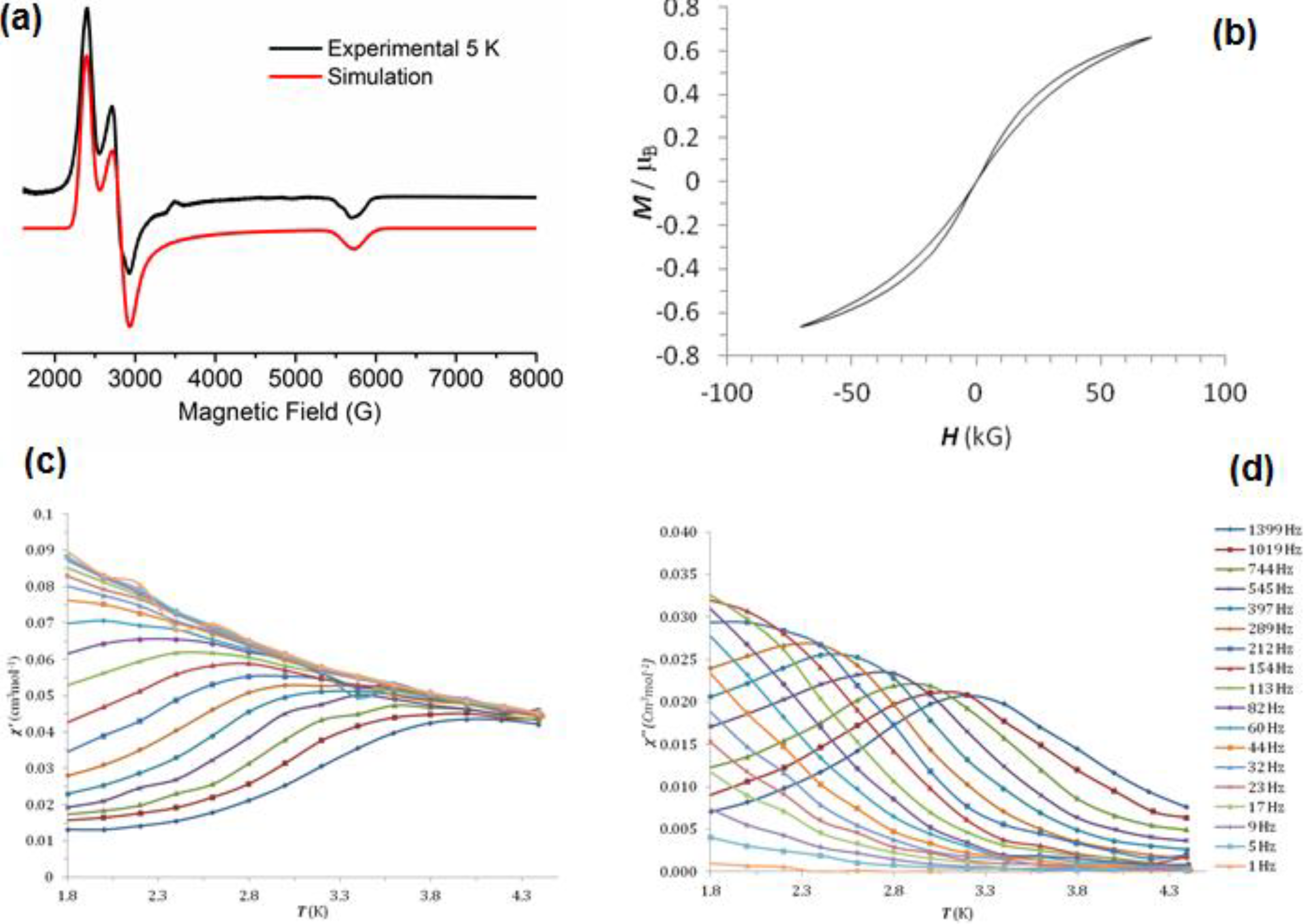
2.5. Magnetism and EPR Spectroscopy
3. Discussion
4. Materials and Methods
General Information
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mills, D.P.; Liddle, S.T. Ligand Design in Modern Lanthanide Chemistry. In Ligand Design in Metal Chemistry: Reactivity and Catalysis; Lundgren, R., Stradiotto, M., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2016; pp. 330–363. [Google Scholar] [CrossRef]
- Cotton, S. Lanthanide and Actinide Chemistry; John Wiley & Sons, Ltd.: Chichester, UK, 2006. [Google Scholar] [CrossRef]
- Ephritikhine, M. The vitality of uranium molecular chemistry at the dawn of the XXIst century. Dalton Trans. 2006, 2501–2516. [Google Scholar] [CrossRef] [PubMed]
- Bart, S.C.; Meyer, K. Highlights in Uranium Coordination Chemistry. Struct. Bond. 2008, 127, 119–176. [Google Scholar] [CrossRef]
- Meyer, K.; Bart, S.C. Tripodal carbene and aryloxide ligands for small-molecule activation at electron-rich uranium and transition metal centers. Adv. Inorg. Chem. 2008, 60, 1–30. [Google Scholar] [CrossRef]
- Liddle, S.T. The Renaissance of Non-Aqueous Uranium Chemistry. Angew. Chem. Int. Ed. 2015, 54, 8604–8641. [Google Scholar] [CrossRef]
- Gardner, B.M.; Liddle, S.T. Uranium Triamidoamine Chemistry. Chem. Commun. 2015, 51, 10589–10607. [Google Scholar] [CrossRef] [Green Version]
- Maria, L.; Santos, I.C.; Sousa, V.R.; Marçalo, J. Uranium(III) Redox Chemistry Assisted by a Hemilabile Bis(phenolate) Cyclam Ligand: Uranium–Nitrogen Multiple Bond Formation Comprising a trans-{RN=U(VI)=NR}2+ Complex. Inorg. Chem. 2015, 54, 9115–9126. [Google Scholar] [CrossRef]
- Maria, L.; Bandeira, N.A.G.; Santos, I.C.; Marçalo, J.; Gibson, J.K. CO2 conversion to phenyl isocyanates by uranium(vi) bis(imido) complexes. Chem. Commun. 2020, 56, 431–434. [Google Scholar] [CrossRef]
- Xin, T.; Wang, X.; Yang, K.; Liang, J.; Huang, W. Rare Earth Metal Complexes Supported by a Tripodal Tris(amido) Ligand System Featuring an Arene Anchor. Inorg. Chem. 2021, 60, 15321–15329. [Google Scholar] [CrossRef]
- Halter, D.P.; Heinemann, F.W.; Bachman, J.; Meyer, K. Uranium-mediated electrocatalytic dihydrogen production from water. Nature 2016, 530, 317–321. [Google Scholar] [CrossRef]
- Halter, D.P.; Heinemann, F.W.; Maron, L.; Meyer, K. The role of uranium–arene bonding in H2O reduction catalysis. Nat. Chem. 2018, 10, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Halter, D.P.; Palumbo, C.T.; Ziller, J.W.; Gembicky, M.; Rheingold, A.L.; Evans, W.J.; Meyer, K. Electrocatalytic H2O Reduction with f-Elements: Mechanistic Insight and Overpotential Tuning in a Series of Lanthanide Complexes. J. Am. Chem. Soc. 2018, 140, 2587–2594. [Google Scholar] [CrossRef] [PubMed]
- Castro-Rodríguez, I.; Meyer, K. Carbon Dioxide Reduction and Carbon Monoxide Activation Employing a Reactive Uranium(III) Complex. J. Am. Chem. Soc. 2005, 127, 11242–11243. [Google Scholar] [CrossRef] [PubMed]
- Castro-Rodríguez, I.; Nakai, H.; Zacharov, L.N.; Rheingold, A.L.; Meyer, K. A Linear, O-Coordinated η1-CO2 Bound to Uranium. Science 2004, 305, 1757–1760. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, B.; Roitershtein, D.; Ferreira, H.; Ascenso, J.R.; Martins, A.M.; Domingos, A.; Marques, N. Triamidotriazacyclononane Complexes of Group 3 Metals. Synthesis and Crystal Structures. Inorg. Chem. 2003, 42, 4223–4231. [Google Scholar] [CrossRef] [PubMed]
- Antunes, M.A.; Dias, M.; Monteiro, B.; Domingos, A.; Santos, I.C.; Marques, N. Synthesis and reactivity of uranium(iv) amide complexes supported by a triamidotriazacyclononane ligand. Dalton Trans. 2006, 3368–3374. [Google Scholar] [CrossRef]
- Camp, C.; Antunes, M.A.; García, G.; Ciofini, I.; Santos, I.C.; Pécaut, J.; Almeida, M.; Marçalo, J.; Mazzanti, M. Two-electron versus one-electron reduction of chalcogens by uranium(iii): Synthesis of a terminal U(v) persulfide complex. Chem. Sci. 2014, 5, 841–850. [Google Scholar] [CrossRef]
- Antunes, M.A.; Coutinho, J.T.; Santos, I.C.; Marçalo, J.; Almeida, M.; Baldoví, J.J.; Pereira, L.C.J.; Gaita-Ariño, A.; Coronado, E. A Mononuclear Uranium(IV) Single-Molecule Magnet with an Azobenzene Radical Ligand. Chem. Eur. J. 2015, 21, 17817–17826. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, J.T.; Perfetti, M.; Baldoví, J.J.; Antunes, M.A.; Hallmen, P.P.; Bamberger, H.; Crassee, I.; Orlita, M.; Almeida, M.; van Slageren, J.; et al. Spectroscopic Determination of the Electronic Structure of a Uranium Single-Ion Magnet. Chem. Eur. J. 2019, 25, 1758–1766. [Google Scholar] [CrossRef]
- Dias, A.R.; Martins, A.M.; Ascenso, J.R.; Ferreria, H.; Duarte, M.T.; Henriques, R.T. Li, Ti(III), and Ti(IV) Trisamidotriazacyclononane Complexes. Syntheses, Reactivity, and Structures. Inorg. Chem. 2003, 42, 2675–2682. [Google Scholar] [CrossRef]
- Izod, K.; Liddle, S.T.; Clegg, W. A Convenient Route to Lanthanide Triiodide THF Solvates. Crystal Structures of LnI3(THF)4 [Ln = Pr] and LnI3(THF)3.5 [Ln = Nd, Gd, Y]. Inorg. Chem. 2004, 43, 214–218. [Google Scholar] [CrossRef]
- Avens, L.R.; Bott, S.G.; Clark, D.L.; Sattelberger, A.P.; Watkin, J.G.; Zwick, B.D. A Convenient Entry into Trivalent Actinide Chemistry: Synthesis and Characterization of AnI3(THF)4 and An[N(SiMe3)2]3 (An = U, Np, Pu). Inorg. Chem. 1994, 33, 2248–2256. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Patel, D.; Wooles, A.J.; Hashem, E.; Omorodion, H.; Baker, R.J.; Liddle, S.T. Comments on reactions of oxide derivatives of uranium with hexachloropropene to give UCl4. New J. Chem. 2015, 39, 7559–7562. [Google Scholar] [CrossRef] [Green Version]
- Kiplinger, J.L.; Morris, D.E.; Scott, B.L.; Burns, C.J. Convenient Synthesis, Structure, and Reactivity of (C5Me5)U(CH2C6H5)3: A Simple Strategy for the Preparation of Monopentamethylcyclopentadienyl Uranium(IV) Complexes. Organometallics 2002, 21, 5978–5982. [Google Scholar] [CrossRef]
- Hashem, E.; Swinburne, A.N.; Schulzke, C.; Evans, R.C.; Platts, J.A.; Kerridge, A.; Natrajan, L.S.; Baker, R.J. Emission spectroscopy of uranium(IV) compounds: A combined synthetic, spectroscopic and computational study. RSC Adv. 2013, 3, 4350–4361. [Google Scholar] [CrossRef] [Green Version]
- Windorff, C.J.; Evans, W.J. 29Si NMR Spectra of Silicon-Containing Uranium Complexes. Organometallics 2014, 33, 3786–3791. [Google Scholar] [CrossRef]
- Carnall, W.T. A systematic analysis of the spectra of trivalent actinide chlorides in D3h site symmetry. J. Chem. Phys. 1992, 96, 8713. [Google Scholar] [CrossRef] [Green Version]
- Sur, S.K. Measurement of magnetic susceptibility and magnetic moment of paramagnetic molecules in solution by high-field fourier transform NMR spectroscopy. J. Magn. Reson. 1989, 82, 169–173. [Google Scholar] [CrossRef]
- Jones, E.R., Jr.; Hendricks, M.E.; Stone, J.A.; Karraker, D.G. Magnetic properties of the trichlorides, tribromides, and triiodides of U(III), Np(III), and Pu(III). J. Chem. Phys. 1974, 60, 2088. [Google Scholar] [CrossRef]
- Kindra, D.R.; Evans, W.J. Magnetic Susceptibility of Uranium Complexes. Chem. Rev. 2014, 114, 8865–8882. [Google Scholar] [CrossRef]
- Liddle, S.T.; van Slageren, J. Actinide Single Molecule Magnets. In Lanthanides and Actinides in Molecular Magnetism; Layfield, R.A., Murugesu, M., Eds.; Wiley-VCH: Weinheim, Germany, 2015; pp. 315–340. [Google Scholar] [CrossRef]
- Liddle, S.T.; van Slageren, J. Improving f-element single-molecule magnets. Chem. Soc. Rev. 2015, 44, 6655–6670. [Google Scholar] [CrossRef] [Green Version]
- Moro, F.; Mills, D.P.; Liddle, S.T.; van Slageren, J. The Inherent Single-Molecule Magnet Character of Trivalent Uranium. Angew. Chem. Int. Ed. 2013, 52, 3430–3433. [Google Scholar] [CrossRef]
- Goodwin, C.A.P.; Tuna, F.; McInnes, E.J.L.; Liddle, S.T.; McMaster, J.; Vitorica-Yrezabal, I.J.; Mills, D.P. [UIII{N(SiMe2tBu)2}3]: A Structurally Authenticated Trigonal Planar Actinide Complex. Chem. Eur. J. 2014, 20, 14579–14582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CrysAlisPRO; Agilent Technologies UK Ltd.: Yarnton, UK, 2010.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- POV-Ray; Persistence of Vision Raytracer Pty., Ltd.: Williamstown, Australia, 2004.
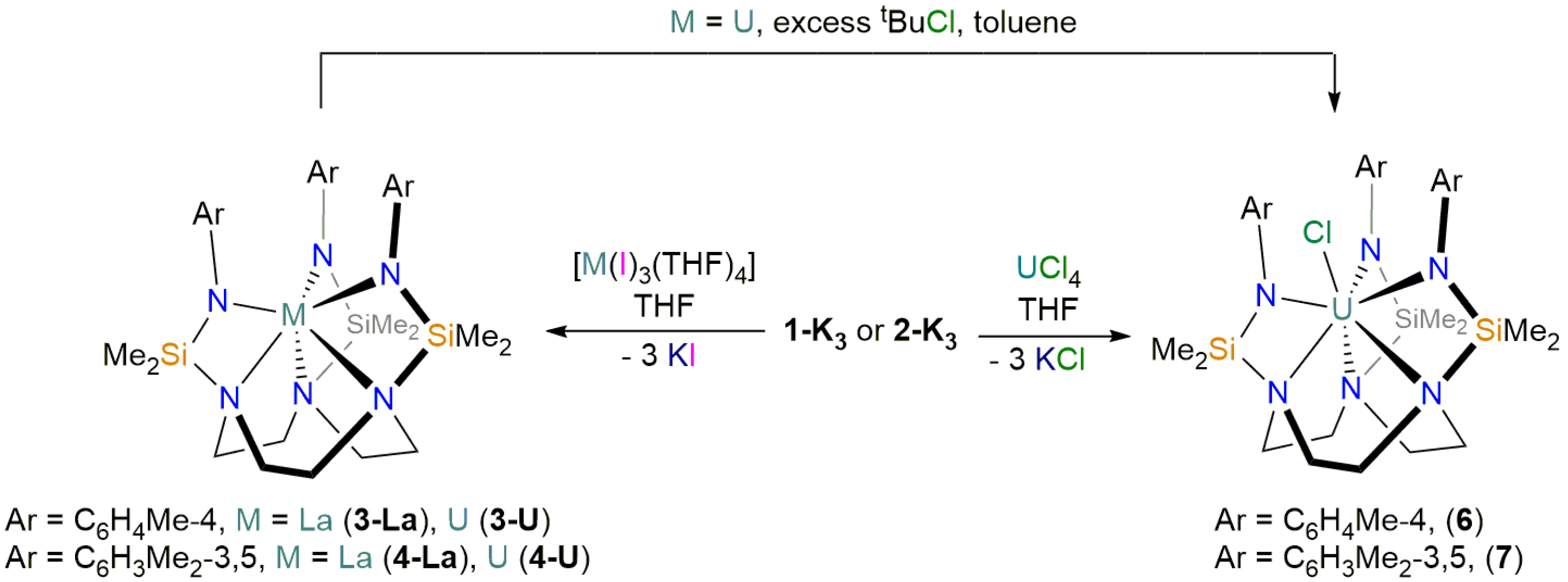

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | M–Namine | M–Nanilide | N–M–Namine | N–M–Nanilide | M–X |
|---|---|---|---|---|---|
| 3-La·0.5C7H8 | 2.705(3) | 2.395(3) | 66.21(8) | 120.06(9) | |
| 2.705(3) | 2.414(3) | 68.00(9) | 113.46(8) | - | |
| 2.654(3) | 2.436(3) | 67.54(7) | 115.46(10) | ||
| 3-U·0.5C7H8 | 2.637(9) | 2.381(8) | 68.2(2) | 119.0(3) | |
| 2.659(8) | 2.364(7) | 67.4(3) | 116.6(3) | - | |
| 2.658(6) | 2.396(10) | 67.5(3) | 110.8(3) | ||
| 4-La | 2.674(2) | 2.421(3) | 67.80(7) | 115.74(9) | |
| 2.676(3) | 2.426(2) | 67.09(8) | 116.64(9) | - | |
| 2.707(3) | 2.400(3) | 68.02(8) | 115.99(8) | ||
| 4-U | 2.616(12) | 2.348(10) | 68.2(4) | 117.9(6) | |
| 2.646(10) | 2.31(2) | 68.3(4) | 115.6(4) | - | |
| 2.63(2) | 2.418(14) | 68.6(5) | 112.9(5) | ||
| 5 | 2.74(4) | 2.36(2) | 66.0(1) | 96.6(4) | |
| 2.67(2) | 2.32(2) | 64.8.(8) | 154.5(7) | 3.143(2) | |
| 2.59(4) | 2.32(2) i | 67.7(9) | 96.6(4) i | ||
| 6·THF | 2.659(8) | 2.285(7) | 66.6(3) | 95.4(3) | |
| 2.683(7) | 2.295(8) | 67.1(2) | 160.3(3) | 2.706(3) | |
| 2.635(7) | 2.327(8) | 66.0(2) | 94.1(3) | ||
| 7 | 2.69(2) | 2.305(5) | 66.8(5) | 95.77(10) | |
| 2.667(7) | 2.286(5) | 67.8(5) | 158.2(3)(10) | 2.687(2) | |
| 2.62(2) | 2.305(5) ii | 65.4(6) | 95.77(10) ii |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Formanuik, A.; Ortu, F.; Vitorica-Yrezabal, I.J.; Tuna, F.; McInnes, E.J.L.; Natrajan, L.S.; Mills, D.P. Functionalized Tris(anilido)triazacyclononanes as Hexadentate Ligands for the Encapsulation of U(III), U(IV) and La(III) Cations. Inorganics 2021, 9, 86. https://doi.org/10.3390/inorganics9120086
Formanuik A, Ortu F, Vitorica-Yrezabal IJ, Tuna F, McInnes EJL, Natrajan LS, Mills DP. Functionalized Tris(anilido)triazacyclononanes as Hexadentate Ligands for the Encapsulation of U(III), U(IV) and La(III) Cations. Inorganics. 2021; 9(12):86. https://doi.org/10.3390/inorganics9120086
Chicago/Turabian StyleFormanuik, Alasdair, Fabrizio Ortu, Iñigo J. Vitorica-Yrezabal, Floriana Tuna, Eric J. L. McInnes, Louise S. Natrajan, and David P. Mills. 2021. "Functionalized Tris(anilido)triazacyclononanes as Hexadentate Ligands for the Encapsulation of U(III), U(IV) and La(III) Cations" Inorganics 9, no. 12: 86. https://doi.org/10.3390/inorganics9120086
APA StyleFormanuik, A., Ortu, F., Vitorica-Yrezabal, I. J., Tuna, F., McInnes, E. J. L., Natrajan, L. S., & Mills, D. P. (2021). Functionalized Tris(anilido)triazacyclononanes as Hexadentate Ligands for the Encapsulation of U(III), U(IV) and La(III) Cations. Inorganics, 9(12), 86. https://doi.org/10.3390/inorganics9120086