
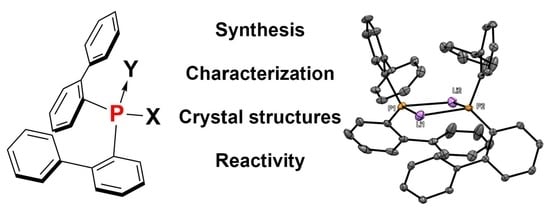
The bis(Biphenyl)phosphorus Fragment in Trivalent and Tetravalent P-Environments



Abstract
:
1. Introduction
2. Results
2.1. Syntheses
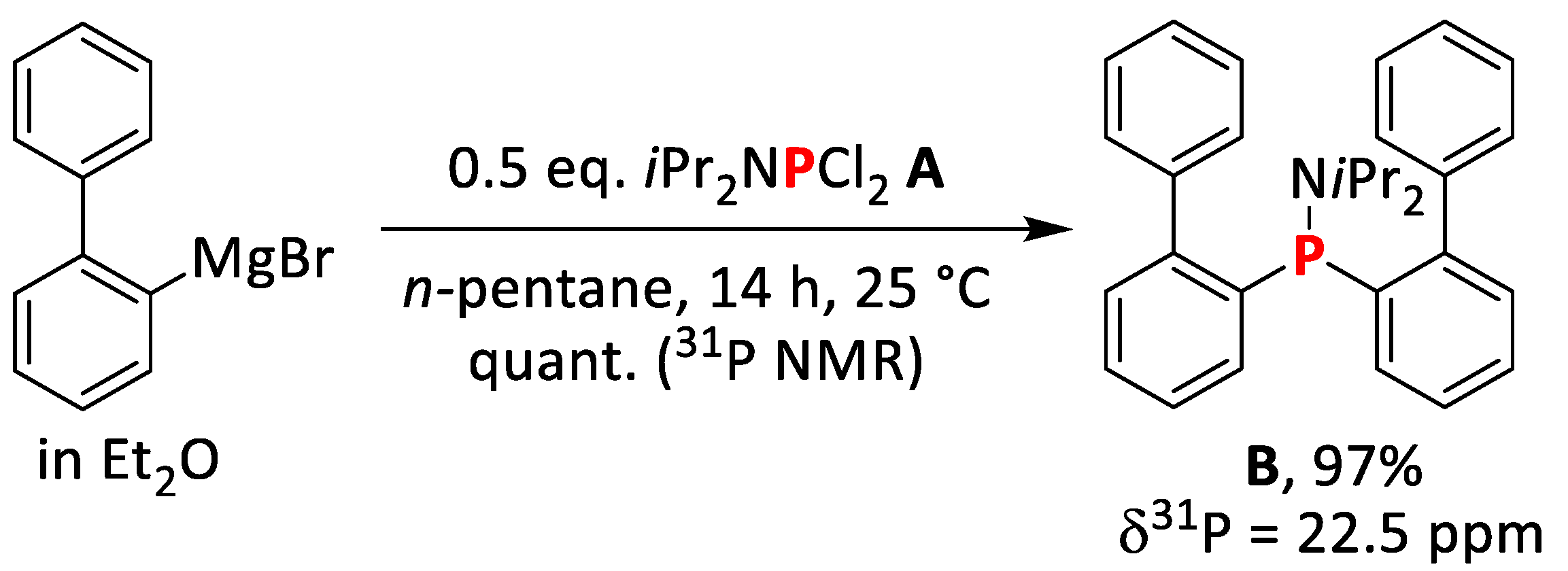
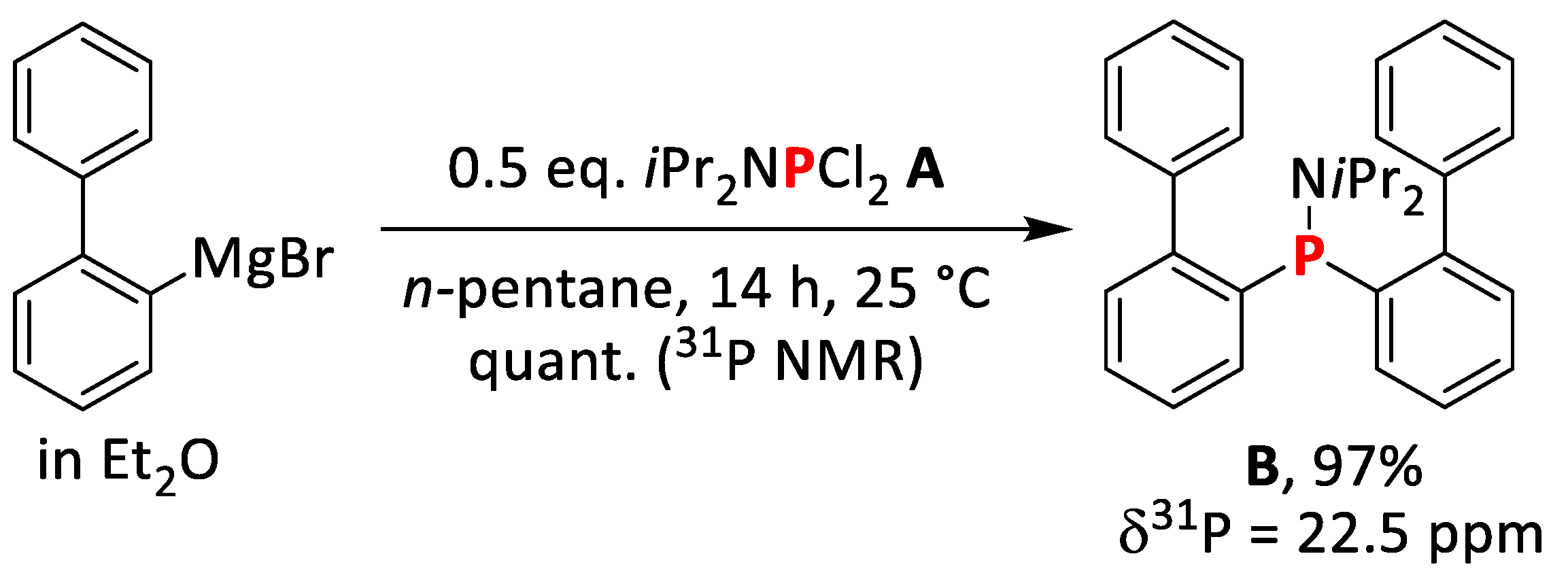
2.1.1. Synthesis of bis(Biphenyl)diisopropylamino Phosphine (A)
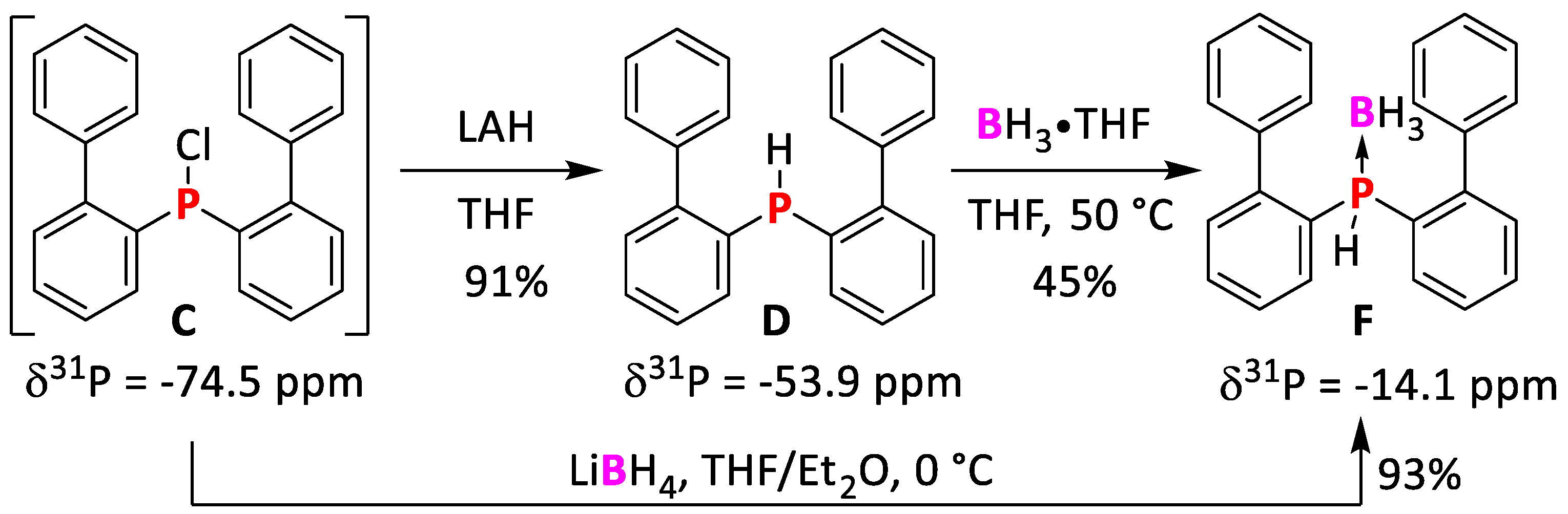
2.1.2. Synthesis of bis(Biphenyl)chlorophosphine
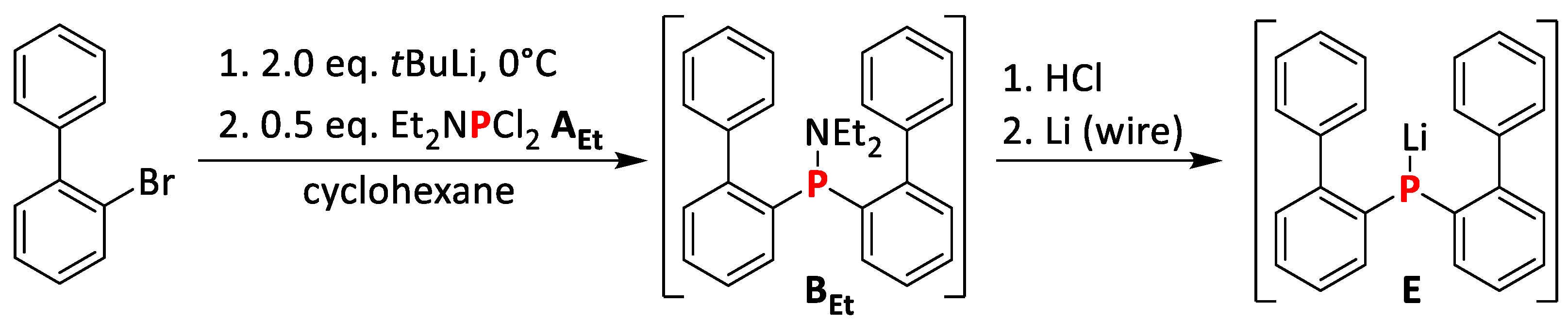
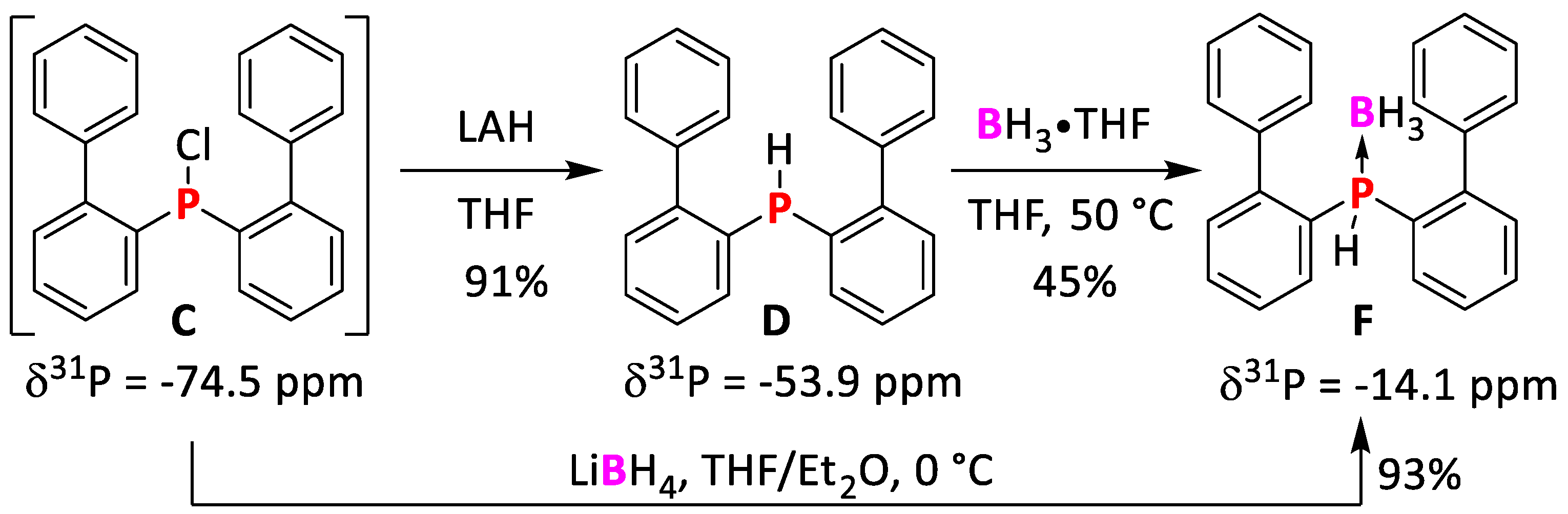
2.1.3. Synthesis of bis(Biphenyl)phosphine
2.1.4. Synthesis of bis(Biphenyl)phosphine Borane Adduct F
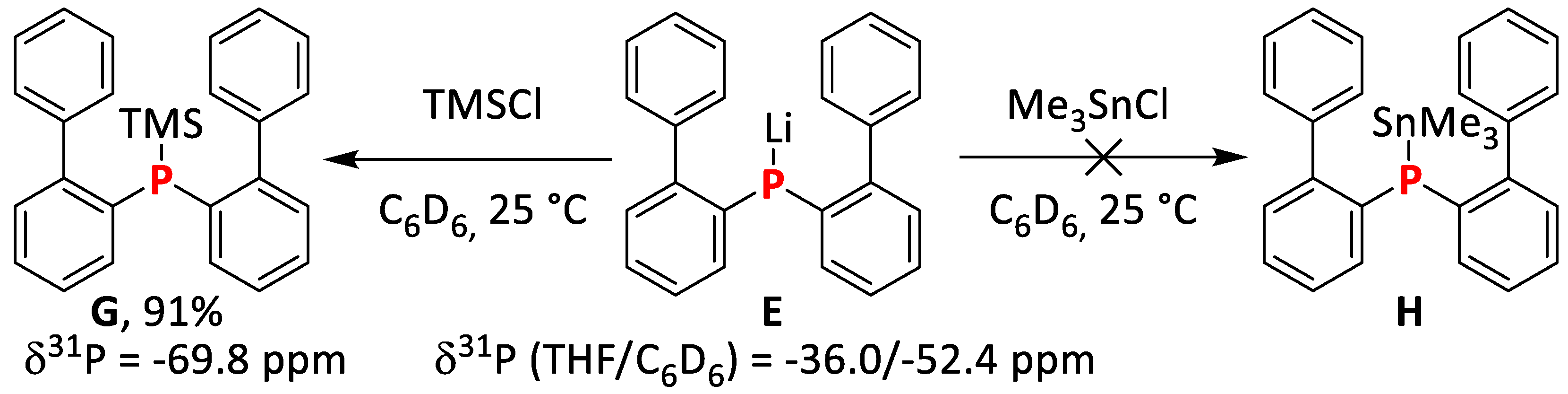
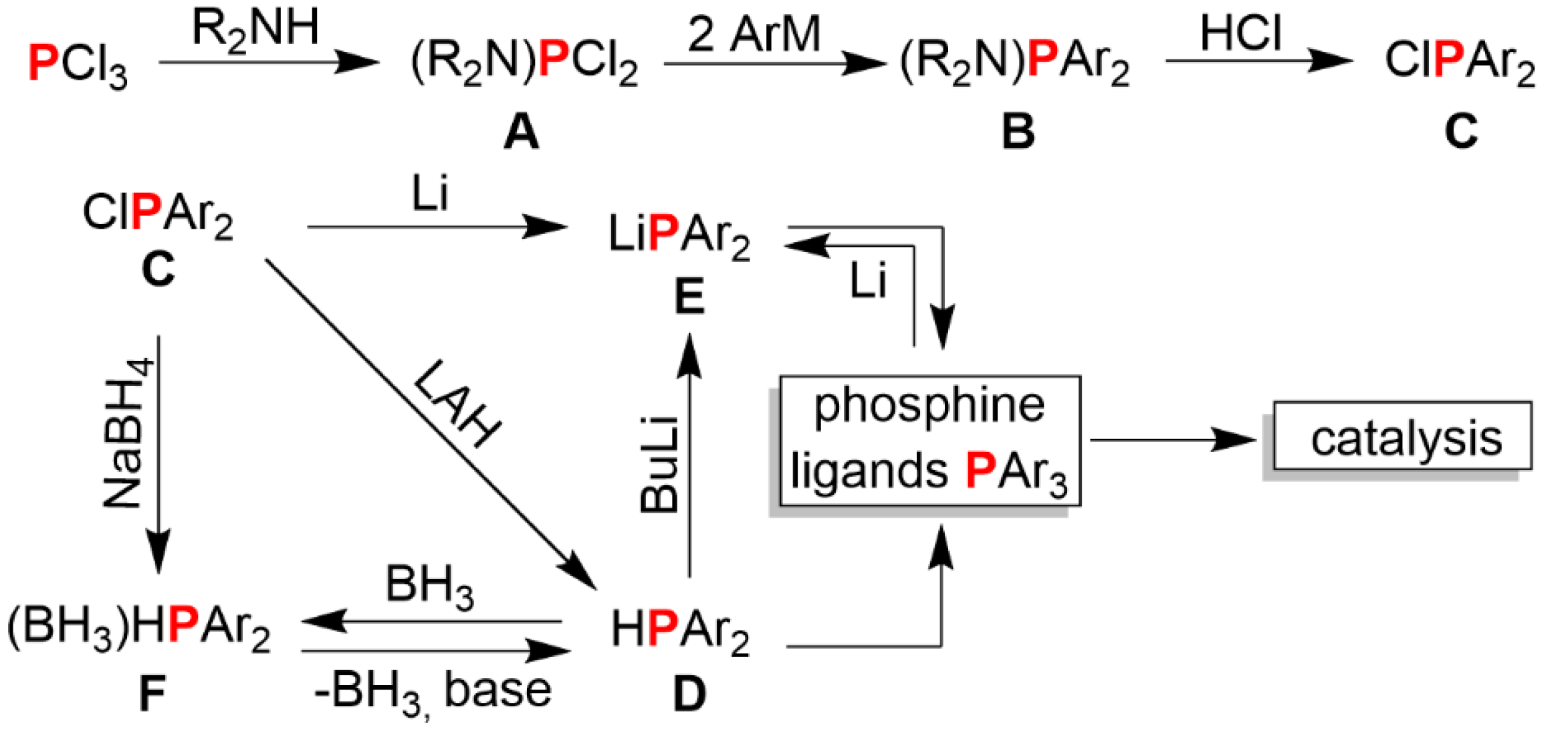
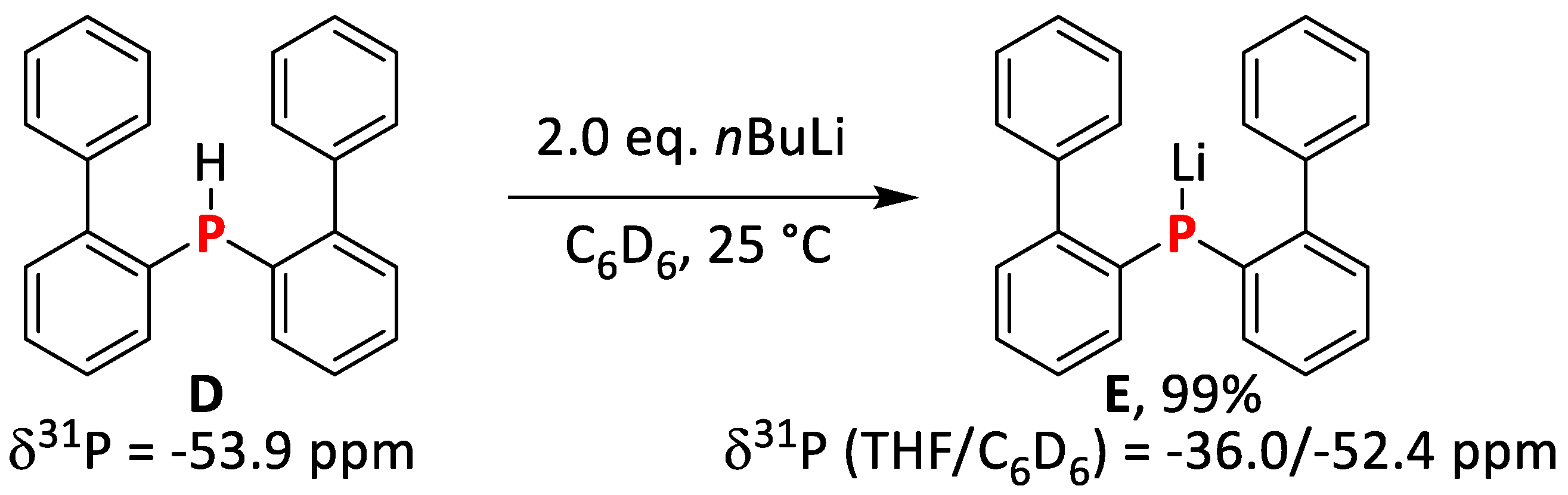
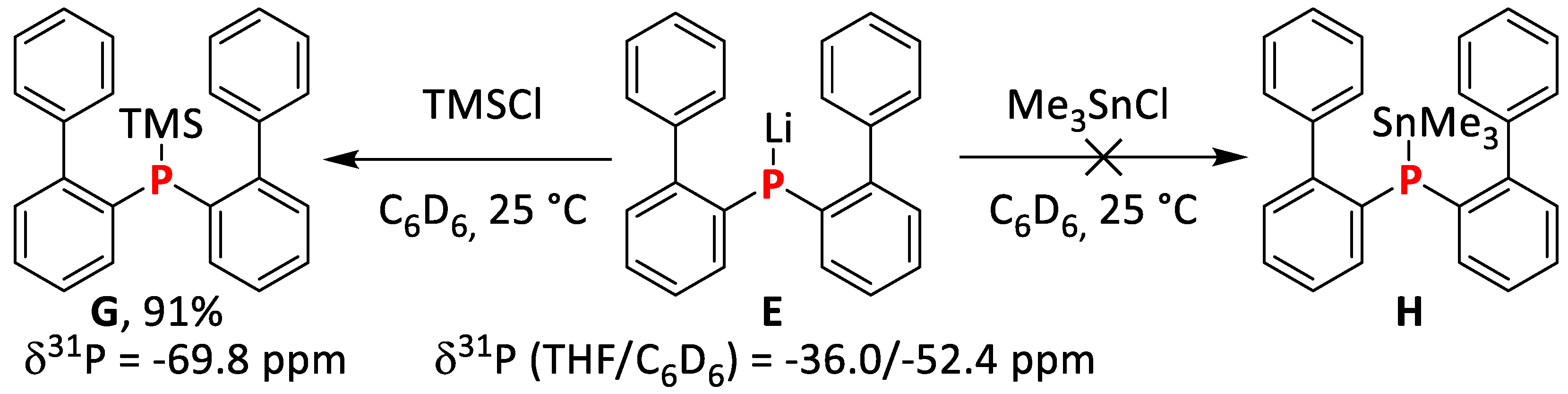
2.1.5. Synthesis and Reactivity of Lithium bis(Biphenyl)phosphide
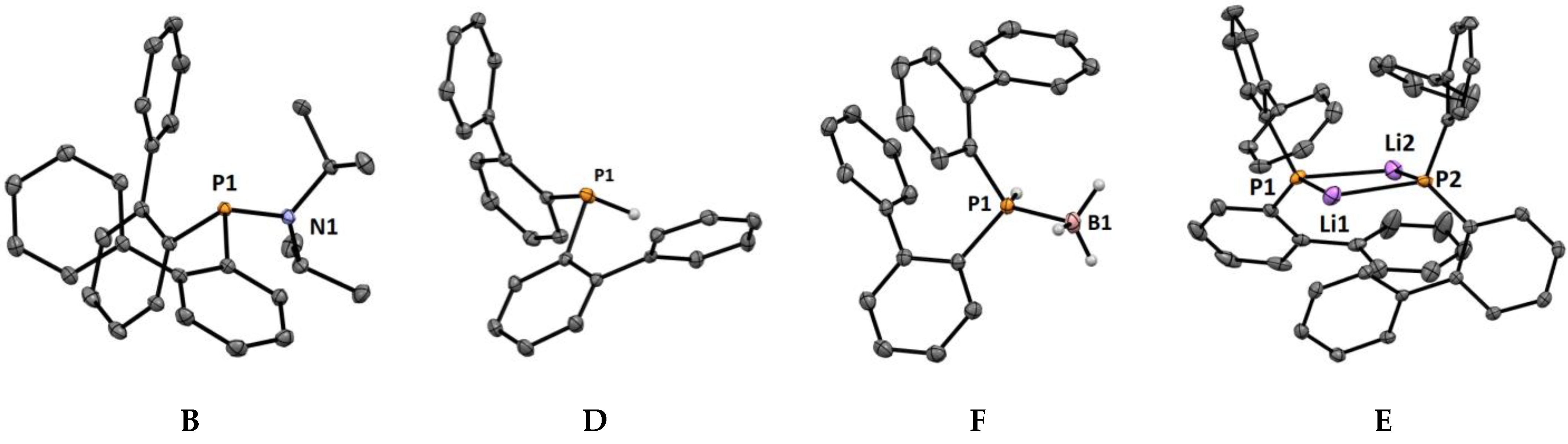
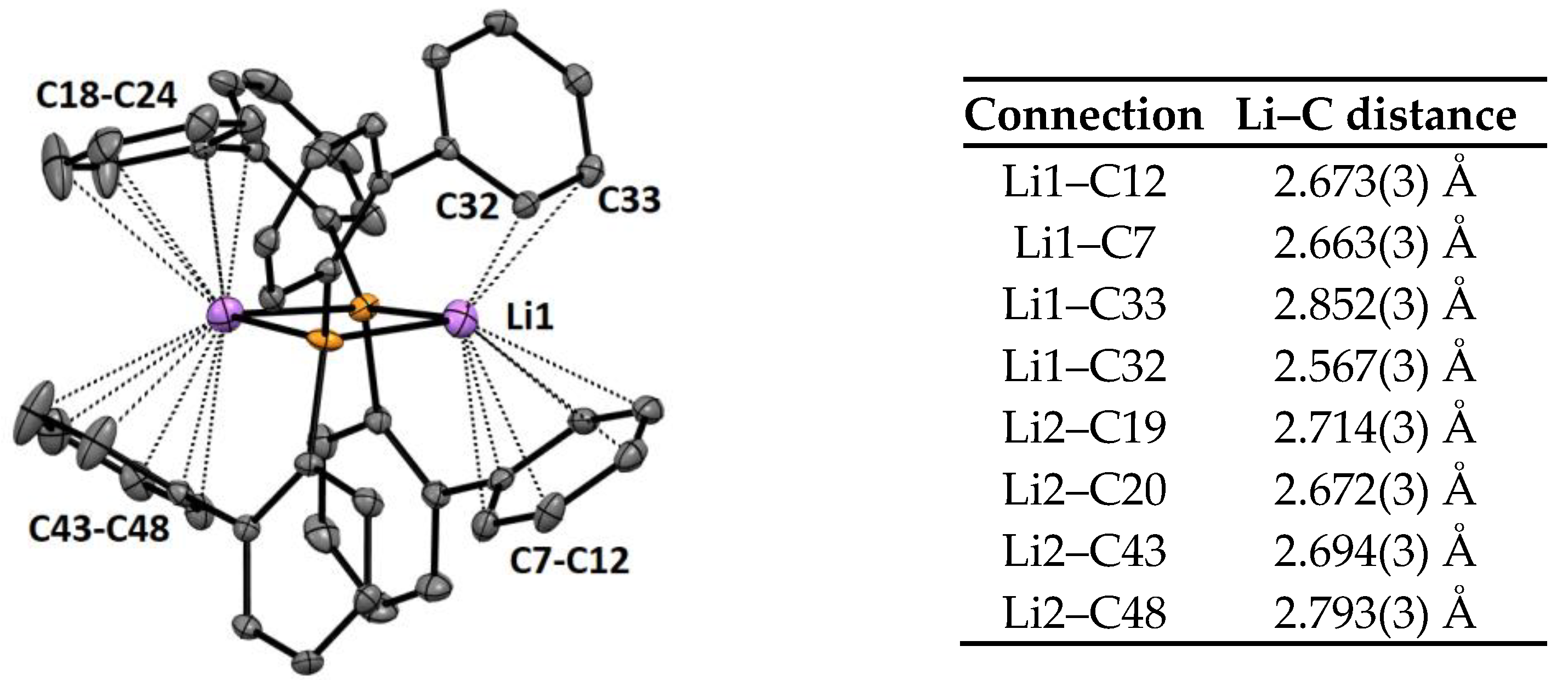
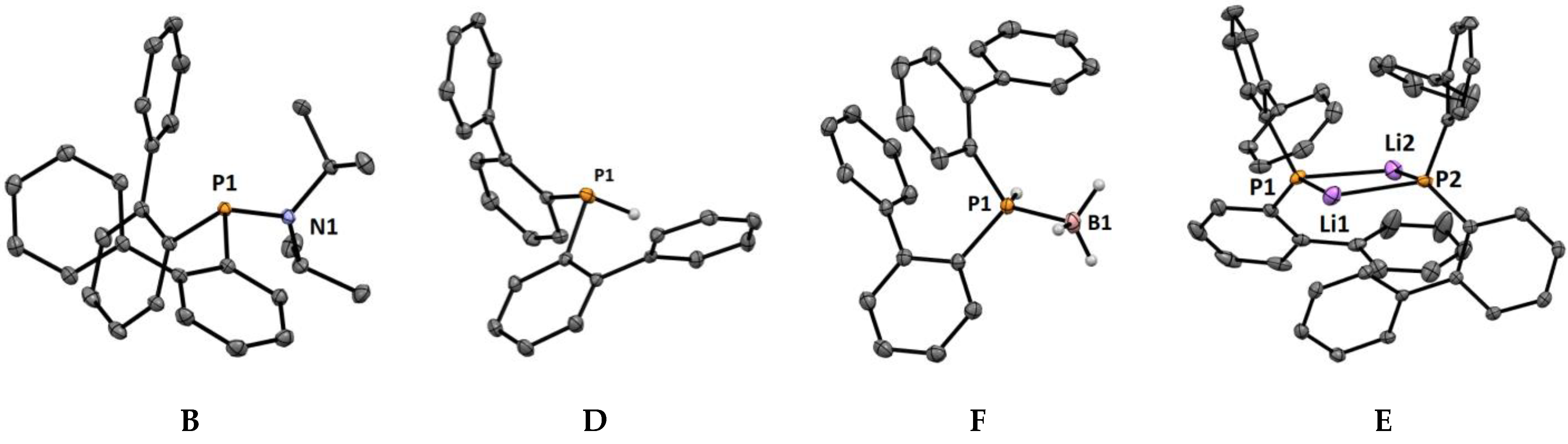
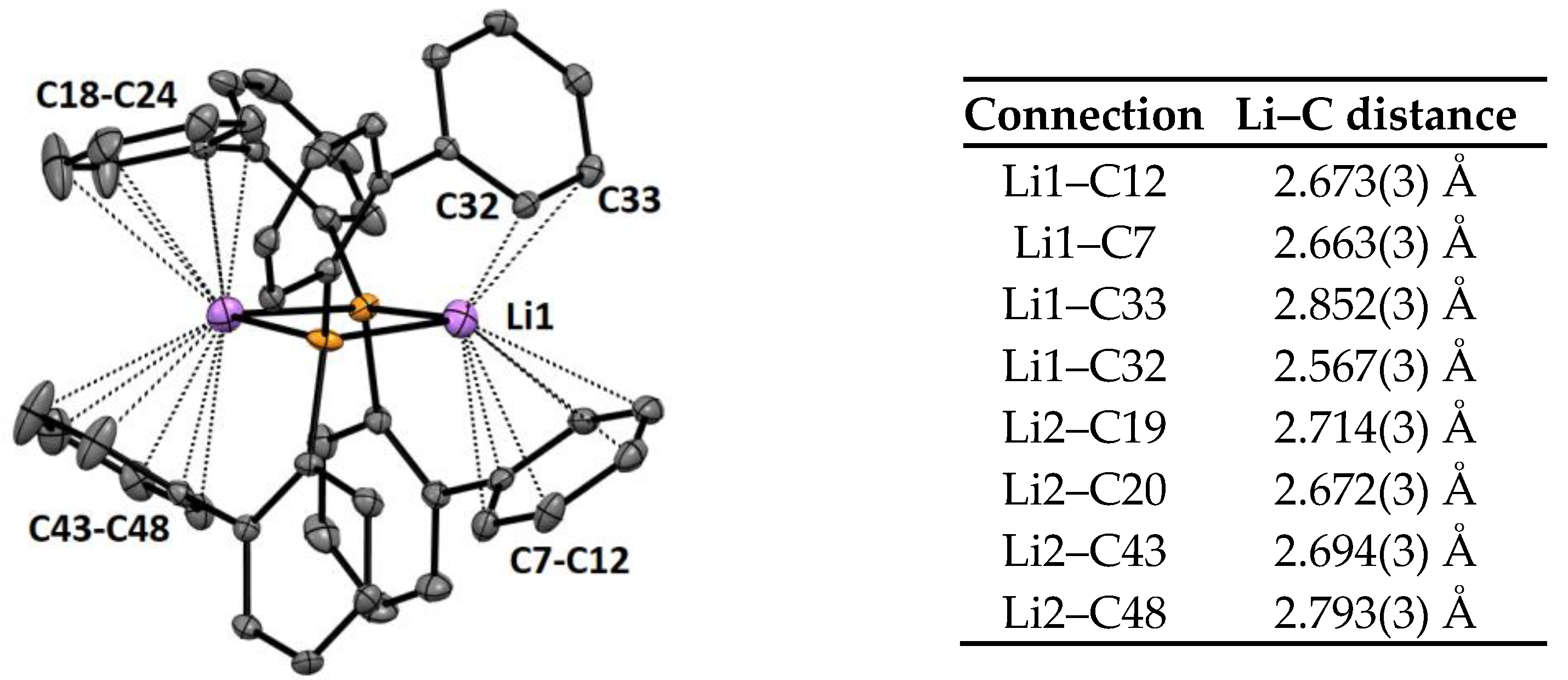
2.2. Crystal Structures
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Staubitz, A.; Hoffmann, J.; Gliese, P. Group 13-Group 15 Element Bonds Replacing Carbon-Carbon Bonds in Main Group Polyolefin Analogs in Smart Inorganic Polymers; Hey-Hawkins, E., Hissler, M., Eds.; Wiley VCH: Weinheim, Germany, 2019; pp. 17–39. [Google Scholar]
- Dorn, H.; Singh, R.A.; Massey, J.A.; Lough, A.J.; Manners, I. Rhodium-catalyzed formation of phosphorus-boron bonds: Synthesis of the first high molecular weight poly (phosphinoborane). Angew. Chem. Int. Ed. 1999, 38, 3321–3323. [Google Scholar] [CrossRef]
- Dorn, H.; Singh, R.A.; Massey, J.A.; Nelson, J.M.; Jaska, C.A.; Lough, A.J.; Manners, I. Transition metal-catalyzed formation of phosphorus−boron bonds: A new route to phosphinoborane rings, chains, and macromolecules. J. Am. Chem. Soc. 2000, 122, 6669–6678. [Google Scholar] [CrossRef]
- Gopalakrishnan, J. Aminophosphines: Their chemistry and role as ligands and synthons. Appl. Organomet. Chem. 2009, 23, 291–318. [Google Scholar] [CrossRef]
- Zubiri, M.R.I.; Woollins, J.D. Synthesis and uses of phosphines containing P-N bonds. Comments Inorg. Chem. 2003, 24, 189–252. [Google Scholar] [CrossRef]
- Dogan, J.; Schulte, J.B.; Swiegers, G.F.; Wild, S.B. Mechanism of phosphorus-carbon bond cleavage by lithium in tertiary phosphines. An optimized synthesis of 1,2-bis (phenylphosphino)ethane. J. Org. Chem. 2000, 65, 951–957. [Google Scholar] [CrossRef]
- Bianco, V.D.; Doronzo, S.; Chan, J.; Bennett, M.A. Diphenylphosphine. In Inorganic Syntheses; Basolo, F., Ed.; McGraw Hill Book Company: New York, NY, USA, 1976; pp. 161–163. [Google Scholar]
- van Doorn, J.A.; Frijns, J.H.G.; Meijboom, N. Formation and reactions of bis (phosphino) succinic anhydrides. J. Chem. Soc. Perkin Trans. 2 1990, 3, 479–485. [Google Scholar] [CrossRef]
- Dingwall, J.G.; Tuck, B. Free radical catalysed additions to the double bond of diketene: A synthesis of novel oxetan-2-ones. J. Chem. Soc. Perkin Trans. 1 1986, 2081–2090. [Google Scholar] [CrossRef]
- Cristau, H.-J.; Chêne, A.; Christol, H. Arylation catalytique d’organophosphorés. Produits de l’arylation, catalysée par les sels de nickel (II), de composés du phosphore tricoordiné. J. Organomet. Chem. 1980, 185, 283–295. [Google Scholar] [CrossRef]
- Gilbertson, S.R.; Starkey, G.W. Palladium-catalyzed synthesis of phosphine-containing amino acids. J. Org. Chem. 1996, 61, 2922–2923. [Google Scholar] [CrossRef]
- Martorell, G.; Garcías, X.; Janura, M.; Saá, J.M. Direct palladium-catalyzed phosphinylation of aryl triflates with secondary phosphines. Its scope and limitations: The synthesis of optically active carboxylated 2-(diphenylphosphino)-1,1‘-binaphthalenes. J. Org. Chem. 1998, 63, 3463–3467. [Google Scholar] [CrossRef]
- Zhao, Y.L.; Wu, G.J.; Li, Y.; Gao, L.X.; Han, F.S. [NiCl2(dppp)]—Catalyzed cross-coupling of aryl halides with dialkyl phosphite, diphenylphosphine oxide, and diphenylphosphine. Chemistry 2012, 18, 9622–9627. [Google Scholar] [CrossRef]
- Leone-Bay, A. Lithium diphenylphosphide as a reagent for the dehydroxylation of alpha-hydroxy ketones. J. Org. Chem. 2002, 51, 2378–2379. [Google Scholar] [CrossRef]
- Mann, F.G.; Tong, B.P.; Wystrach, V.P. 213. The abnormal hydrolysis of certain β-diarylphosphinopropionic esters. Part II. Correction and extension. J. Chem. Soc. 1963, 1155–1167. [Google Scholar] [CrossRef]
- Mann, F.G.; Pragnell, M.J. 761. The dealkylation of alkyl aryl ethers and sulphides by diaryl-phosphide and -arsenide ions. J. Chem. Soc. 1965, 4120–4127. [Google Scholar] [CrossRef]
- Staubitz, A.; Robertson, A.P.; Sloan, M.E.; Manners, I. Amine- and phosphine-borane adducts: New interest in old molecules. Chem. Rev. 2010, 110, 4023–4078. [Google Scholar] [CrossRef] [PubMed]
- Carboni, B.; Monnier, L. Recent developments in the chemistry of amine- and phosphine-boranes. Tetrahedron 1999, 55, 1197–1248. [Google Scholar] [CrossRef]
- Carroll, L.; Boldon, S.; Bejot, R.; Moore, J.E.; Declerck, J.; Gouverneur, V. The traceless staudinger ligation for indirect 18F-radiolabelling. Org. Biomol. Chem. 2011, 9, 136–140. [Google Scholar] [CrossRef]
- Juge, S.; Stephan, M.; Laffitte, J.A.; Genet, J.P. Efficient asymmetric synthesis of optically pure tertiary mono and diphosphine ligands. Tetrahedron Lett. 1990, 31, 6357–6360. [Google Scholar] [CrossRef]
- Bernoud, E.; Alayrac, C.; Delacroix, O.; Gaumont, A.C. Copper-catalyzed synthesis of alkynylphosphine derivatives: Unprecedented use of nucleophilic phosphorus compounds. Chem. Commun. 2011, 47, 3239–3241. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Buzard, D.J.; Yun, C.S. Pd (0)-mediated couplings of aryl nonaflates and triflates with diphenylphosphine-borane. Preparation of bh3-stabilized, unsymmetrical triarylphosphines. Tetrahedron Lett. 1999, 40, 201–204. [Google Scholar] [CrossRef]
- Julienne, D.; Lohier, J.F.; Delacroix, O.; Gaumont, A.C. Palladium-catalyzed C-P coupling reactions between vinyl triflates and phosphine-boranes: Efficient access to vinylphosphine-boranes. J. Org. Chem. 2007, 72, 2247–2250. [Google Scholar] [CrossRef]
- Gilbertson, S.R.; Fu, Z.; Starkey, G.W. Palladium-catalyzed synthesis of vinyl phosphines from ketones. Tetrahedron Lett. 1999, 40, 8509–8512. [Google Scholar] [CrossRef]
- Ueda, S.; Ali, S.; Fors, B.P.; Buchwald, S.L. Me3(OMe)tBuXPhos: A surrogate ligand for Me4 t BuXPhos in palladium-catalyzed C-N and C-O bond-forming reactions. J. Org. Chem. 2012, 77, 2543–2547. [Google Scholar] [CrossRef] [Green Version]
- Hoshiya, N.; Buchwald, S.L. An improved synthesis of BrettPhos and RockPhos-type biarylphosphine ligands. Adv. Synth. Catal. 2012, 354, 2031–2037. [Google Scholar] [CrossRef] [Green Version]
- Su, M.; Buchwald, S.L. A bulky biaryl phosphine ligand allows for palladium-catalyzed amidation of five-membered heterocycles as electrophiles. Angew. Chem. Int. Ed. 2012, 51, 4710–4713. [Google Scholar] [CrossRef] [Green Version]
- Salvi, L.; Davis, N.R.; Ali, S.Z.; Buchwald, S.L. A new biarylphosphine ligand for the Pd-catalyzed synthesis of diaryl ethers under mild conditions. Org. Lett. 2012, 14, 170–173. [Google Scholar] [CrossRef] [Green Version]
- Schweizer, S.; Becht, J.-M.; Le Drian, C. Development of efficient and reusable diarylphosphinopolystyrene-supported palladium catalysts for C-C bond forming cross-coupling reactions. Adv. Synth. Catal. 2007, 349, 1150–1158. [Google Scholar] [CrossRef]
- Siedzielnik, M.; Kaniewska-Laskowska, K.; Szynkiewicz, N.; Chojnacki, J.; Grubba, R. Reactivity of bulky aminophosphanes towards small molecules: Activation of dihydrogen and carbon dioxide by aminophosphane/borane frustrated lewis pairs. Polyhedron 2021, 194, 114930. [Google Scholar] [CrossRef]
- Levin, M.D.; Toste, F.D. Gold-catalyzed allylation of aryl boronic acids: Accessing cross-coupling reactivity with gold. Angew. Chem. Int. Ed. 2014, 53, 6211–6215. [Google Scholar] [CrossRef] [Green Version]
- Alajarín, M.; López-Leonardo, C.; Llamas-Lorente, P. The chemistry of phosphinous amides (aminophosphanes): Old reagents with new applications. Top. Curr. Chem. 2005, 250, 77–106. [Google Scholar]
- Zhang, W.; Wu, H.; Liu, Z.; Zhong, P.; Zhang, L.; Huang, X.; Cheng, J. The use of calcium carbide in one-pot synthesis of symmetric diaryl ethynes. Chem. Commun. 2006, 46, 4826–4828. [Google Scholar] [CrossRef]
- Lamola, J.L.; Shilubana, J.C.; Ngodwana, L.; Vatsha, B.; Adeyinka, A.S.; Maumela, M.C.; Holzapfel, C.W.; Mmutlane, E.M. Easily prepared mono (N, N-dialkylamino) phosphine palladium (II) complexes: Structural and catalytic evaluation. Eur. J. Inorg. Chem. 2021, 2021, 2578–2582. [Google Scholar] [CrossRef]
- Dyer, P.W.; Fawcett, J.; Hanton, M.J.; Kemmitt, R.D.W.; Padda, R.; Singh, N. Exploring the coordination chemistry and reactivity of dialkylamino- and bis(dialkylamino)-phosphines in the coordination sphere of metals. Dalton Trans. 2003, 1, 104–113. [Google Scholar] [CrossRef]
- Li, Y.; Chakrabarty, S.; Muck-Lichtenfeld, C.; Studer, A. Ortho-trialkylstannyl arylphosphanes by C-P and C-Sn bond formation in arynes. Angew. Chem. Int. Ed. 2016, 55, 802–806. [Google Scholar] [CrossRef]
- Balakrishna, M.S.; Abhyankar, R.M.; Mague, J.T. New bis(phosphines) derived from N, N′-substituted ethylenediamine derivatives. Synthesis and transition metal chemistry of X2PN(R)CH2CH2 (R)NPX2 (R = CH2Ph or Ph, X = Ph; R = CH2Ph, X2 = O2C6H4). The crystal and molecular structure of Ph2PN(CH2Ph)CH2CH2(CH2Ph)NPPh2 and cis-[{PtCl2Ph2PN(CH2Ph)CH2CH2(CH2Ph)NPPh2}]. J. Chem. Soc. Dalton Trans. 1999, 9, 1407–1412. [Google Scholar]
- Surry, D.S.; Buchwald, S.L. Dialkylbiaryl phosphines in Pd-catalyzed amination: A user’s guide. Chem. Sci. 2011, 2, 27–50. [Google Scholar] [CrossRef] [Green Version]
- Barder, T.E.; Buchwald, S.L. Rationale behind the resistance of dialkylbiaryl phosphines toward oxidation by molecular oxygen. J. Am. Chem. Soc. 2007, 129, 5096–5101. [Google Scholar] [CrossRef]
- MacInnis, M.C.; McDonald, R.; Turculet, L. Synthesis and characterization of palladium complexes supported by an NPN-phosphido ancillary ligand. Organometallics 2011, 30, 6408–6415. [Google Scholar] [CrossRef]
- Izod, K.; Evans, P.; Waddell, P.G. Desolvation and aggregation of sterically demanding alkali metal diarylphosphides. Dalton Trans. 2017, 46, 13824–13834. [Google Scholar] [CrossRef] [Green Version]
- Kurz, S.; Hey-Hawkins, E. Synthesis and crystal structure of [{Li(2,4,6-tert-Bu3C6H2)}{LiP(H)(2,4,6-tert-Bu3C6H2)}]2: A compound with an unusual (lithium-phosphorus-lithium-carbon)2 eight-membered ring. Organometallics 2002, 11, 2729–2732. [Google Scholar] [CrossRef]
- Rivard, E.; Sutton, A.D.; Fettinger, J.C.; Power, P.P. Synthesis of the sterically congested diarylphosphines ArTrip2P(Ph) H (ArTrip2 = C6H3–2,6(C6H2-2,4,6-Pri3)) and ArMes2P(Ph) H (ArMes2 = C6H3-2,6(C6H2-2,4,6-Me3)) and the monomeric Sn (II)-diphosphide [ArMes2P(Ph)]2Sn. Inorg. Chim. Acta 2007, 360, 1278–1286. [Google Scholar] [CrossRef]
- Bodach, A.; Hebestreit, R.; Bolte, M.; Fink, L. Syntheses and crystal structures of phenyl-lithium derivatives. Inorg. Chem. 2018, 57, 9079–9085. [Google Scholar] [CrossRef]
- Hey, E.; Hitchcock, P.B.; Lappert, M.F.; Rai, A.K. Bis (trimethylsilyl) phosphidometal complexes. J. Organomet. Chem. 1987, 325, 1–12. [Google Scholar] [CrossRef]
- Mulvey, E.R.; Wade, K.; Armstrong, R.D.; Walker, T.G.; Snaith, R.; Clegg, W.; Reed, D. X-ray crystallographic and solution studies of the pentamethyldiethylenetriamine and tetramethylethylenediamine adducts of lithium diphenylphosphide. Polyhedron 1987, 6, 987–993. [Google Scholar] [CrossRef]
- Campbell, I.G.M.; Fowles, G.W.A.; Nixon, L.A. 267. Organometallic compounds containing a tin–phosphorus bond. J. Chem. Soc. 1964, 1389–1396. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Harris, C.M.; Harris, T.M.; Kim, H.-Y.H.; Kim, S.J. Synthesis of deoxyadenosine 3′-phosphates bearingcisandtransadducts of 7β,8α-dihydroxy-9α,10α-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene: Standards for 32P—Postlabeling assays. J. Org. Chem. 1996, 61, 174–178. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
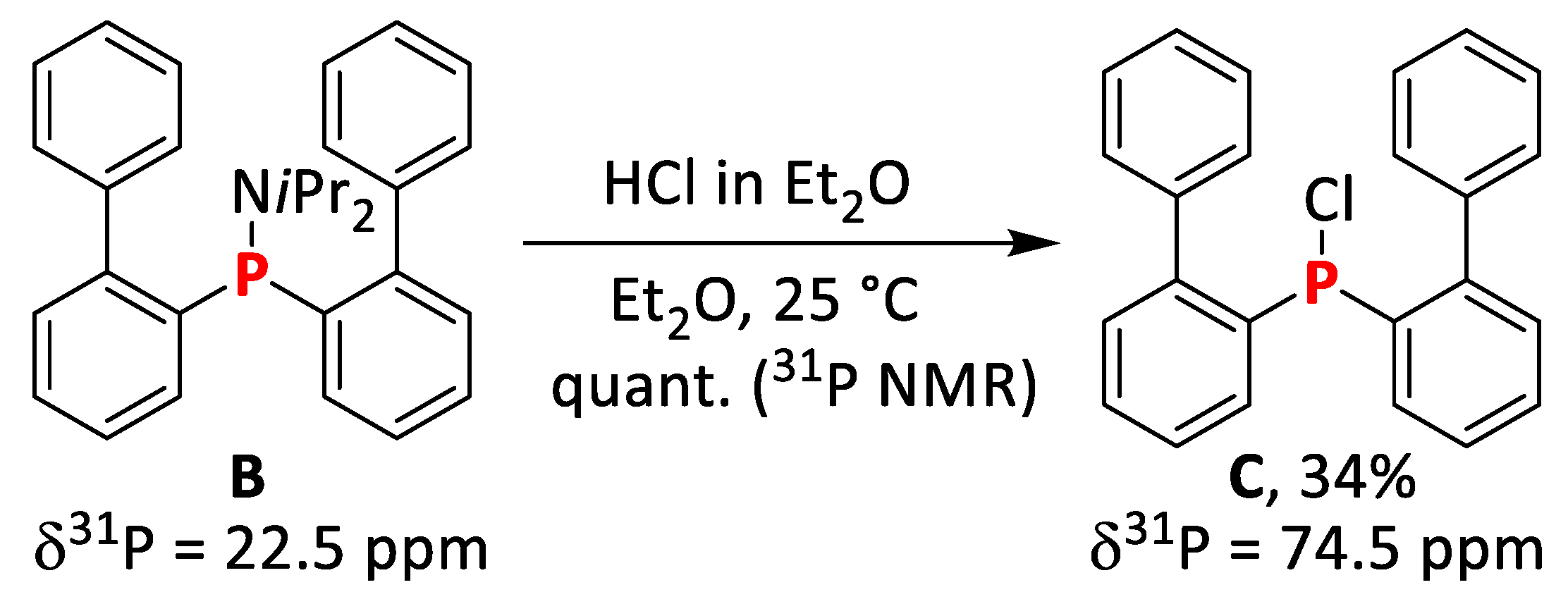{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
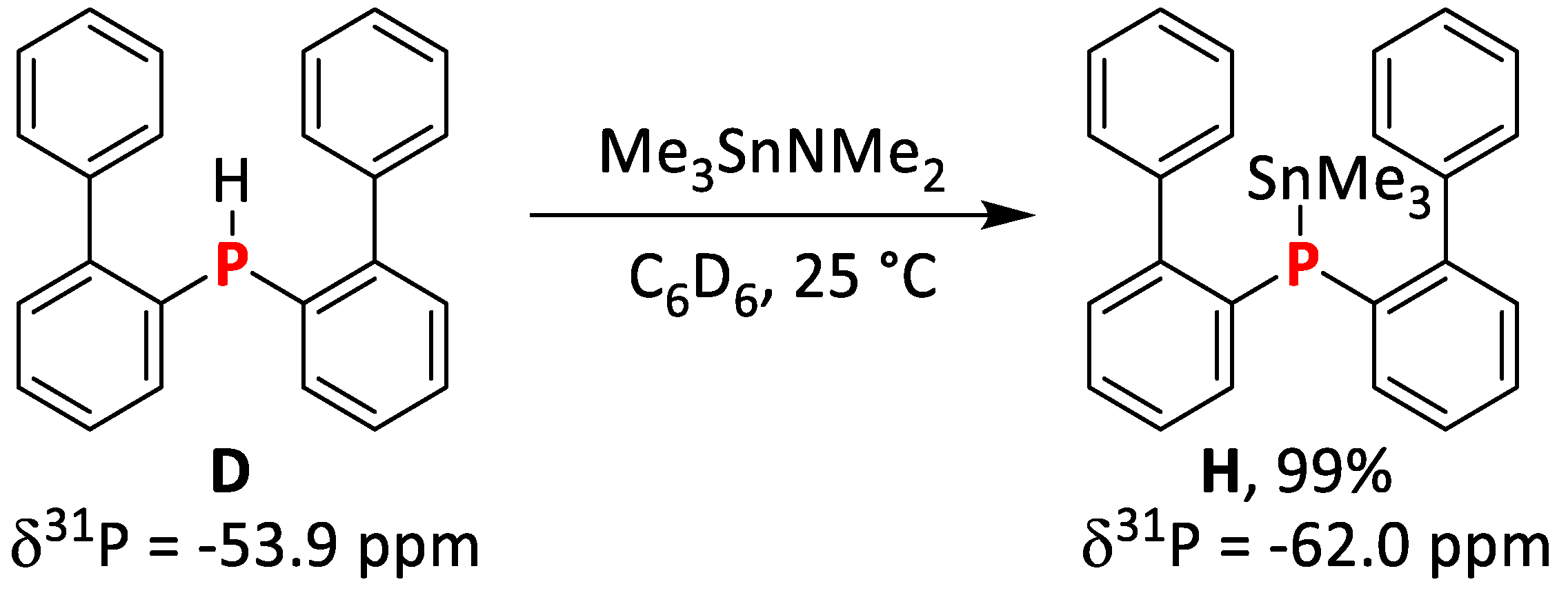{kind=link}
| B | D | F | E | |
|---|---|---|---|---|
| crystal system | triclinic | monoclinic | monoclinic | monoclinic |
| space group | P-1 | P21/c | P21/c | P21/n |
| P–X bond length | 1.6917(7) Å | 1.32(2) Å | 1.30(1) Å/ 1.926(1) Å | 2.503(2)/2.490(3) Å 2.507(3)/2.567(2) Å |
| P–C bond length | 1.8491(6) Å | 1.837(1) Å | 1.817(1) Å | 1.820(1)/1.822(1) Å |
| P–C bond length | 1.8546(9) Å | 1.847(1) Å | 1.812(1) Å | 1.821(1)/1.824(1) Å |
| ∠C-P-C | 99.39° | 102.30° | 104.93° | 104.33°/103.58° |
| ΣP | 307.71° | 297.23° | 316.13° | 326.29° |
| ϕbiaryl1 | −56.2(1)° | −65.2(2)° | −49.7(1)° | 66.2(2)°/−69.3(2)° |
| ϕbiaryl2 | −55.6(1)° | −65.6(2)° | −113.3(1)° | −76.3(2)°/−58.6(2)° |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, J.; Duvinage, D.; Lork, E.; Staubitz, A. The bis(Biphenyl)phosphorus Fragment in Trivalent and Tetravalent P-Environments. Inorganics 2021, 9, 82. https://doi.org/10.3390/inorganics9110082
Hoffmann J, Duvinage D, Lork E, Staubitz A. The bis(Biphenyl)phosphorus Fragment in Trivalent and Tetravalent P-Environments. Inorganics. 2021; 9(11):82. https://doi.org/10.3390/inorganics9110082
Chicago/Turabian StyleHoffmann, Jonas, Daniel Duvinage, Enno Lork, and Anne Staubitz. 2021. "The bis(Biphenyl)phosphorus Fragment in Trivalent and Tetravalent P-Environments" Inorganics 9, no. 11: 82. https://doi.org/10.3390/inorganics9110082
APA StyleHoffmann, J., Duvinage, D., Lork, E., & Staubitz, A. (2021). The bis(Biphenyl)phosphorus Fragment in Trivalent and Tetravalent P-Environments. Inorganics, 9(11), 82. https://doi.org/10.3390/inorganics9110082