
Cytochrome c: Using Biological Insight toward Engineering an Optimized Anticancer Biodrug


 , and
, and  add
Show full author list
add
Show full author list
Abstract
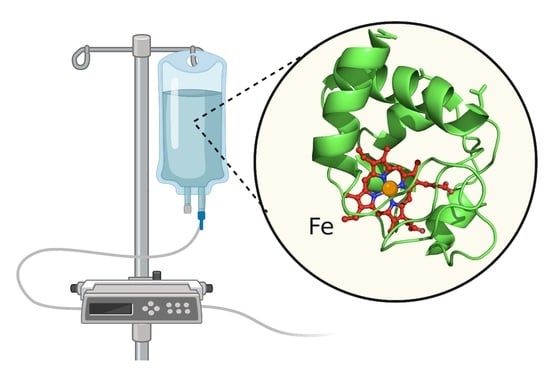
:
1. Introduction
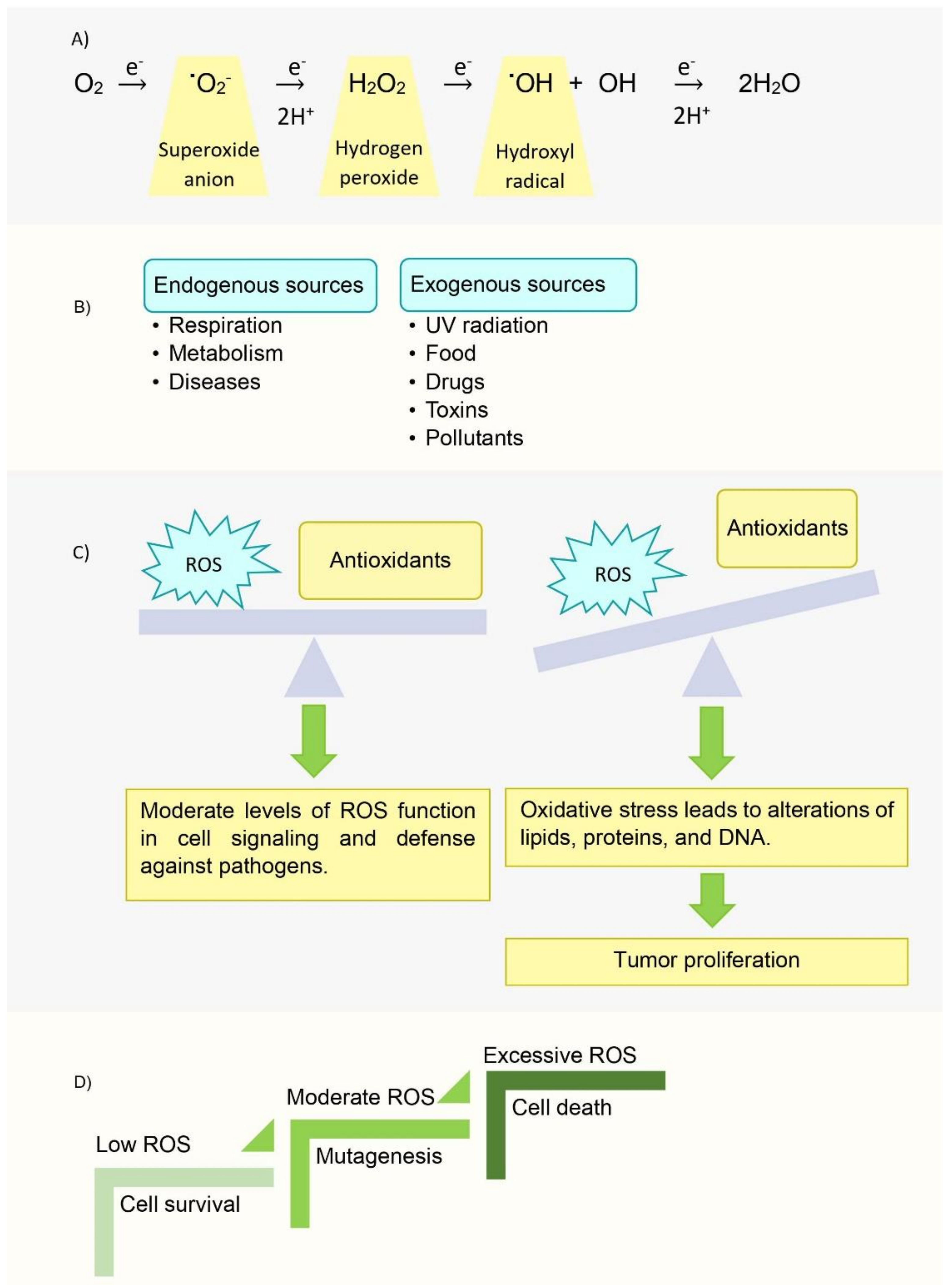
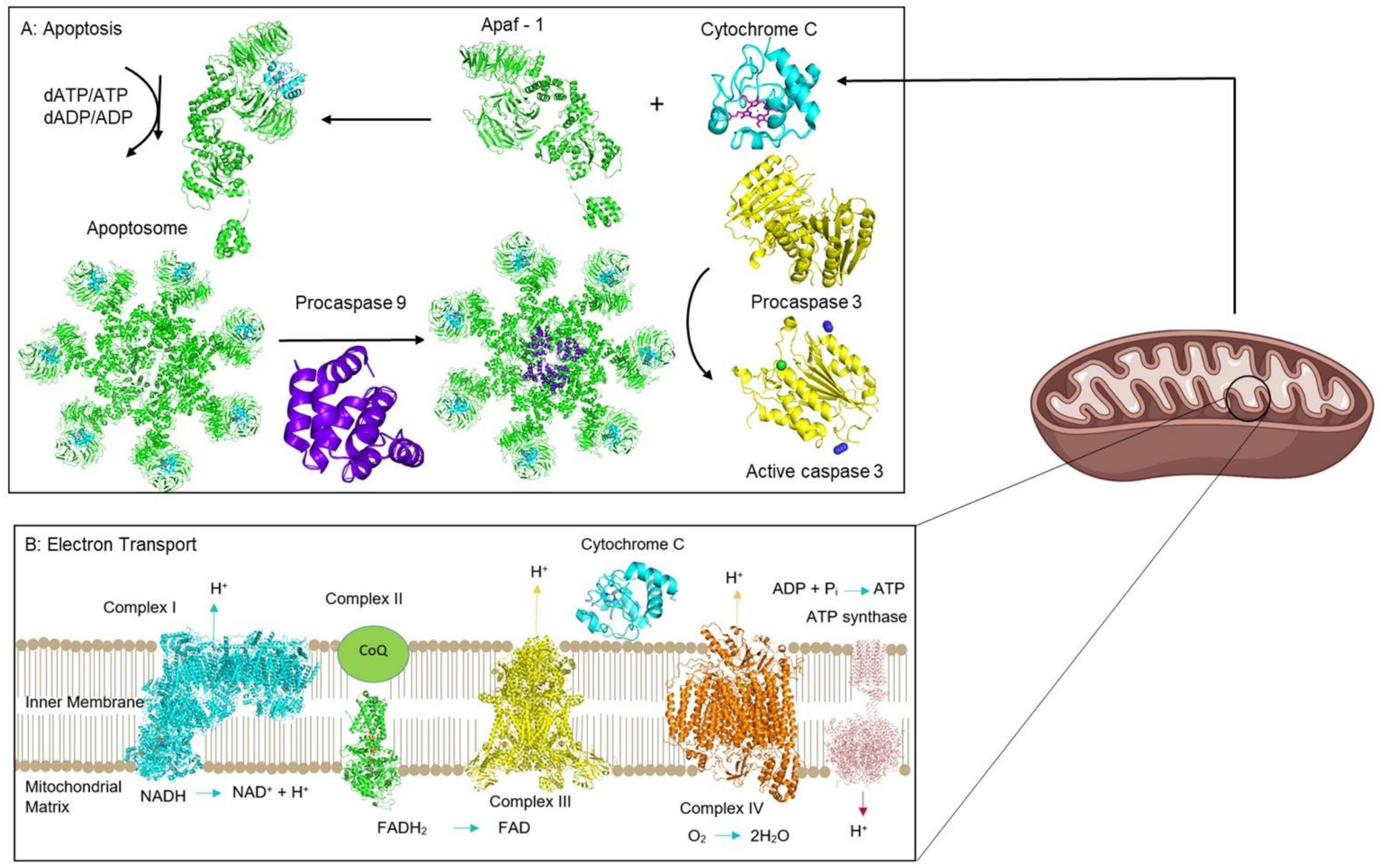
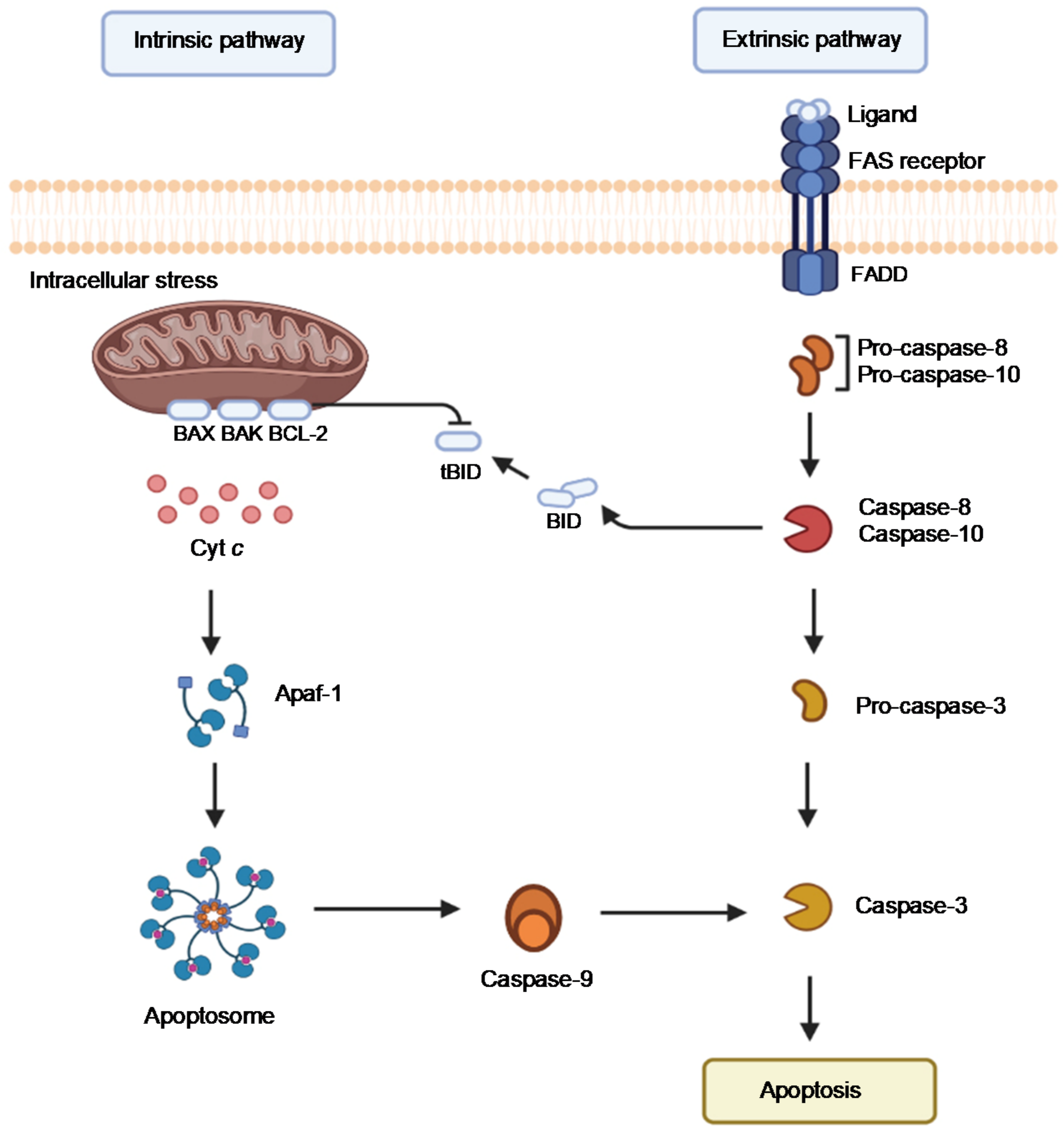
2. Examining the Biological Functions of Cyt c
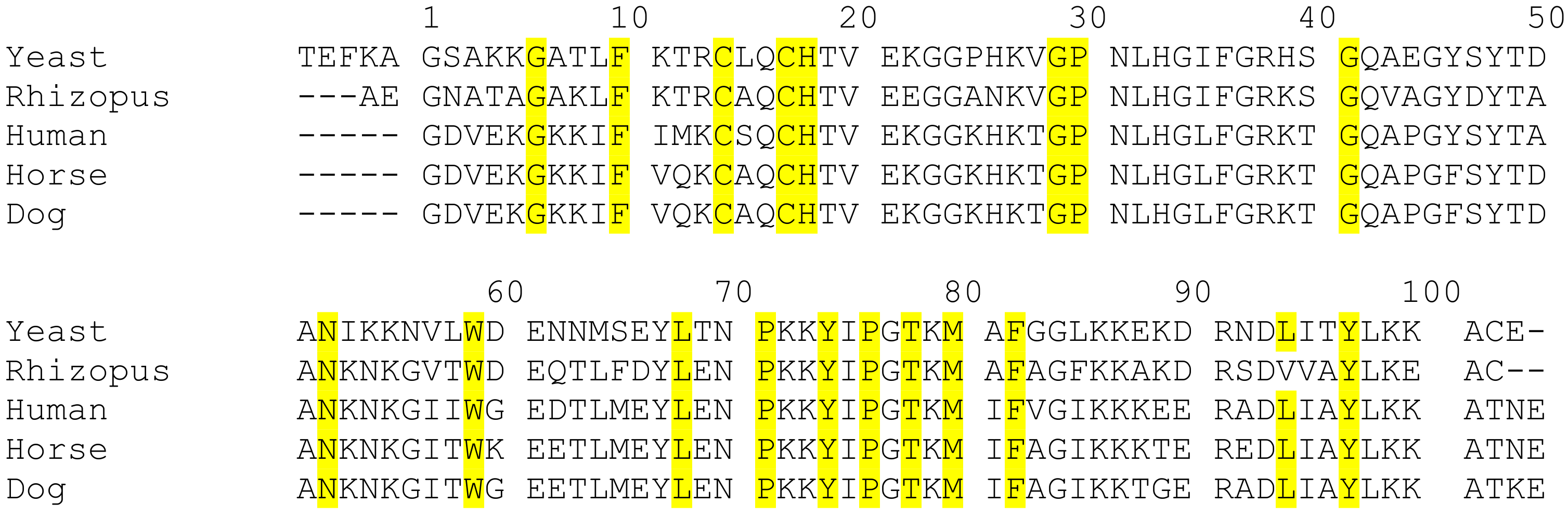
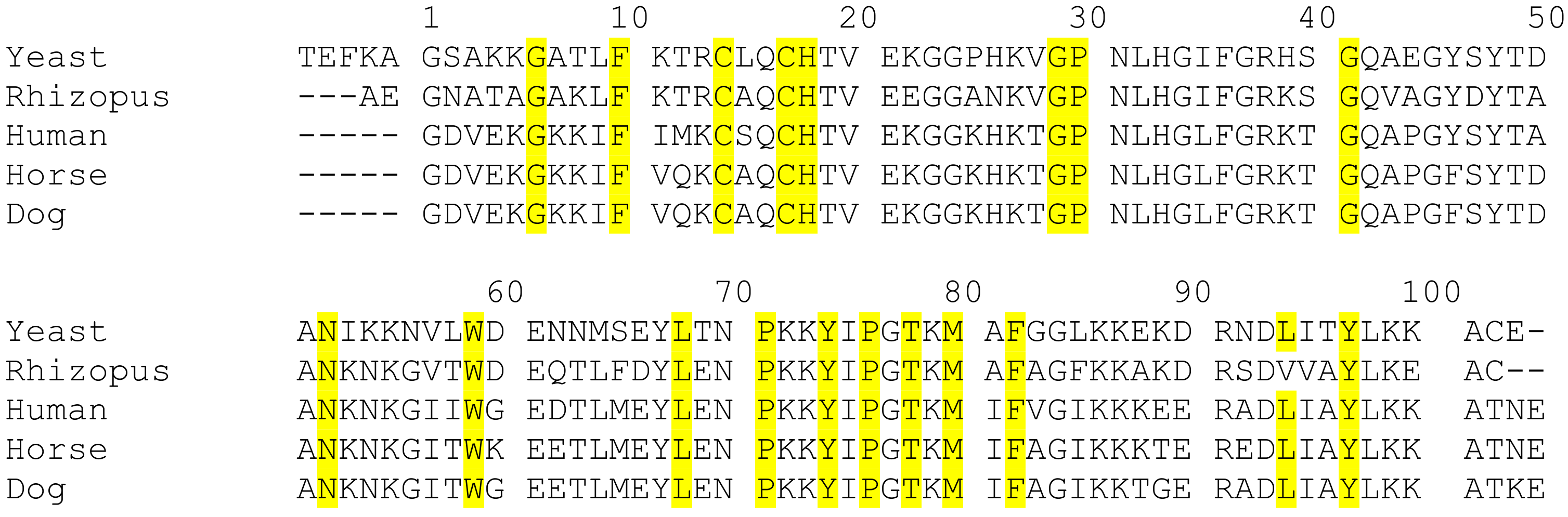
2.1. Evolutionary Insight of Cyt c
2.2. Classes of Cyt c
2.3. Cyt c as a Multifunctional Protein
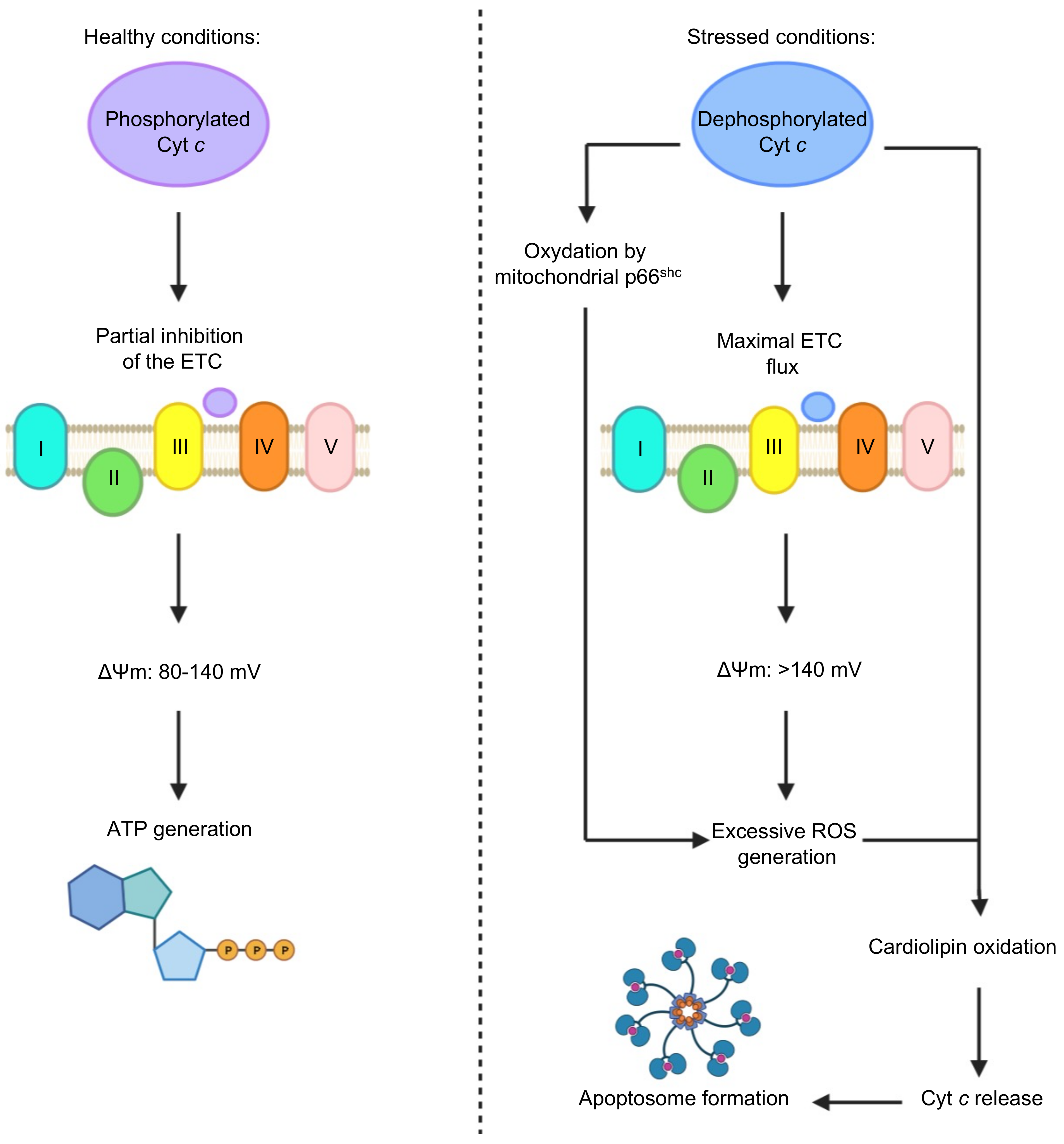
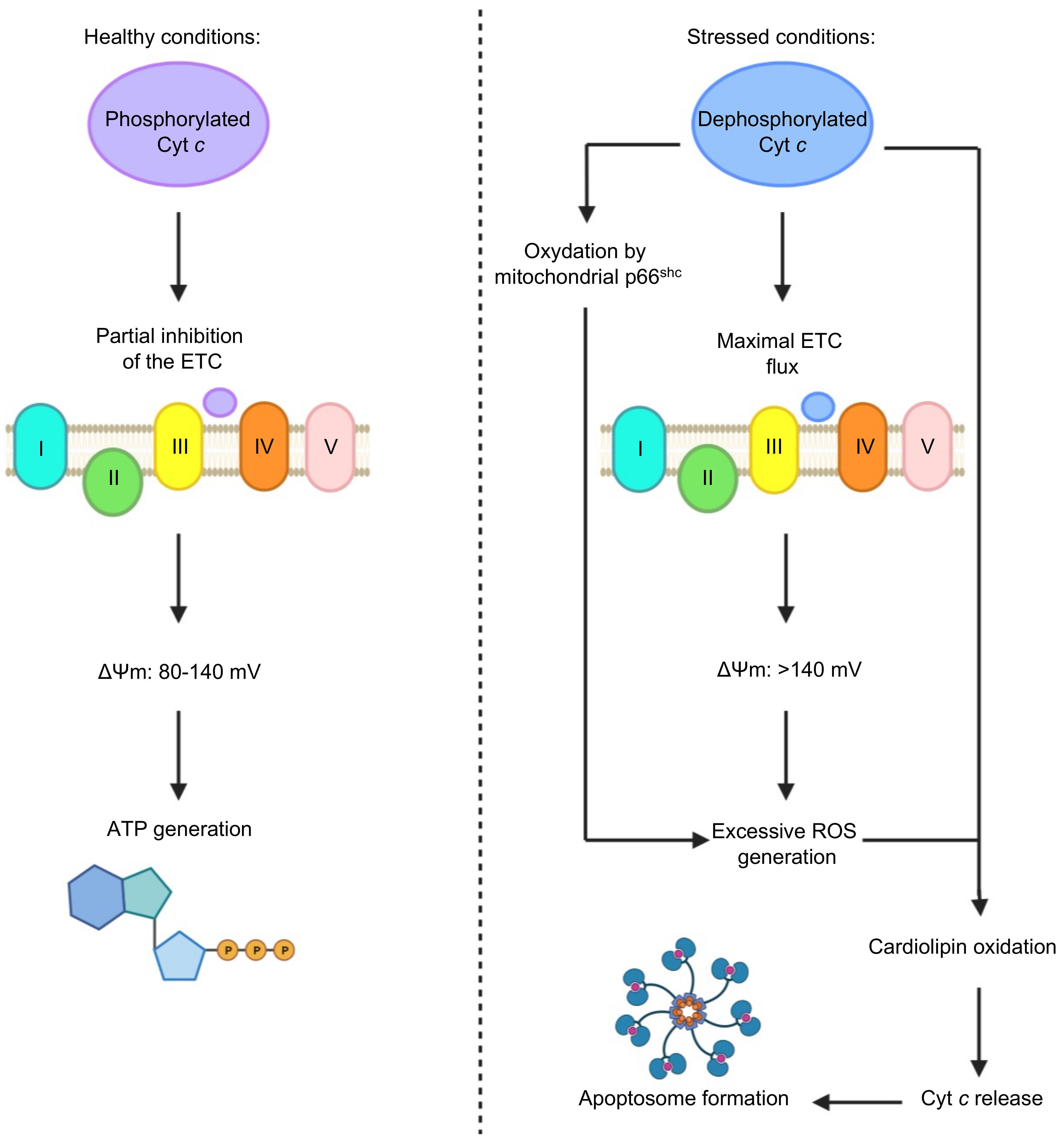
2.4. Cyt c Phosphorylation as a Regulator of Cyt c Activity
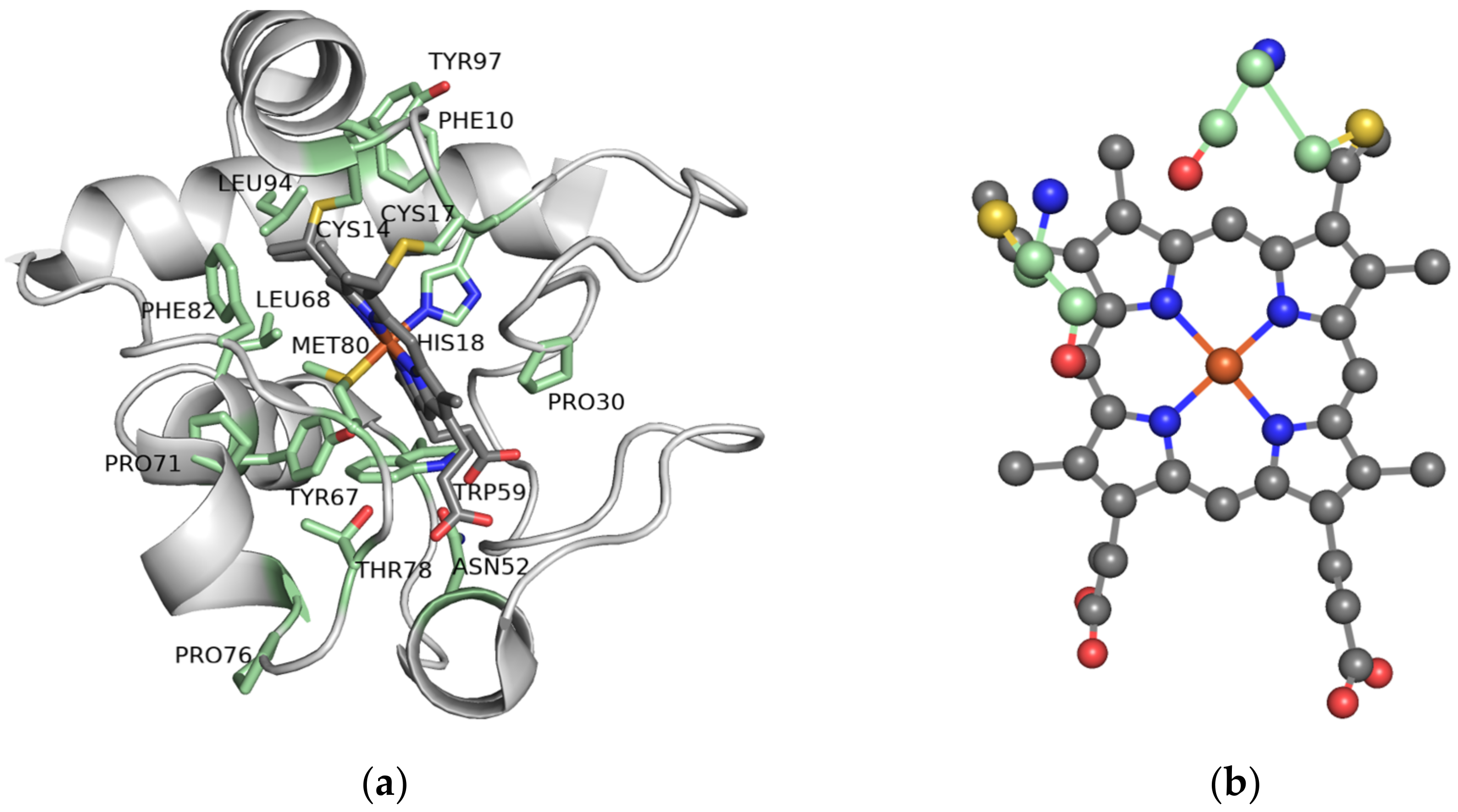
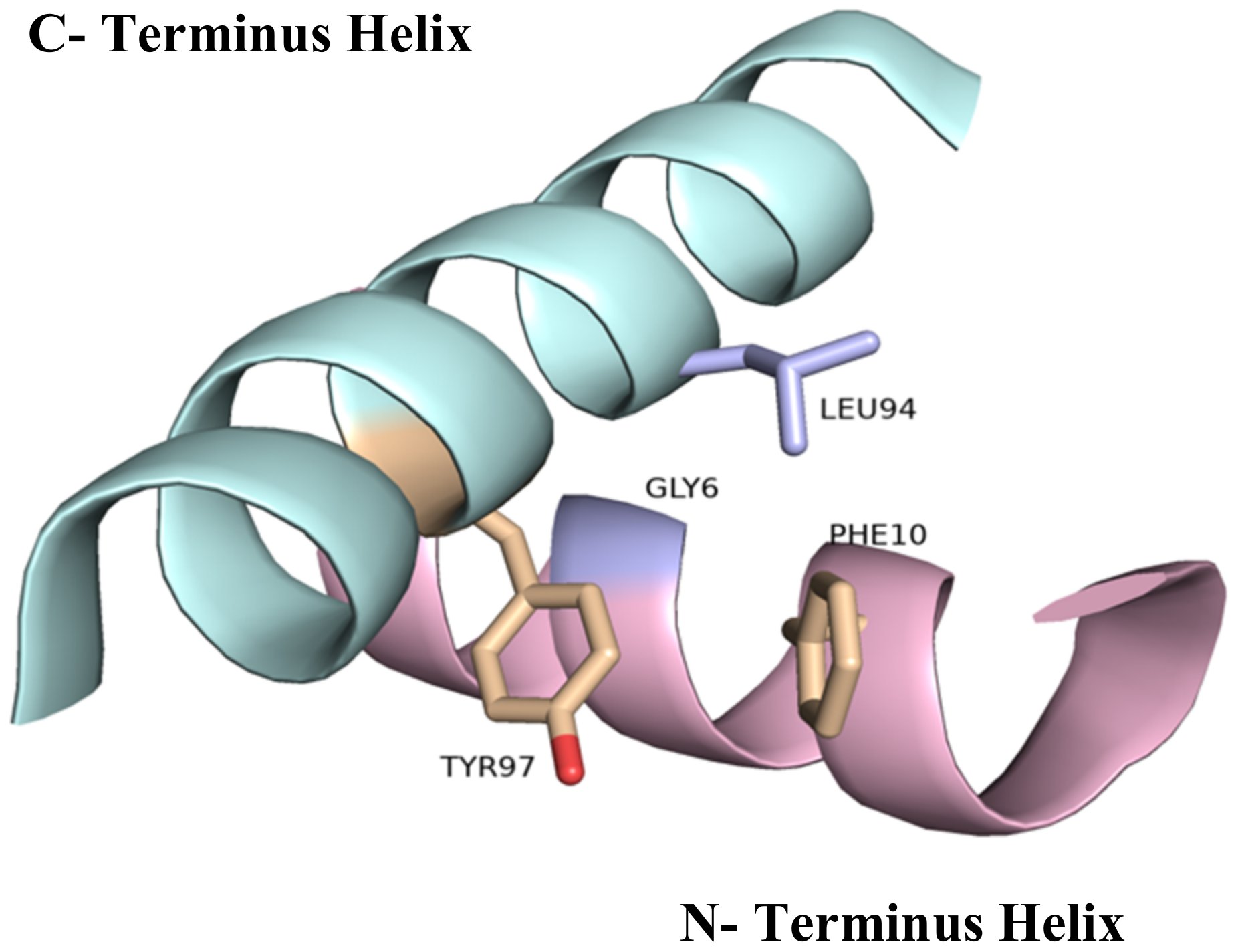
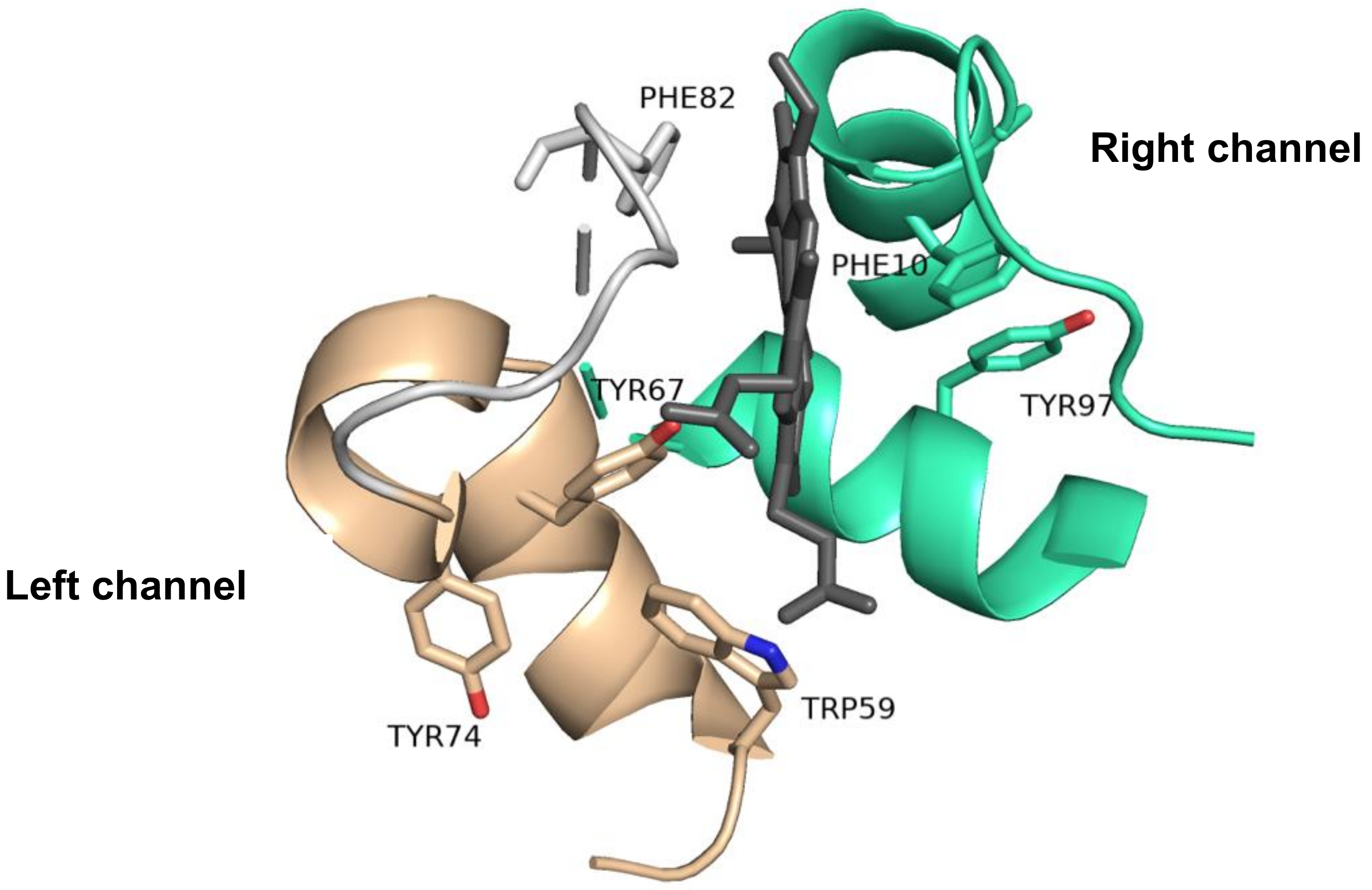
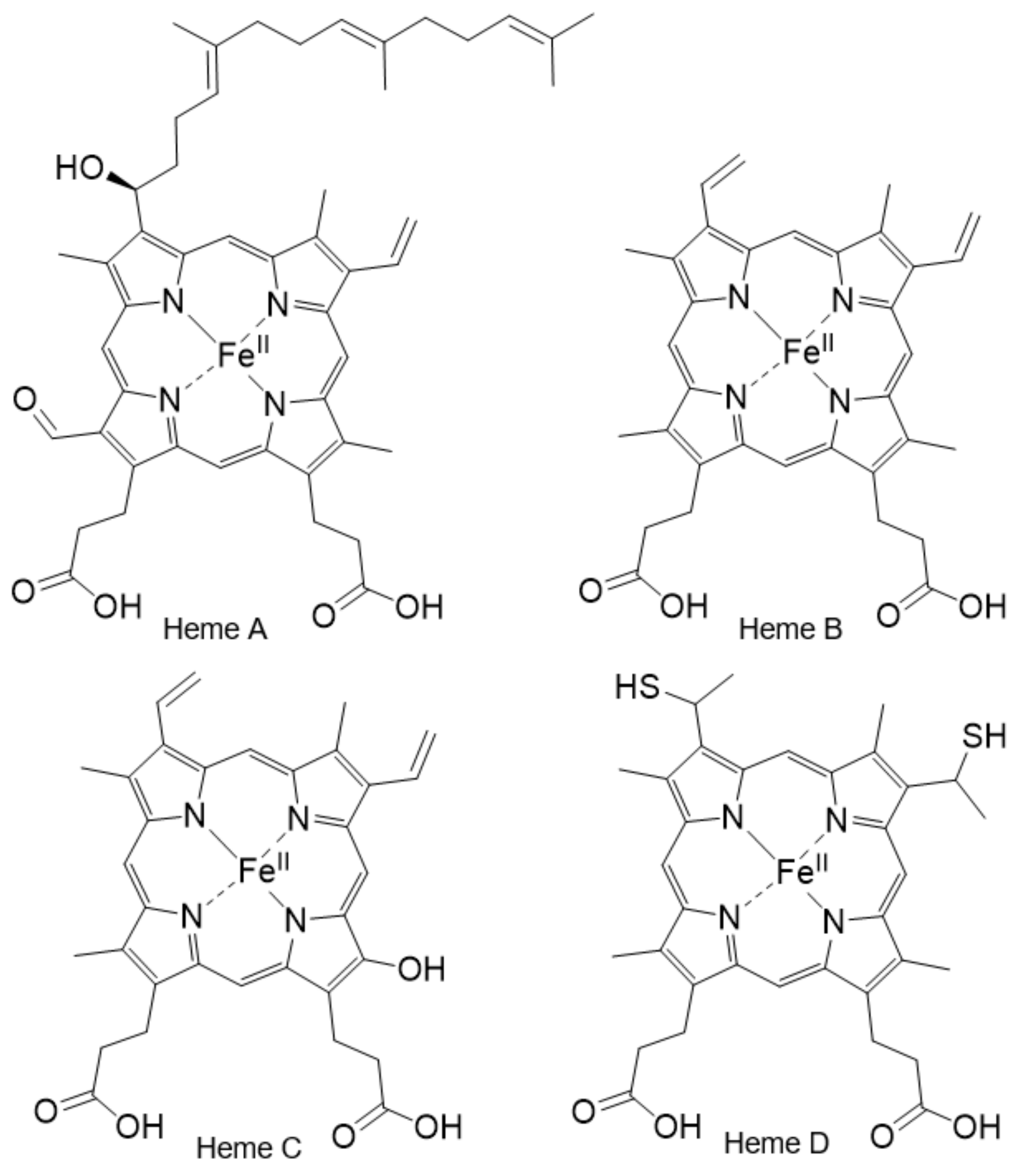
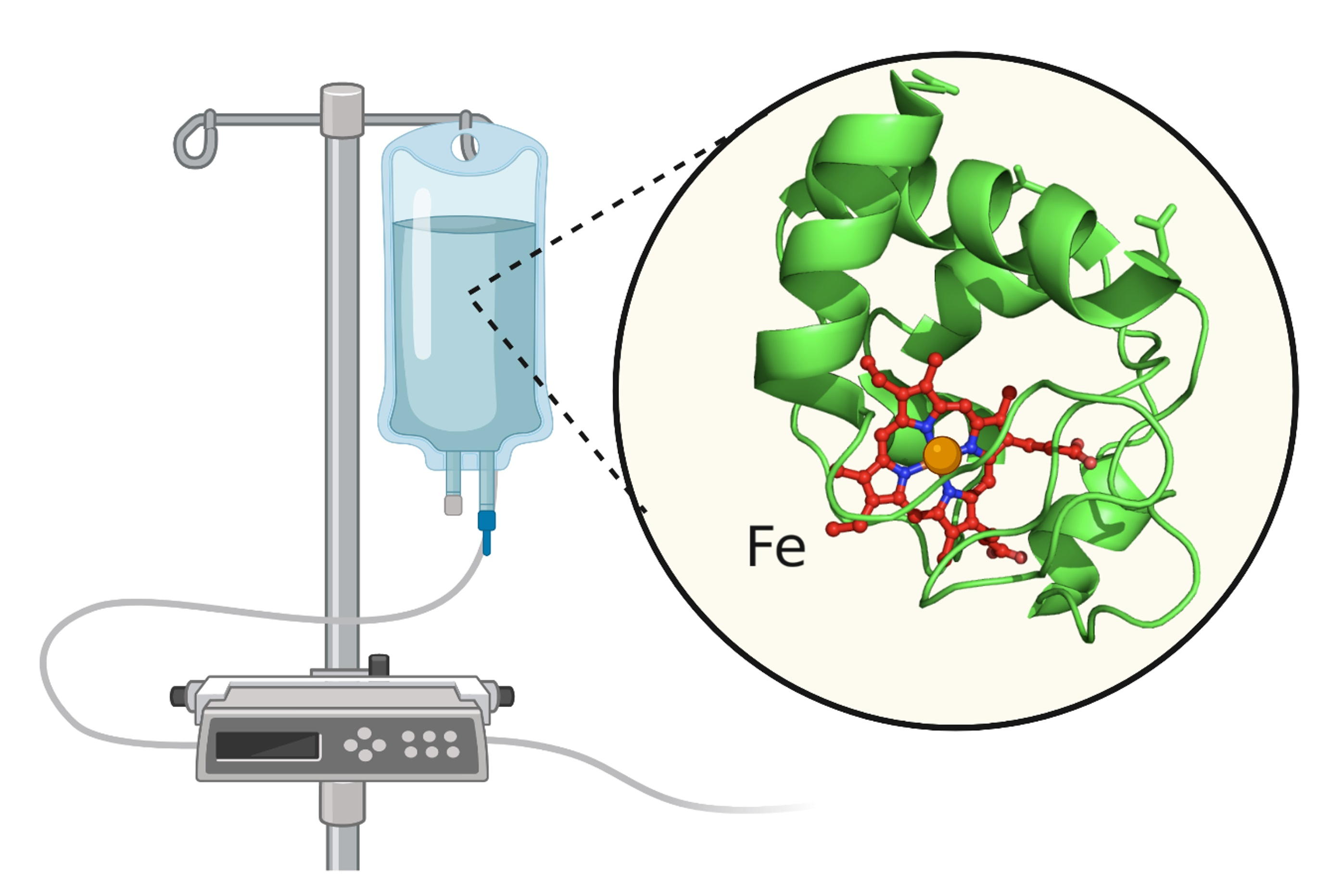
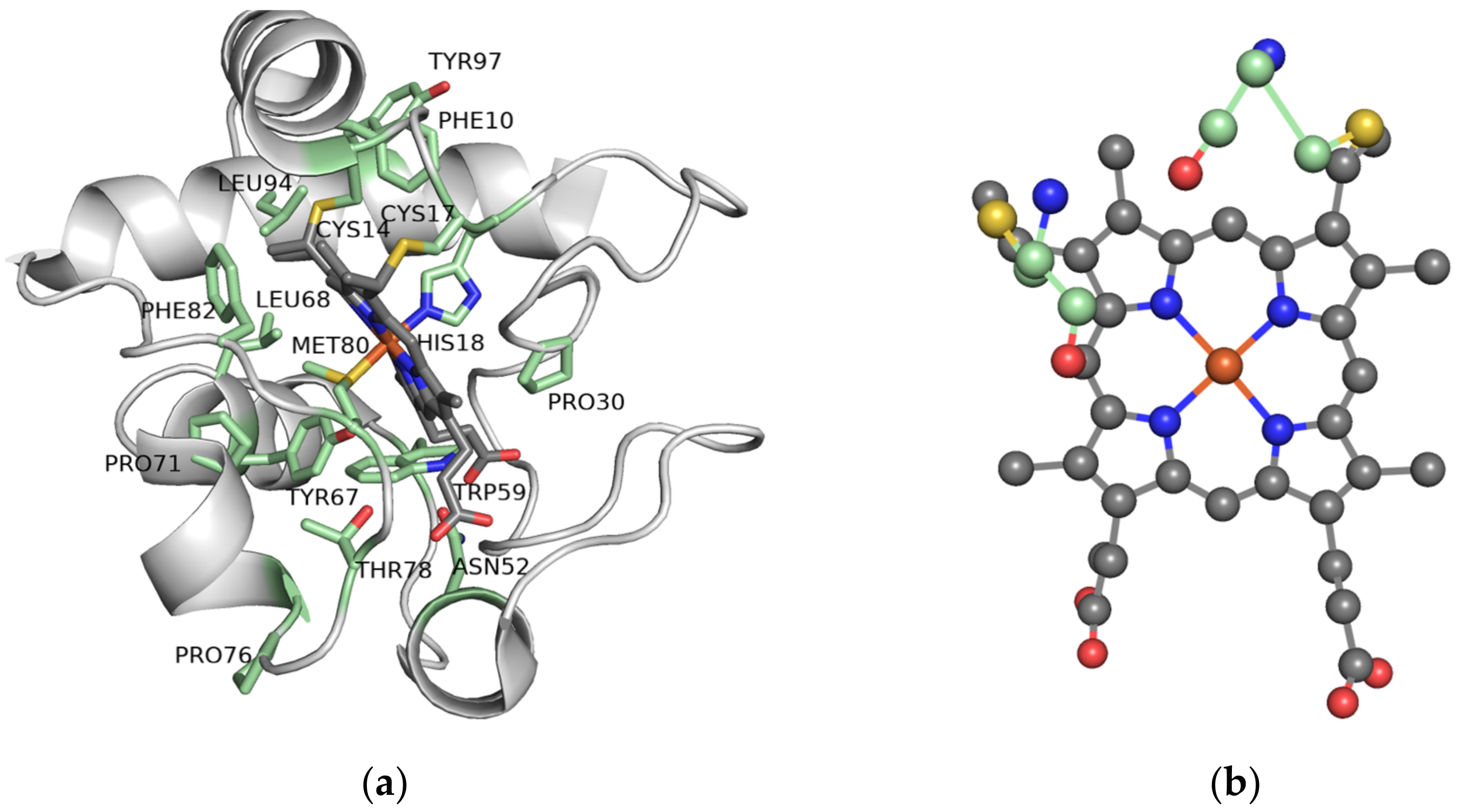
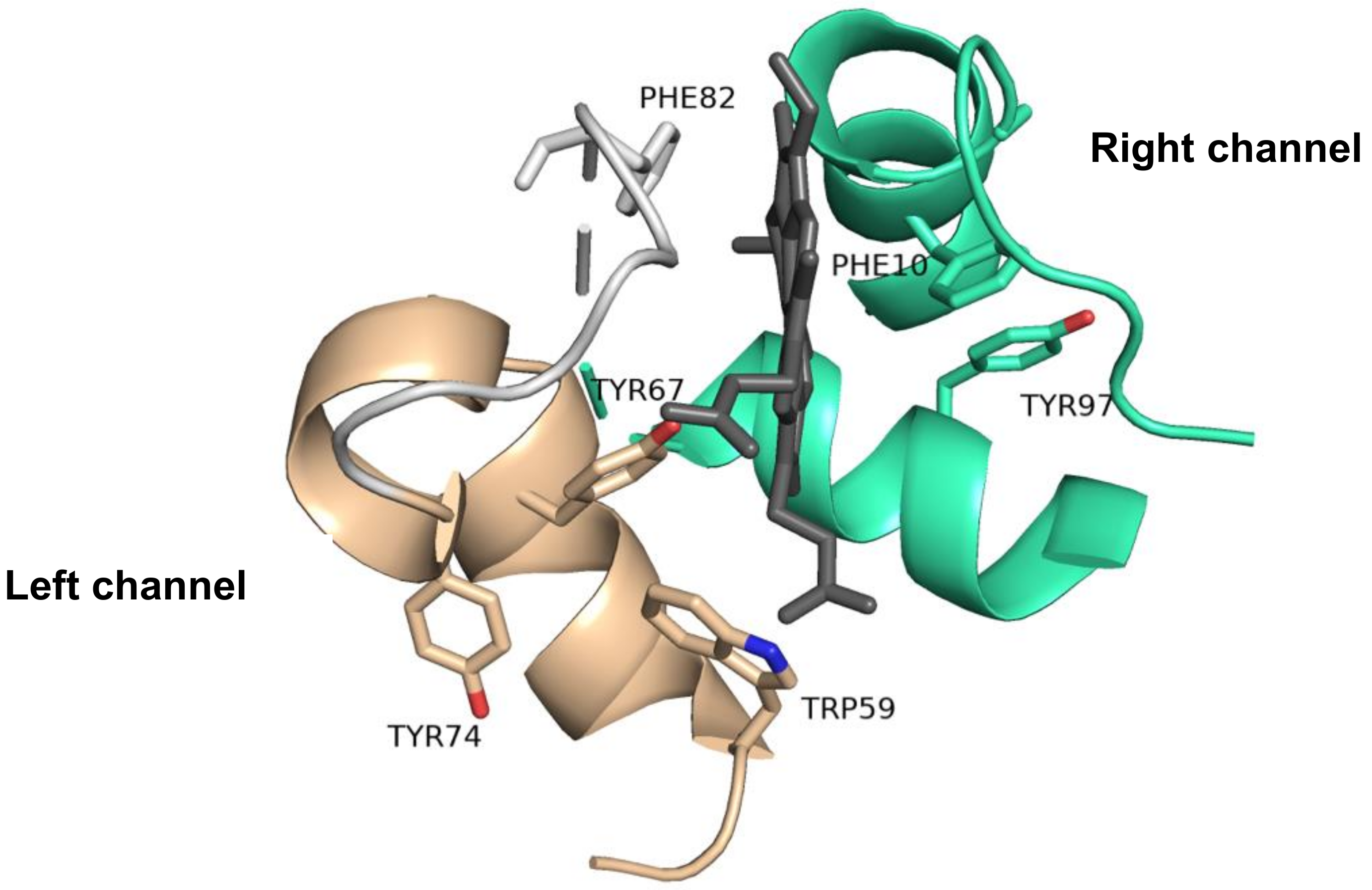
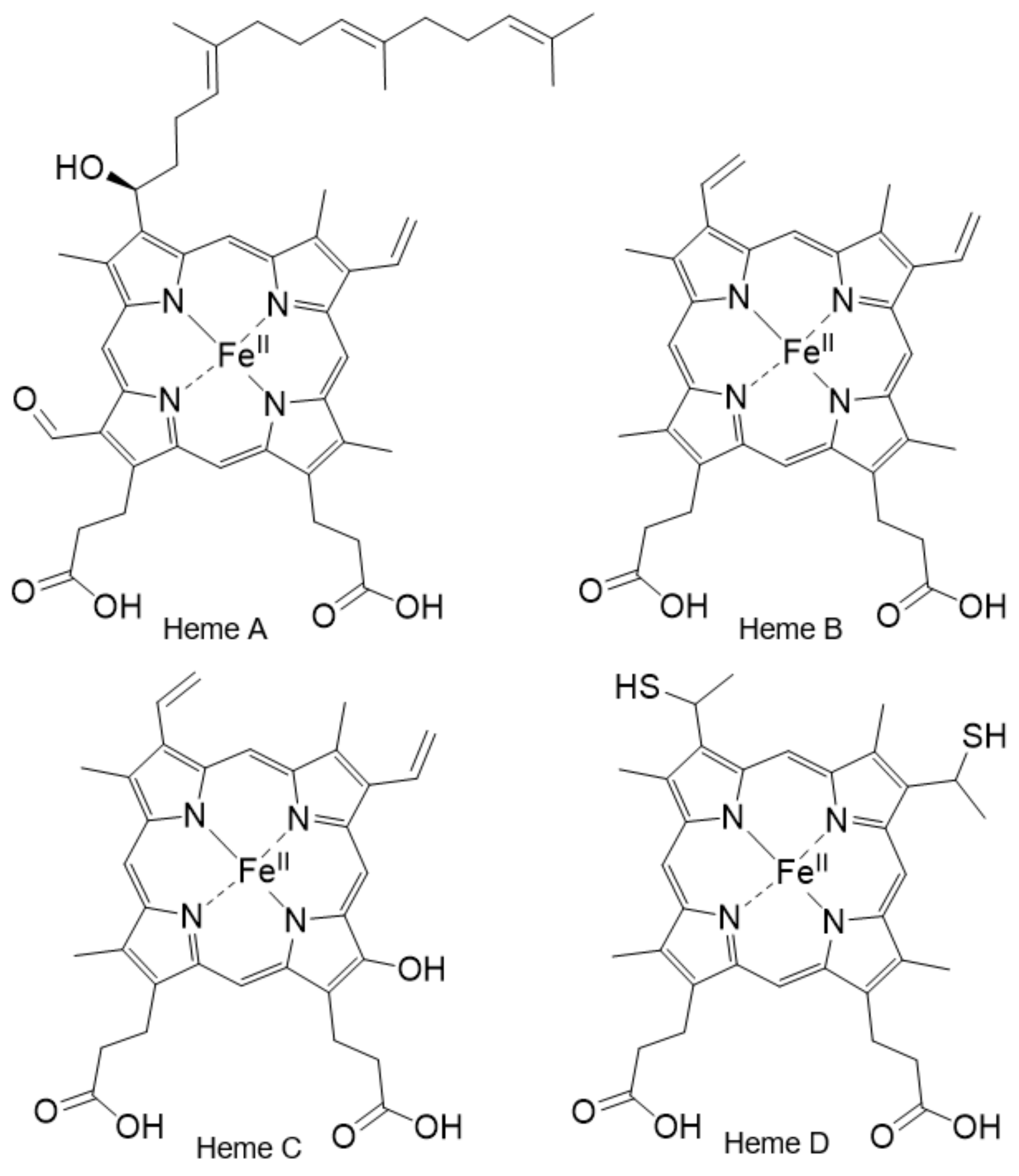
2.5. The Importance of the Heme Fe Center to Cyt c Activity
3. Biomedical Applications of Cyt c in Cancer Research
3.1. Cyt c as a Biomarker
3.2. Cyt c in Anticancer Therapies
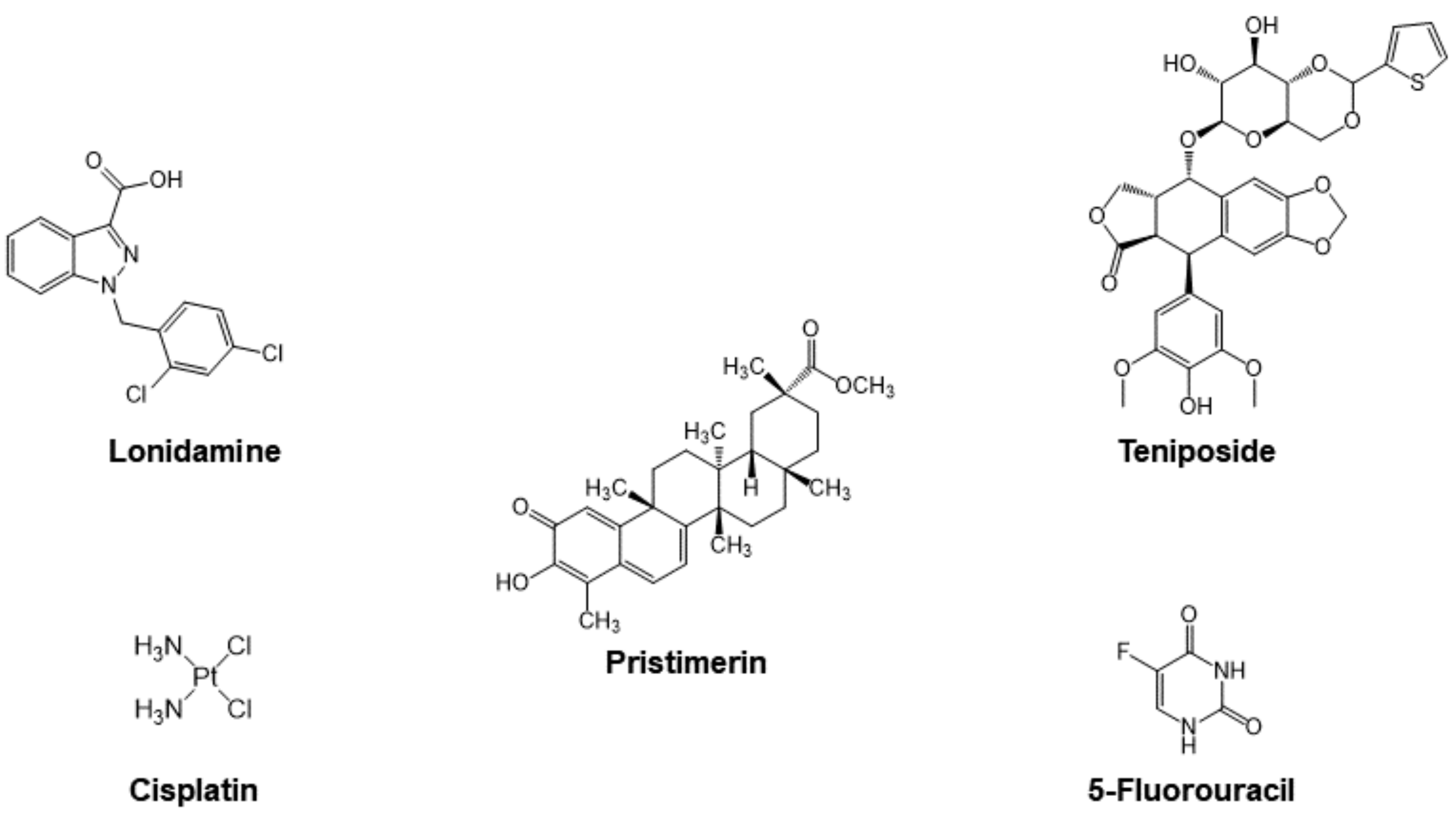
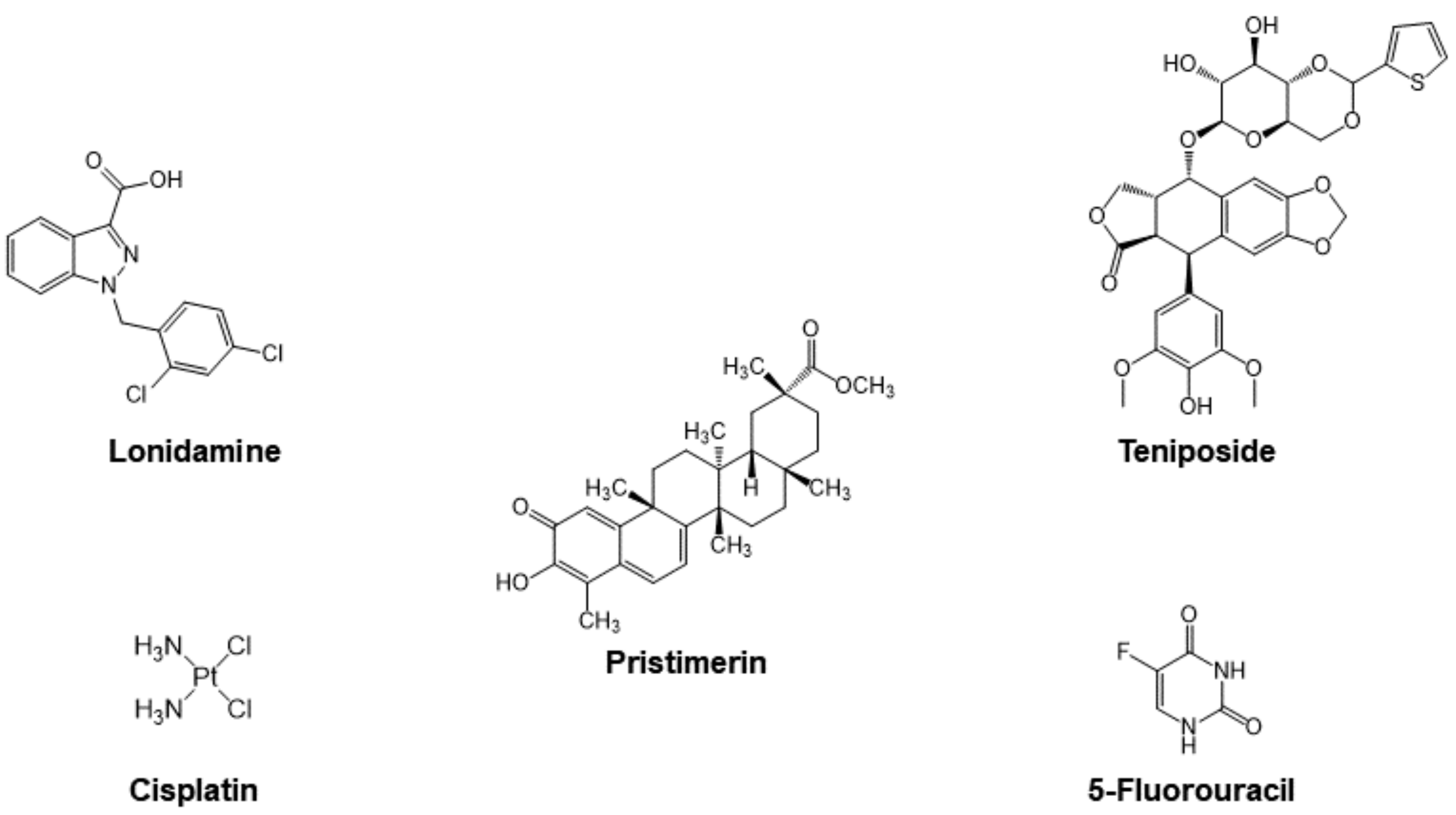
3.2.1. Cyt c as Part of the Mechanism of Small Molecule Anticancer Drugs
Cisplatin
Organic Drugs
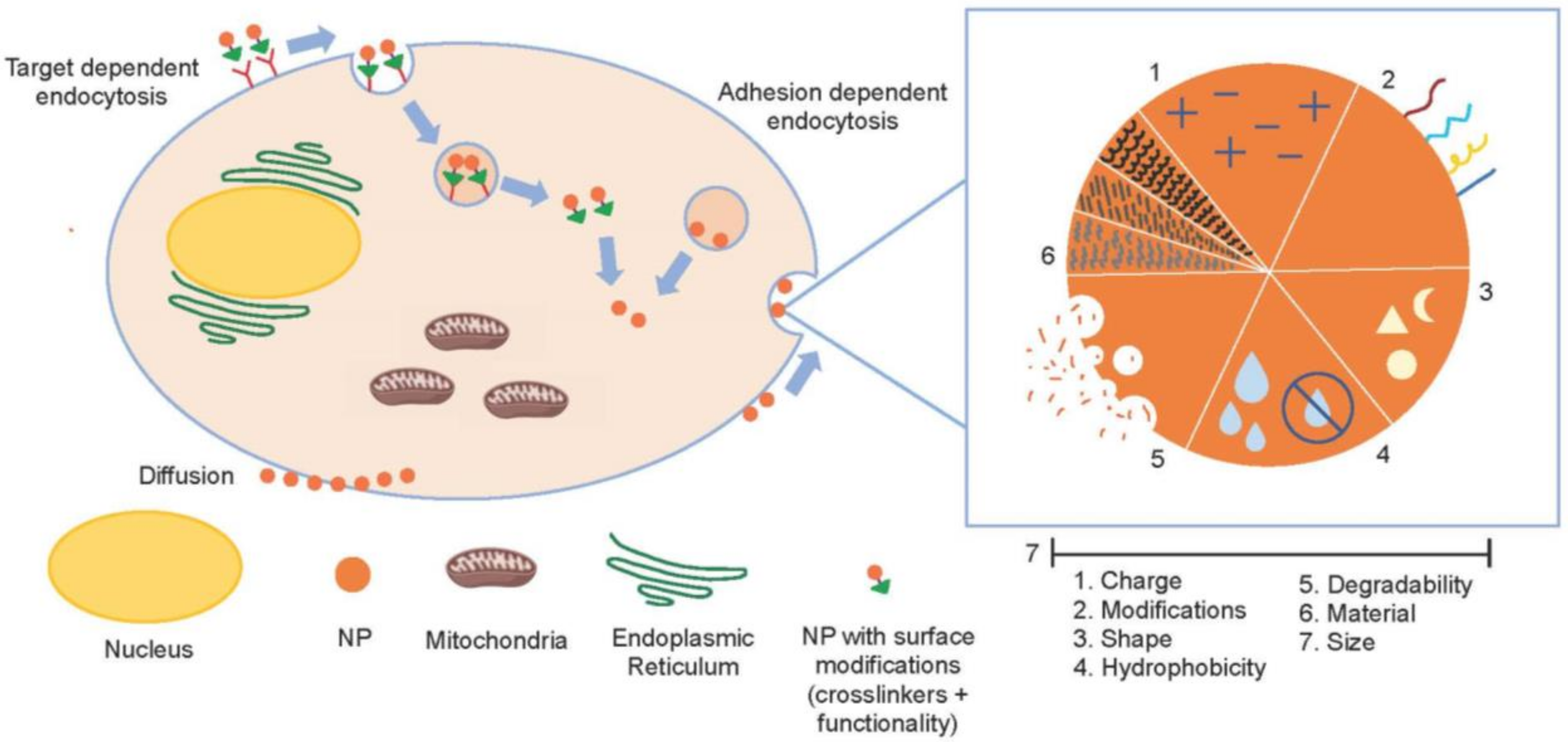
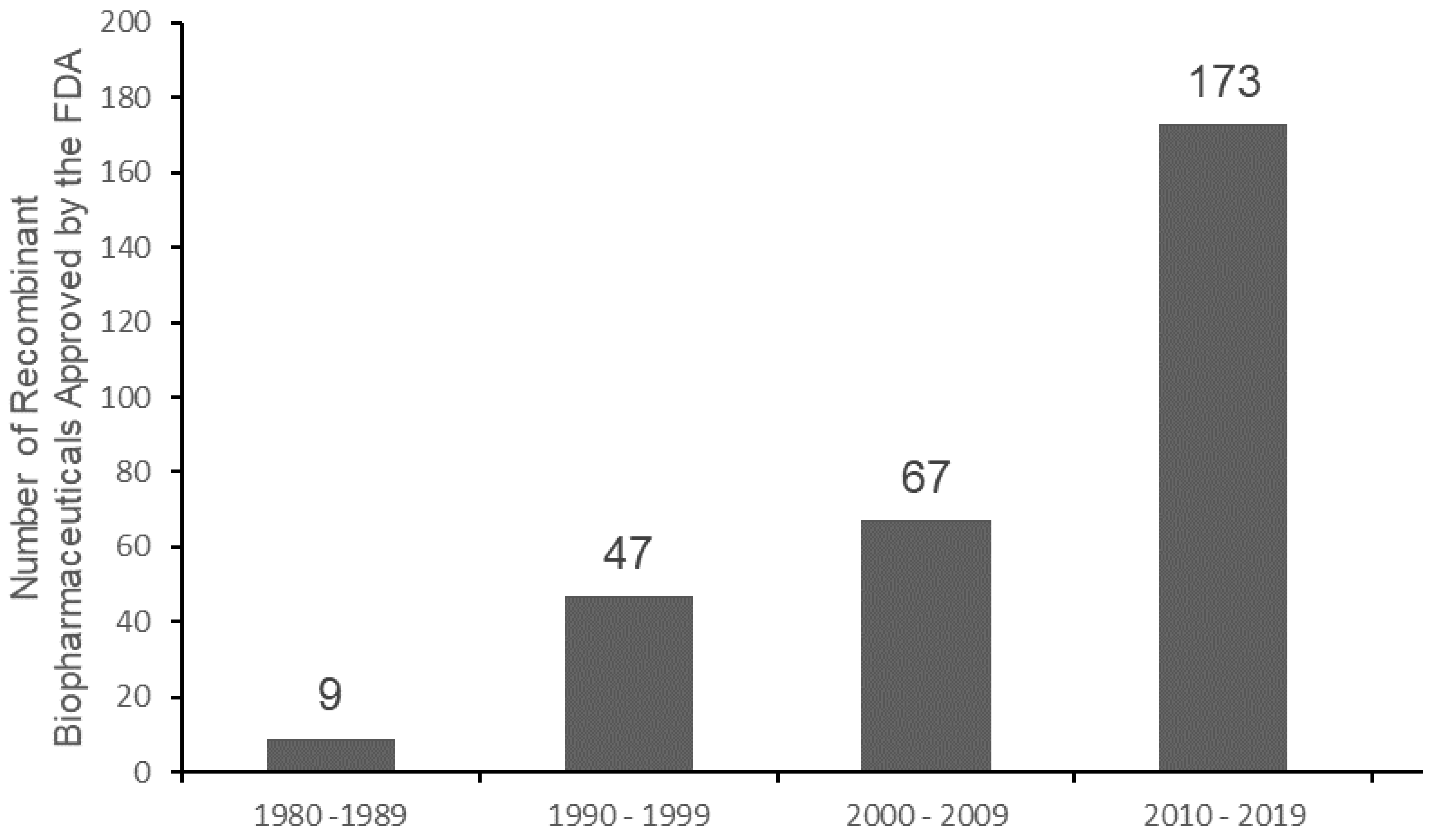
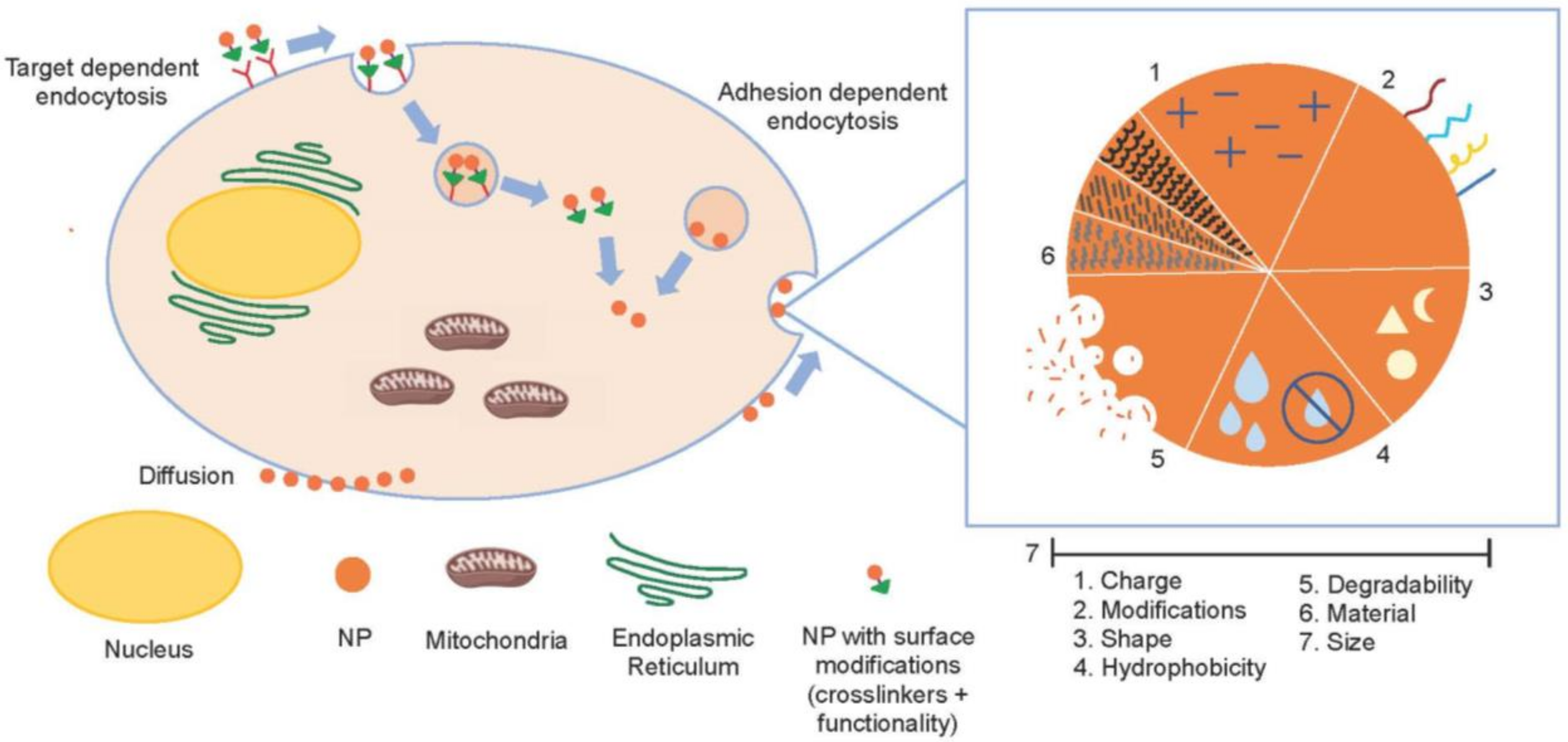
3.2.2. Drug Delivery Systems for the Application of Cyt c as an Anticancer Biodrug
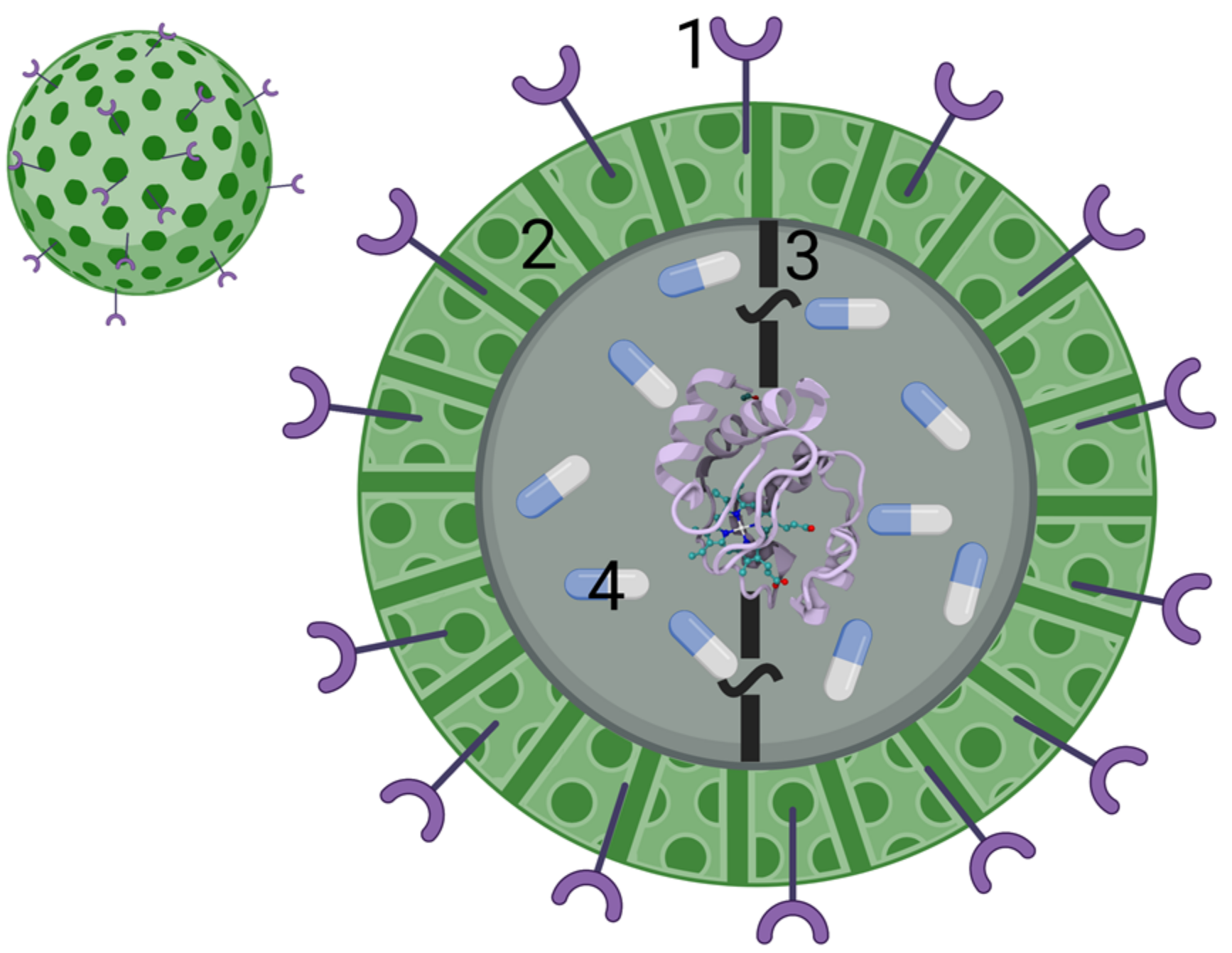
Nanoparticle-Based Cyt c Delivery Systems
Protein-Based Cyt c Delivery Systems
4. New Insight for Optimizing Cyt c Anticancer Drug Design
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Patt, D.; Gordan, L.; Diaz, M.; Okon, T.; Grady, L.; Harmison, M.; Markward, N.; Sullivan, M.; Peng, J.; Zhou, A. Impact of COVID-19 on Cancer Care: How the Pandemic Is Delaying Cancer Diagnosis and Treatment for American Seniors. JCO Clin. Cancer Inform. 2020, 4, 1059–1071. [Google Scholar] [CrossRef]
- Maringe, C.; Spicer, J.; Morris, M.; Purushotham, A.; Nolte, E.; Sullivan, R.; Rachet, B.; Aggarwal, A. The impact of the COVID-19 pandemic on cancer deaths due to delays in diagnosis in England, UK: A national, population-based, modelling study. Lancet Oncol. 2020, 21, 1023–1034. [Google Scholar] [CrossRef]
- Yashavantha Rao, H.C.; Siddeeqh, S.; Taqui, S.N. Caring for cancer patients during COVID-19 global pandemic: Risk factors and precautionary principle. Int. J. Clin. Pract. 2021, 75, e14141. [Google Scholar] [CrossRef]
- Chakraborty, S.; Hosen, M.I.; Ahmed, M.; Shekhar, H.U. Onco-Multi-OMICS Approach: A New Frontier in Cancer Research. Biomed Res. Int. 2018, 2018, 9836256. [Google Scholar] [CrossRef] [Green Version]
- Dunn, B.K.; Meerzaman, D. Editorial: Using Cancer ‘Omics’ to Understand Cancer. Front. Oncol. 2020, 10, 1201. [Google Scholar] [CrossRef]
- Yu, K.H.; Snyder, M. Omics Profiling in Precision Oncology. Mol. Cell. Proteom. 2016, 15, 2525–2536. [Google Scholar] [CrossRef] [Green Version]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, 6481. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Leader, B.; Baca, Q.J.; Golan, D.E. Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 2008, 7, 21–39. [Google Scholar] [CrossRef]
- Murray, J.; Laurieri, N.; Delgoda, R. Proteins. In Pharmacognosy Fundamentals, Applications and Strategies; Protein Therapeutics: A Summary and Pharmacological Classification; Badal, S., Delgoda, R., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2017; pp. 477–494. [Google Scholar] [CrossRef]
- Serna, N.; Sánchez-García, L.; Unzueta, U.; Díaz, R.; Vázquez, E.; Mangues, R.; Villaverde, A. Protein-Based Therapeutic Killing for Cancer Therapies. Trends Biotechnol. 2018, 36, 318–335. [Google Scholar] [CrossRef]
- Patil, S.; Narvekar, A.; Puranik, A.; Jain, R.; Dandekar, P. Formulation of Therapeutic Proteins: Strategies for Developing Oral Protein Formulations. In Innovative Dosage Forms: Design and Development at Early Stage; Bachhav, Y., Ed.; Wiley-VCH: Hoboken, NJ, USA, 2019; Volume 76, pp. 391–432. [Google Scholar]
- Dimitrov, D.S. Therapeutic Proteins. Methods Mol. Biol. 2012, 899, 1–26. [Google Scholar] [CrossRef]
- Usmani, S.S.; Bedi, G.; Samuel, J.S.; Singh, S.; Kalra, S.; Kumar, P.; Ahuja, A.A.; Sharma, M.; Gautam, A.; Raghava, G.P.S. THPdb: Database of FDA-approved peptide and protein therapeutics. PLoS ONE 2017, 12, e0181748. [Google Scholar] [CrossRef] [Green Version]
- Rader, R.A. Biosimilars/Biobetters Pipeline Directory. Available online: http://www.biosimilarspipeline.com/ (accessed on 6 August 2021).
- Lu, T.C.; Zhao, G.-H.; Chen, Y.Y.; Chien, C.-Y.; Huang, C.-H.; Lin, K.H.; Chen, S.L. Transduction of Recombinant M3-p53-R12 Protein Enhances Human Leukemia Cell Apoptosis. J. Cancer 2016, 7, 1360–1373. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.; Jia, Q.; Kang, X.; Lou, Y.; Zou, L.; Yang, J.; Gao, J.; Han, L.; Li, X. Antitumor activity of recombinant RGD-IFN-α2a-core fusion protein in vitro. Anticancer Drugs 2017, 28, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Sokolova, E.; Guryev, E.; Yudintsev, A.; Vodeneev, V.; Deyev, S.; Balalaeva, I. HER2-specific recombinant immunotoxin 4D5scFv-PE40 passes through retrograde trafficking route and forces cells to enter apoptosis. Oncotarget 2017, 8, 22048–22058. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Yi, C.; Huang, X.-Z.; Yan, J.; Wei, L.-J.; Tang, W.-J.; Chen, S.-C.; Huang, Y. Recombinant protein TRAIL-Mu3 enhances the antitumor effects in pancreatic cancer cells by strengthening the apoptotic signaling pathway. Oncol. Lett. 2021, 21, 438. [Google Scholar] [CrossRef]
- Korsmeyer, S.J. BCL-2 gene family and the regulation of programmed cell death. Cancer Res. 1999, 59, 1693s–1700s. [Google Scholar] [CrossRef]
- Bruckheimer, E.; Cho, S.; Sarkiss, M.; Herrmann, J.; McDonnell, T.J. The Bcl-2 gene family and apoptosis. Adv. Biochem. Engin./Biotechnol. 1998, 62, 75–105. [Google Scholar] [CrossRef]
- Wang, Z.-B.; Liu, Y.-Q.; Cui, Y.-F. Pathways to caspase activation. Cell Biol. Int. 2005, 29, 489–496. [Google Scholar] [CrossRef]
- Dickerson, R.E. The structures of cytochrome c and the rates of molecular evolution. J. Mol. Evol. 1971, 1, 26–45. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.; Hassan, M.I.; Islam, A.; Ahmad, F. The role of key residues in structure, function, and stability of cytochrome-c. Cell. Mol. Life Sci. 2014, 71, 229–255. [Google Scholar] [CrossRef] [PubMed]
- Rydberg, P.; Sigfridsson, E.; Ryde, U. On the role of the axial ligand in heme proteins: A theoretical study. J. Biol. Inorg. Chem. 2004, 9, 203–223. [Google Scholar] [CrossRef] [PubMed]
- Senn, H.; Wüthrich, K. Amino acid sequence, haem-iron co-ordination geometry and functional properties of mitochondrial and bacterial c-type cytochromes. Q. Rev. Biophys. 1985, 18, 111–134. [Google Scholar] [CrossRef] [Green Version]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef] [Green Version]
- Olteanu, A.; Patel, C.N.; Dedmon, M.M.; Kennedy, S.; Linhoff, M.W.; Minder, C.M.; Potts, P.R.; Deshmukh, M.; Pielak, G.J. Stability and apoptotic activity of recombinant human cytochrome c. Biochem. Biophys. Res. Commun. 2003, 312, 733–740. [Google Scholar] [CrossRef]
- Yu, T.; Wang, X.; Purring-Koch, C.; Wei, Y.; McLendon, G.L. A mutational epitope for cytochrome C binding to the apoptosis protease activation factor-1. J. Biol. Chem. 2001, 276, 13034–13038. [Google Scholar] [CrossRef] [Green Version]
- Banci, L.; Bertini, I.; Rosato, A.; Varani, G. Mitochondrial cytochromes c: A comparative analysis. J. Biol. Inorg. Chem. 1999, 4, 824–837. [Google Scholar] [CrossRef]
- Morison, I.M.; Cramer Bordé, E.M.; Cheesman, E.J.; Cheong, P.L.; Holyoake, A.J.; Fichelson, S.; Weeks, R.J.; Lo, A.; Davies, S.M.; Wilbanks, S.M.; et al. A mutation of human cytochrome c enhances the intrinsic apoptotic pathway but causes only thrombocytopenia. Nat. Genet. 2008, 40, 387–389. [Google Scholar] [CrossRef]
- Liu, J.; Chakraborty, S.; Hosseinzadeh, P.; Yu, Y.; Tian, S.; Petrik, I.; Bhagi, A.; Lu, Y. Metalloproteins Containing Cytochrome, Iron–Sulfur, or Copper Redox Centers. Chem. Rev. 2014, 114, 4366–4469. [Google Scholar] [CrossRef]
- Yamanaka, T.; Okunuki, K. Chapter 14 Cytochromes. In Microbial Iron Metabolism: A Comprehensive Treatise; Neilands, J.B., Ed.; Academic Press: Cambridge, MA, USA, 1974. [Google Scholar]
- Kranz, R.G.; Richard-Fogal, C.; Taylor, J.S.; Frawley, E.R. Cytochrome c Biogenesis: Mechanisms for Covalent Modifications and Trafficking of Heme and for Heme-Iron Redox Control. Microbiol. Mol. Biol. Rev. 2009, 73, 510–528. [Google Scholar] [CrossRef] [Green Version]
- Santucci, R.; Sinibaldi, F.; Cozza, P.; Polticelli, F.; Fiorucci, L. Cytochrome c: An extreme multifunctional protein with a key role in cell fate. Int. J. Biol. Macromol. 2019, 136, 1237–1246. [Google Scholar] [CrossRef]
- Rumbley, J.; Hoang, L.; Mayne, L.; Englander, S.W. An amino acid code for protein folding. Proc. Natl. Acad. Sci. USA 2001, 98, 105–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maity, H.; Maity, M.; Englander, S.W. How Cytochrome c Folds, and Why: Submolecular Foldon Units and their Stepwise Sequential Stabilization. J. Mol. Biol. 2004, 343, 223–233. [Google Scholar] [CrossRef]
- Hüttemann, M.; Pecina, P.; Rainbolt, M.; Sanderson, T.H.; Kagan, V.E.; Samavati, L.; Doan, J.W.; Lee, I. The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: From respiration to apoptosis. Mitochondrion 2011, 11, 369–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
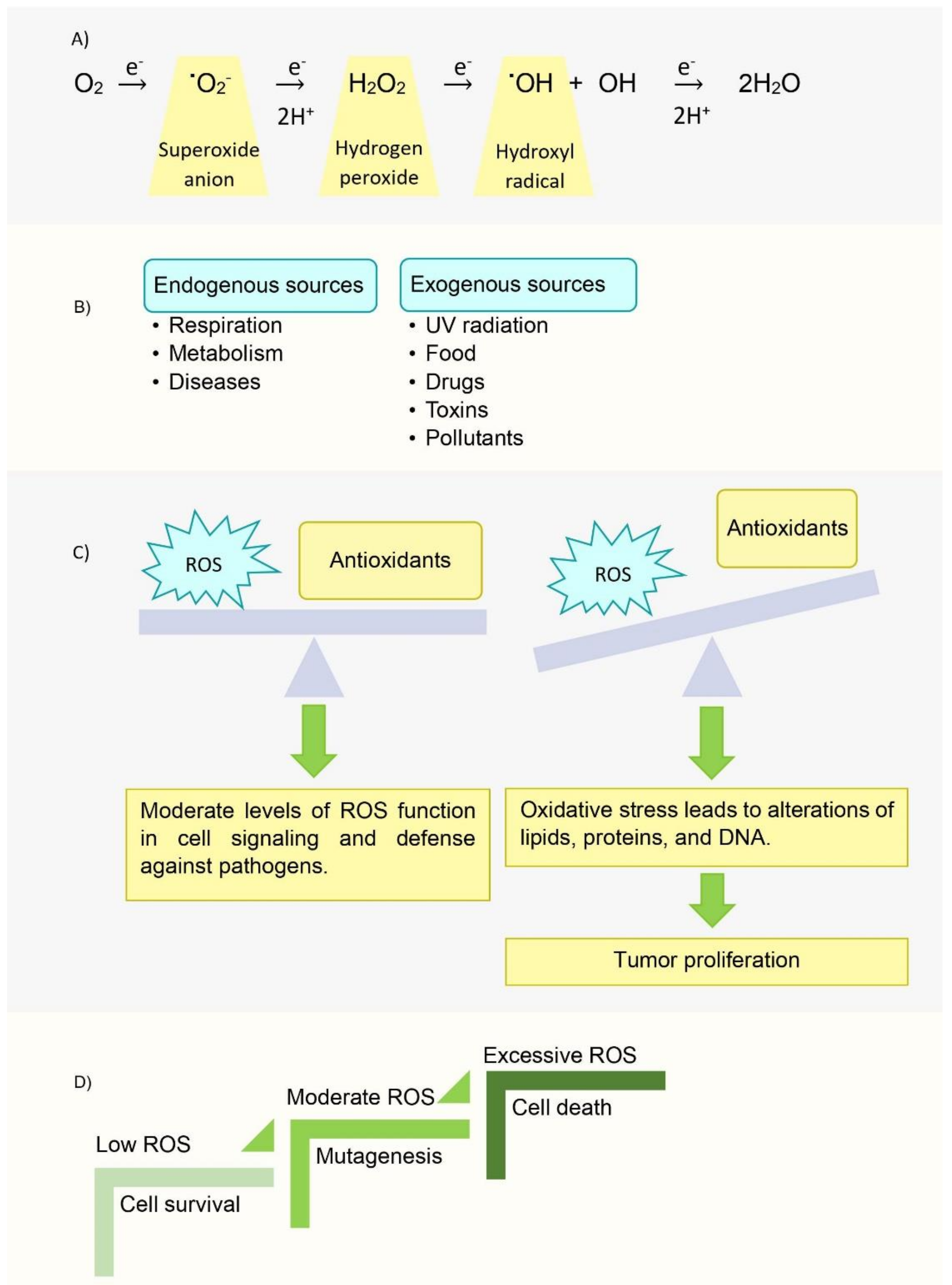
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002, 80, 780–787. [Google Scholar] [CrossRef]
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Mitsunaga, M.; Ogawa, M.; Kosaka, N.; Rosenblum, L.T.; Choyke, P.L.; Kobayashi, H. Cancer cell–selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat. Med. 2011, 17, 1685–1691. [Google Scholar] [CrossRef] [Green Version]
- Kalpage, H.A.; Wan, J.; Morse, P.T.; Zurek, M.P.; Turner, A.A.; Khobeir, A.; Yazdi, N.; Hakim, L.; Liu, J.; Vaishnav, A.; et al. Cytochrome c phosphorylation: Control of mitochondrial electron transport chain flux and apoptosis. Int. J. Biochem. Cell Biol. 2020, 121. [Google Scholar] [CrossRef] [PubMed]
- Pasdois, P.; Parker, J.E.; Griffiths, E.J.; Halestrap, A.P. The role of oxidized cytochrome c in regulating mitochondrial reactive oxygen species production and its perturbation in ischaemia. Biochem. J. 2011, 436, 493–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loreto, C.; La Rocca, G.; Anzalone, R.; Caltabiano, R.; Vespasiani, G.; Castorina, S.; Ralph, D.J.; Cellek, S.; Musumeci, G.; Giunta, S.; et al. The role of intrinsic pathway in apoptosis activation and progression in Peyronie’s disease. Biomed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Pinedo, C. Signaling pathways that regulate life and cell death: Evolution of apoptosis in the context of self-defense. Adv. Exp. Med. Biol. 2012, 738, 124–143. [Google Scholar] [CrossRef] [PubMed]
- Caltabiano, R.; Leonardi, R.; Musumeci, G.; Bartoloni, G.; Rusu, M.C.; Almeida, L.E.; Loreto, C. Apoptosis in temporomandibular joint disc with internal derangement involves mitochondrial-dependent pathways. An in vivo study. Acta Odontol. Scand. 2013, 71, 577–583. [Google Scholar] [CrossRef]
- Guerra-Castellano, A.; Márquez, I.; Pérez-Mejías, G.; Díaz-Quintana, A.; De la Rosa, M.A.; Díaz-Moreno, I. Post-Translational Modifications of Cytochrome c in Cell Life and Disease. Int. J. Mol. Sci. 2020, 21, 8483. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, L.C.; Chien, J.C. Cobalt-cytochrome c. I. Preparation, properties, and enzymic activity. Biochemistry 1975, 14, 3526–3534. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, L.C.; Chien, J.C. Cobalt-cytochrome c. II. Magnetic resonance spectra and conformational transitions. Biochemistry 1975, 14, 3534–3542. [Google Scholar] [CrossRef]
- Findlay, M.C.; Dickinson, L.C.; Chien, J.C.W. Copper-cytochrome c. J. Am. Chem. Soc. 1977, 99, 5168–5173. [Google Scholar] [CrossRef]
- Qian, C.; Yao, Y.; Tong, Y.; Wang, J.; Tang, W. Structural analysis of zinc-substituted cytochrome c. J. Biol. Inorg. Chem. 2003, 8, 394–400. [Google Scholar] [CrossRef]
- Chien, J.C.; Gibson, H.L.; Dickinson, L.C. Ferricytochrome c oxidation of cobaltocytochrome c. Comparison of experiments with electron-transfer theories. Biochemistry 1978, 17, 2579–2584. [Google Scholar] [CrossRef]
- Sun, J.; Su, C.; Wishart, J.F. Intramolecular Electron Transfer in Pentaammineruthenium(III)-Modified Cobaltocytochrome c. Inorg. Chem. 1996, 35, 5893–5901. [Google Scholar] [CrossRef]
- Anni, H.; Vanderkooi, J.M.; Mayne, L. Structure of zinc-substituted cytochrome c: Nuclear magnetic resonance and optical spectroscopic studies. Biochemistry 1995, 34, 5744–5753. [Google Scholar] [CrossRef]
- Zhou, J.S.; Nocek, J.M.; DeVan, M.L.; Hoffman, B.M. Inhibitor-Enhanced Electron Transfer: Copper Cytochrome c as a Redox-Inert Probe of Ternary Complexes. Science 1995, 269, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Kluck, R.M.; Martin, S.J.; Hoffman, B.M.; Zhou, J.S.; Green, D.R.; Newmeyer, D.D. Cytochrome c activation of CPP32-like proteolysis plays a critical role in a Xenopus cell-free apoptosis system. EMBO J. 1997, 16, 4639–4649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feifel, S.C.; Stieger, K.R.; Kapp, A.; Weber, D.; Allegrozzi, M.; Piccioli, M.; Turano, P.; Lisdat, F. Insights into Interprotein Electron Transfer of Human Cytochrome c Variants Arranged in Multilayer Architectures by Means of an Artificial Silica Nanoparticle Matrix. ACS Omega 2016, 1, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Watkins, D.W.; Armstrong, C.T.; Beesley, J.L.; Marsh, J.E.; Jenkins, J.M.X.; Sessions, R.B.; Mann, S.; Ross Anderson, J.L. A suite of de novo c-type cytochromes for functional oxidoreductase engineering. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 493–502. [Google Scholar] [CrossRef] [Green Version]
- Aghamiri, Z.S.; Mohsennia, M.; Rafiee-Pour, H.A. Immobilization of cytochrome c and its application as electrochemical biosensors. Talanta 2018, 176, 195–207. [Google Scholar] [CrossRef]
- Pinck, S.; Xu, M.; Clement, R.; Lojou, E.; Jorand, F.P.A.; Etienne, M. Influence of cytochrome charge and potential on the cathodic current of electroactive artificial biofilms. Bioelectrochemistry 2018, 124, 185–194. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, X.; Huang, C.; Li, J.; Shen, X. Artificial Cytochrome c Mimics: Graphene Oxide–Fe(III) Complex-Coated Molecularly Imprinted Colloidosomes for Selective Photoreduction of Highly Toxic Pollutants. ACS Appl. Mater. Interfaces 2020, 12, 6615–6626. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Liakopoulos, V.; Stefanidis, I. Cytochrome c as a Potentially Clinical Useful Marker of Mitochondrial and Cellular Damage. Front. Immunol. 2016, 7, 279. [Google Scholar] [CrossRef] [Green Version]
- Afify, M.; Kamel, R.R.; Elhosary, Y.A.; Hegazy, A.E.; Fahim, H.H.; Ezzat, W.M. The possible role of Cytochrome c and programmed cell death protein 4 (PDCD4) on pathogenesis of hepatocellular carcinoma. J. Genet. Eng. Biotechnol. 2015, 13, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthi, B.V.; Nepal, S.; Varambally, S. Genomic and Epigenomic Alterations in Cancer. Am. J. Pathol. 2016, 186, 1724–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chittenden, T.; Flemington, C.; Houghton, A.B.; Ebb, R.G.; Gallo, G.J.; Elangovan, B.; Chinnadurai, G.; Lutz, R.J. A conserved domain in Bak, distinct from BH1 and BH2, mediates cell death and protein binding functions. EMBO J. 1995, 14, 5589–5596. [Google Scholar] [CrossRef] [PubMed]
- Zha, H.; Aimé-Sempé, C.; Sato, T.; Reed, J.C. Proapoptotic Protein Bax Heterodimerizes with Bcl-2 and Homodimerizes with Bax via a Novel Domain (BH3) Distinct from BH1 and BH2. J. Biol. Chem. 1996, 271, 7440–7444. [Google Scholar] [CrossRef] [Green Version]
- Boyd, J.M.; Gallo, G.J.; Elangovan, B.; Houghton, A.B.; Malstrom, S.; Avery, B.J.; Ebb, R.G.; Subramanian, T.; Chittenden, T.; Lutz, R.J.; et al. Bik, a novel death-inducing protein shares a distinct sequence motif with Bcl-2 family proteins and interacts with viral and cellular survival-promoting proteins. Oncogene 1995, 11, 1921–1928. [Google Scholar]
- Sattler, M.; Liang, H.; Nettesheim, D.; Meadows, R.P.; Harlan, J.E.; Eberstadt, M.; Yoon, H.S.; Shuker, S.B.; Chang, B.S.; Minn, A.J.; et al. Structure of Bcl-xL-Bak peptide complex: Recognition between regulators of apoptosis. Science 1997, 275, 983–986. [Google Scholar] [CrossRef]
- Lovell, J.F.; Billen, L.P.; Bindner, S.; Shamas-Din, A.; Fradin, C.; Leber, B.; Andrews, D.W. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 2008, 135, 1074–1084. [Google Scholar] [CrossRef] [Green Version]
- Kuwana, T.; Bouchier-Hayes, L.; Chipuk, J.E.; Bonzon, C.; Sullivan, B.A.; Green, D.R.; Newmeyer, D.D. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 2005, 17, 525–535. [Google Scholar] [CrossRef]
- Gorbunova, A.; Denisenko, T.; Yapryntseva, M.; Pivnyuk, A.; Prikazchikova, T.; Gogvadze, V.; Zhivotovsky, B. BNIP3 as a Regulator of Cisplatin-Induced Apoptosis. Biochemistry 2020, 85, 1245–1253. [Google Scholar] [CrossRef]
- Marchetti, P.; Galla, D.A.; Russo, F.P.; Ricevuto, E.; Flati, V.; Porzio, G.; Ficorella, C.; Cifone, M.G. Apoptosis induced by oxaliplatin in human colon cancer HCT15 cell line. Anticancer Res. 2004, 24, 219–226. [Google Scholar] [PubMed]
- Kleih, M.; Böpple, K.; Dong, M.; Gaißler, A.; Heine, S.; Olayioye, M.A.; Aulitzky, W.E.; Essmann, F. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Dabrowiak, J.C. Metals in Medicine, 2nd ed.; Wiley: Hoboken, NJ, USA, 2017; pp. 91–146. [Google Scholar]
- MacLaine, N.J.; Hupp, T.R. How phosphorylation controls p53. Cell Cycle 2011, 10, 916–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravagnan, L.; Marzo, I.; Costantini, P.; Susin, S.A.; Zamzami, N.; Petit, P.X.; Hirsch, F.; Goulbern, M.; Poupon, M.-F.; Miccoli, L. Lonidamine triggers apoptosis via a direct, Bcl-2-inhibited effect on the mitochondrial permeability transition pore. Oncogene 1999, 18, 2537–2546. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, E.; Armour, S.M.; Harris, M.H.; Thompson, C.B. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 2003, 10, 709–717. [Google Scholar] [CrossRef]
- Belzacq, A.-S.; El Hamel, C.; Vieira, H.L.; Cohen, I.; Haouzi, D.; Métivier, D.; Marchetti, P.; Brenner, C.; Kroemer, G. Adenine nucleotide translocator mediates the mitochondrial membrane permeabilization induced by lonidamine, arsenite and CD437. Oncogene 2001, 20, 7579–7587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-C.; Chan, M.-L.; Chen, W.-Y.; Tsai, C.-Y.; Chang, F.-R.; Wu, Y.-C. Pristimerin induces caspase-dependent apoptosis in MDA-MB-231 cells via direct effects on mitochondria. Mol. Cancer Ther. 2005, 4, 1277–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Zhang, W.; Yan, Y.-y.; Ma, C.-g.; Wang, X.; Wang, C.; Zhao, J.-l. Triterpenoid pristimerin induced HepG2 cells apoptosis through ROS-mediated mitochondrial dysfunction. JBUON 2013, 18, 477–485. [Google Scholar]
- Teniposide. Available online: https://www.cancerquest.org/node/6455 (accessed on 31 October 2021).
- Gordaliza, M.; Castro, M.d.; Miguel del Corral, J.; Feliciano, A.S. Antitumor properties of podophyllotoxin and related compounds. Curr. Pharm. Des. 2000, 6, 1811–1839. [Google Scholar] [CrossRef]
- Sánchez-Alcázar, J.A.; Khodjakov, A.; Schneider, E. Anticancer drugs induce increased mitochondrial cytochrome c expression that precedes cell death. Cancer Res. 2001, 61, 1038–1044. [Google Scholar]
- Moghimipour, E.; Rezaei, M.; Ramezani, Z.; Kouchak, M.; Amini, M.; Angali, K.A.; Dorkoosh, F.A.; Handali, S. Transferrin targeted liposomal 5-fluorouracil induced apoptosis via mitochondria signaling pathway in cancer cells. Life Sci. 2018, 194, 104–110. [Google Scholar] [CrossRef]
- Yang, L.; Wu, D.; Luo, K.; Wu, S.; Wu, P. Andrographolide enhances 5-fluorouracil-induced apoptosis via caspase-8-dependent mitochondrial pathway involving p53 participation in hepatocellular carcinoma (SMMC-7721) cells. Cancer Lett. 2009, 276, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Chen, J.; Wang, Y.; Wong, Y.-S.; Zhang, Y.; Zheng, W.; Cao, W.; Chen, T. Selenocystine potentiates cancer cell apoptosis induced by 5-fluorouracil by triggering reactive oxygen species-mediated DNA damage and inactivation of the ERK pathway. Free Radic. Biol. Med. 2013, 65, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Al-Shakarchi, W.; Alsuraifi, A.; Abed, M.; Abdullah, M.; Richardson, A.; Curtis, A.; Hoskins, C. Combined Effect of Anticancer Agents and Cytochrome C Decorated Hybrid Nanoparticles for Liver Cancer Therapy. Pharmaceutics 2018, 10, 48. [Google Scholar] [CrossRef] [Green Version]
- Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria in cancer cells: What is so special about them? Trends Cell Biol 2008, 18, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.M.; Tang, H.L. Anastasis: Recovery from the brink of cell death. R. Soc. Open Sci. 2018, 5, 180442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Røise, J.J.; Lee, K.; Li, J.; Murthy, N. Recent developments in intracellular protein delivery. Curr. Opin. Biotechnol. 2018, 52, 25–31. [Google Scholar] [CrossRef]
- Li, F.; Srinivasan, A.; Wang, Y.; Armstrong, R.C.; Tomaselli, K.J.; Fritz, L.C. Cell-specific Induction of Apoptosis by Microinjection of Cytochrome c: Bcl-x L has activity independent of cytochrome c release. J. Biol. Chem. 1997, 272, 30299–30305. [Google Scholar] [CrossRef] [Green Version]
- Devarajan, E.; Sahin, A.A.; Chen, J.S.; Krishnamurthy, R.R.; Aggarwal, N.; Brun, A.-M.; Sapino, A.; Zhang, F.; Sharma, D.; Yang, X.-H.; et al. Down-regulation of caspase 3 in breast cancer: A possible mechanism for chemoresistance. Oncogene 2002, 21, 8843–8851. [Google Scholar] [CrossRef] [Green Version]
- Mooney, L.M.; Al-Sakkaf, K.A.; Brown, B.L.; Dobson, P.R.M. Apoptotic mechanisms in T47D and MCF-7 human breast cancer cells. Br. J. Cancer 2002, 87, 909–917. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Yang, F.; Xiong, F.; Gu, N. The smart drug delivery system and its clinical potential. Theranostics 2016, 6, 1306. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H. Nanoparticle-based drug delivery systems for enhanced tumor-targeting treatment. J. Biomed. Nanotechnol. 2019, 15, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, F.; Ji, Y.; Yin, L. Recent Advances in Anti-cancer Protein/Peptide Delivery. Bioconjug. Chem. 2019, 30, 305–324. [Google Scholar] [CrossRef] [PubMed]
- Greish, K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. In Cancer Nanotechnology; Springer: Berlin/Heidelberg, Germany, 2010; pp. 25–37. [Google Scholar]
- Rappoport, J.; Preece, J.; Chipman, K.; Argatov, I.; Davies, A.; Dyson, L. How do Manufactured Nanoparticles Enter Cells. Micropor. Mesopor. Mat. 2011, 116, 123–130. [Google Scholar]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2020, 20, 101–124. [Google Scholar] [CrossRef]
- Chen, X.; Tian, F.; Zhang, X.; Wang, W. Internalization pathways of nanoparticles and their interaction with a vesicle. Soft Matter 2013, 9, 7592–7600. [Google Scholar] [CrossRef]
- Aubin-Tam, M.E.; Hamad-Schifferli, K. Gold nanoparticle-cytochrome C complexes: The effect of nanoparticle ligand charge on protein structure. Langmuir 2005, 21, 12080–12084. [Google Scholar] [CrossRef]
- Choi, E.; Lim, D.-K.; Kim, S. Hydrolytic surface erosion of mesoporous silica nanoparticles for efficient intracellular delivery of cytochrome c. J. Colloid Interface Sci. 2020, 560, 416–425. [Google Scholar] [CrossRef]
- Guo, C.; Zhang, Y.; Li, Y.; Zhang, L.; Jiang, H.; Tao, J.; Zhu, J. Gold nanoparticle-guarded large-pore mesoporous silica nanocomposites for delivery and controlled release of cytochrome c. J. Colloid Interface Sci. 2021, 589, 34–44. [Google Scholar] [CrossRef]
- Méndez, J.; Morales Cruz, M.; Delgado, Y.; Figueroa, C.M.; Orellano, E.A.; Morales, M.; Monteagudo, A.; Griebenow, K. Delivery of chemically glycosylated cytochrome c immobilized in mesoporous silica nanoparticles induces apoptosis in HeLa cancer cells. Mol. Pharm. 2014, 11, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Morales-Cruz, M.; Figueroa, C.M.; González-Robles, T.; Delgado, Y.; Molina, A.; Méndez, J.; Morales, M.; Griebenow, K. Activation of caspase-dependent apoptosis by intracellular delivery of cytochrome c-based nanoparticles. J. Nanobiotechnol. 2014, 12, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales-Cruz, M.; Cruz-Montañez, A.; Figueroa, C.M.; González-Robles, T.; Davila, J.; Inyushin, M.; Loza-Rosas, S.A.; Molina, A.M.; Muñoz-Perez, L.; Kucheryavykh, L.Y.; et al. Combining stimulus-triggered release and active targeting strategies improves cytotoxicity of cytochrome c nanoparticles in tumor cells. Mol. Pharmaceut. 2016, 13, 2844–2854. [Google Scholar] [CrossRef]
- Barcelo-Bovea, V.; Dominguez-Martinez, I.; Joaquin-Ovalle, F.; Amador, L.A.; Castro-Rivera, E.; Medina-Álvarez, K.; McGoron, A.; Griebenow, K.; Ferrer-Acosta, Y. Optimization and Characterization of Protein Nanoparticles for the Targeted and Smart Delivery of Cytochrome c to Non-Small Cell Lung Carcinoma. Cancers 2020, 12, 1215. [Google Scholar] [CrossRef] [PubMed]
- Kucheryavykh, Y.V.; Davila, J.; Ortiz-Rivera, J.; Inyushin, M.; Almodovar, L.; Mayol, M.; Morales-Cruz, M.; Cruz-Montañez, A.; Barcelo-Bovea, V.; Griebenow, K.; et al. Targeted Delivery of Nanoparticulate Cytochrome C into Glioma Cells Through the Proton-Coupled Folate Transporter. Biomolecules 2019, 9, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Zhang, J.; Deng, C.; Meng, F.; Yu, L.; Zhong, Z. Redox-Sensitive and Intrinsically Fluorescent Photoclick Hyaluronic Acid Nanogels for Traceable and Targeted Delivery of Cytochrome c to Breast Tumor in Mice. ACS Appl. Mater. Interfaces 2016, 8, 21155–21162. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I.K.; Lee, S.C.; Han, B.; Park, K. Analysis on the current status of targeted drug delivery to tumors. J. Control Release 2012, 164, 108–114. [Google Scholar] [CrossRef] [Green Version]
- Macone, A.; Masciarelli, S.; Palombarini, F.; Quaglio, D.; Boffi, A.; Trabuco, M.C.; Baiocco, P.; Fazi, F.; Bonamore, A. Ferritin nanovehicle for targeted delivery of cytochrome C to cancer cells. Sci. Rep. 2019, 9, 11749. [Google Scholar] [CrossRef] [Green Version]
- Yeh, T.-H.; Wu, F.-L.L.; Shen, L.-J. Intracellular delivery of cytochrome c by galactosylated albumin to hepatocarcinoma cells. J. Drug Target. 2014, 22, 528–535. [Google Scholar] [CrossRef]
- Saxena, M.; Delgado, Y.; Sharma, R.K.; Sharma, S.; Guzmán, S.; Tinoco, A.D.; Griebenow, K. Inducing cell death in vitro in cancer cells by targeted delivery of cytochrome c via a transferrin conjugate. PLoS ONE 2018, 13, e0195542. [Google Scholar] [CrossRef]
- Deming, P.B.; Schafer, Z.T.; Tashker, J.S.; Potts, M.B.; Deshmukh, M.; Kornbluth, S. Bcr-Abl-mediated protection from apoptosis downstream of mitochondrial cytochrome c release. Mol. Cell. Biol. 2004, 24, 10289–10299. [Google Scholar] [CrossRef] [Green Version]
- Delinois, L.J.; Peón, H.; Villalobos-Santos, J.C.; Ramírez-Paz, J.; Miller, J.; Griebenow, K.H.; Tinoco, A.D. A Cytochrome c-Chlorotoxin Hybrid Protein as a Possible Antiglioma Drug. ChemMedChem 2020, 15, 2185–2192. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.C.; Sorger, P.K. Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell 2017, 171, 1678–1691.e1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.L.; Tang, H.M.; Mak, K.H.; Hu, S.; Wang, S.S.; Wong, K.M.; Wong, C.S.; Wu, H.Y.; Law, H.T.; Liu, K.; et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol. Biol. Cell. 2012, 23, 2240–2252. [Google Scholar] [CrossRef] [PubMed]
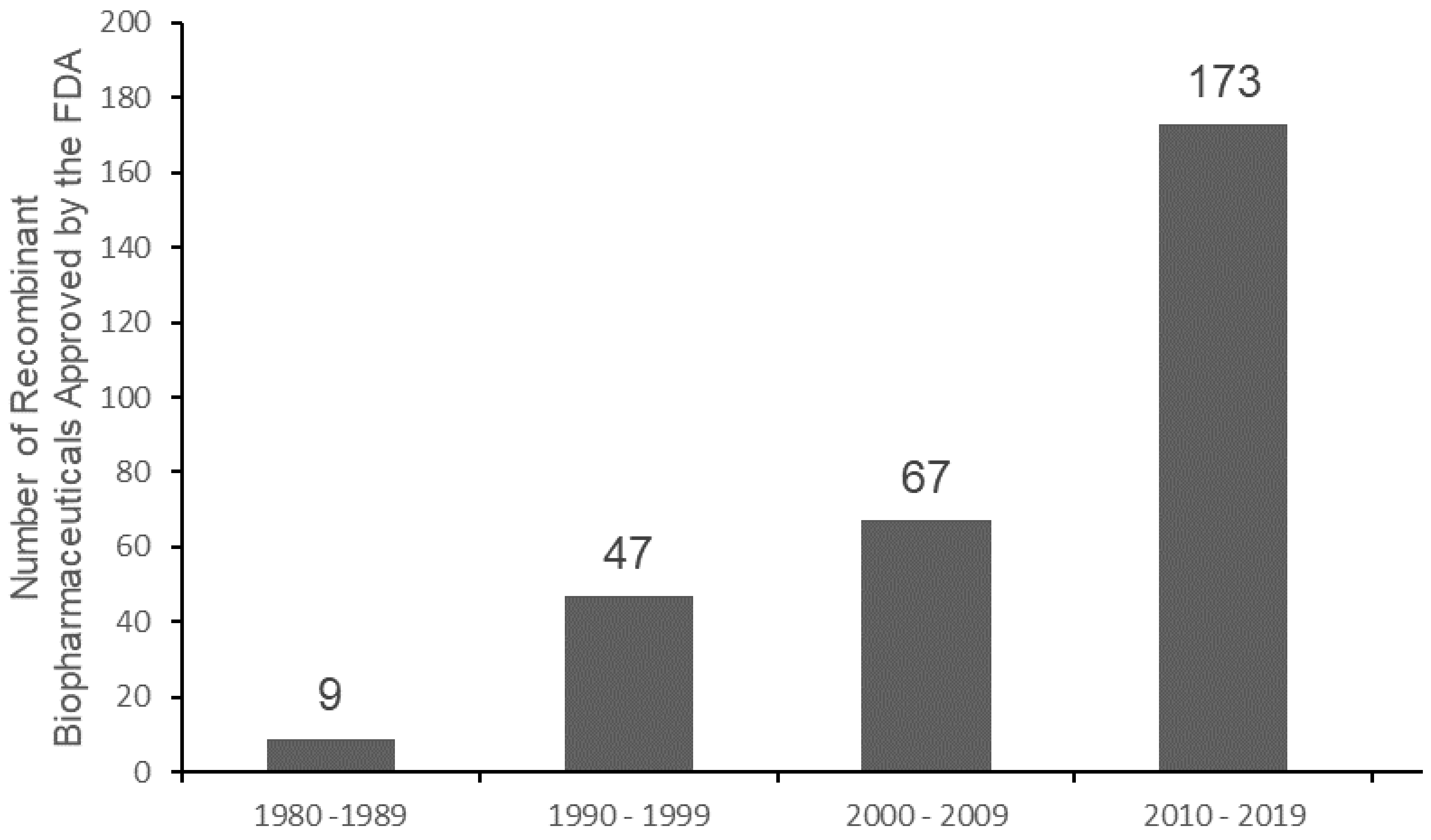



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Source | Mechanism of Action | Recombinant Protein (Producing Organism) | Cancer Tested |
|---|---|---|---|---|
| BID | Homo sapiens | Activator: interacts with high affinity to all antiapoptotic proteins and directly activates BAX and BAK | E. coli | Breast, ovarian, and prostate cancer |
| PUMA | Homo sapiens | Activator: interacts with high affinity to all antiapoptotic proteins and directly activates BAX and BAK | E. coli | Colon cancer |
| BAD | Homo sapiens | Sensitizer: interacts with antiapoptotic protein BCL-2 and BCL-XL with high affinity | E. coli | Glioma, leukemia, and gastrointestinal carcinoma |
| BIK | Homo sapiens | Sensitizer: interacts with antiapoptotic protein BCL-2 and BCL-XL with high affinity | E. coli | Colon adenocarcinoma |
| BAKBH3 | Homo sapiens | Antagonizes antiapoptotic protein function | E. coli | Cervical and colon cancer |
| Cyt c | Homo sapiens | Trigger apoptosis by interacting with Apaf-1 and cleave procaspase-9 | E. coli | Many cancer types |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delinois, L.J.; De León-Vélez, O.; Vázquez-Medina, A.; Vélez-Cabrera, A.; Marrero-Sánchez, A.; Nieves-Escobar, C.; Alfonso-Cano, D.; Caraballo-Rodríguez, D.; Rodriguez-Ortiz, J.; Acosta-Mercado, J.; et al. Cytochrome c: Using Biological Insight toward Engineering an Optimized Anticancer Biodrug. Inorganics 2021, 9, 83. https://doi.org/10.3390/inorganics9110083
Delinois LJ, De León-Vélez O, Vázquez-Medina A, Vélez-Cabrera A, Marrero-Sánchez A, Nieves-Escobar C, Alfonso-Cano D, Caraballo-Rodríguez D, Rodriguez-Ortiz J, Acosta-Mercado J, et al. Cytochrome c: Using Biological Insight toward Engineering an Optimized Anticancer Biodrug. Inorganics. 2021; 9(11):83. https://doi.org/10.3390/inorganics9110083
Chicago/Turabian StyleDelinois, Louis J., Omar De León-Vélez, Adriana Vázquez-Medina, Alondra Vélez-Cabrera, Amanda Marrero-Sánchez, Christopher Nieves-Escobar, Daniela Alfonso-Cano, Delvin Caraballo-Rodríguez, Jael Rodriguez-Ortiz, Jemily Acosta-Mercado, and et al. 2021. "Cytochrome c: Using Biological Insight toward Engineering an Optimized Anticancer Biodrug" Inorganics 9, no. 11: 83. https://doi.org/10.3390/inorganics9110083
APA StyleDelinois, L. J., De León-Vélez, O., Vázquez-Medina, A., Vélez-Cabrera, A., Marrero-Sánchez, A., Nieves-Escobar, C., Alfonso-Cano, D., Caraballo-Rodríguez, D., Rodriguez-Ortiz, J., Acosta-Mercado, J., Benjamín-Rivera, J. A., González-González, K., Fernández-Adorno, K., Santiago-Pagán, L., Delgado-Vergara, R., Torres-Ávila, X., Maser-Figueroa, A., Grajales-Avilés, G., Méndez, G. I. M., ... Tinoco, A. D. (2021). Cytochrome c: Using Biological Insight toward Engineering an Optimized Anticancer Biodrug. Inorganics, 9(11), 83. https://doi.org/10.3390/inorganics9110083