Exploring Serum Transferrin Regulation of Nonferric Metal Therapeutic Function and Toxicity

 ,
,  ,
, 
Abstract

1. Introduction
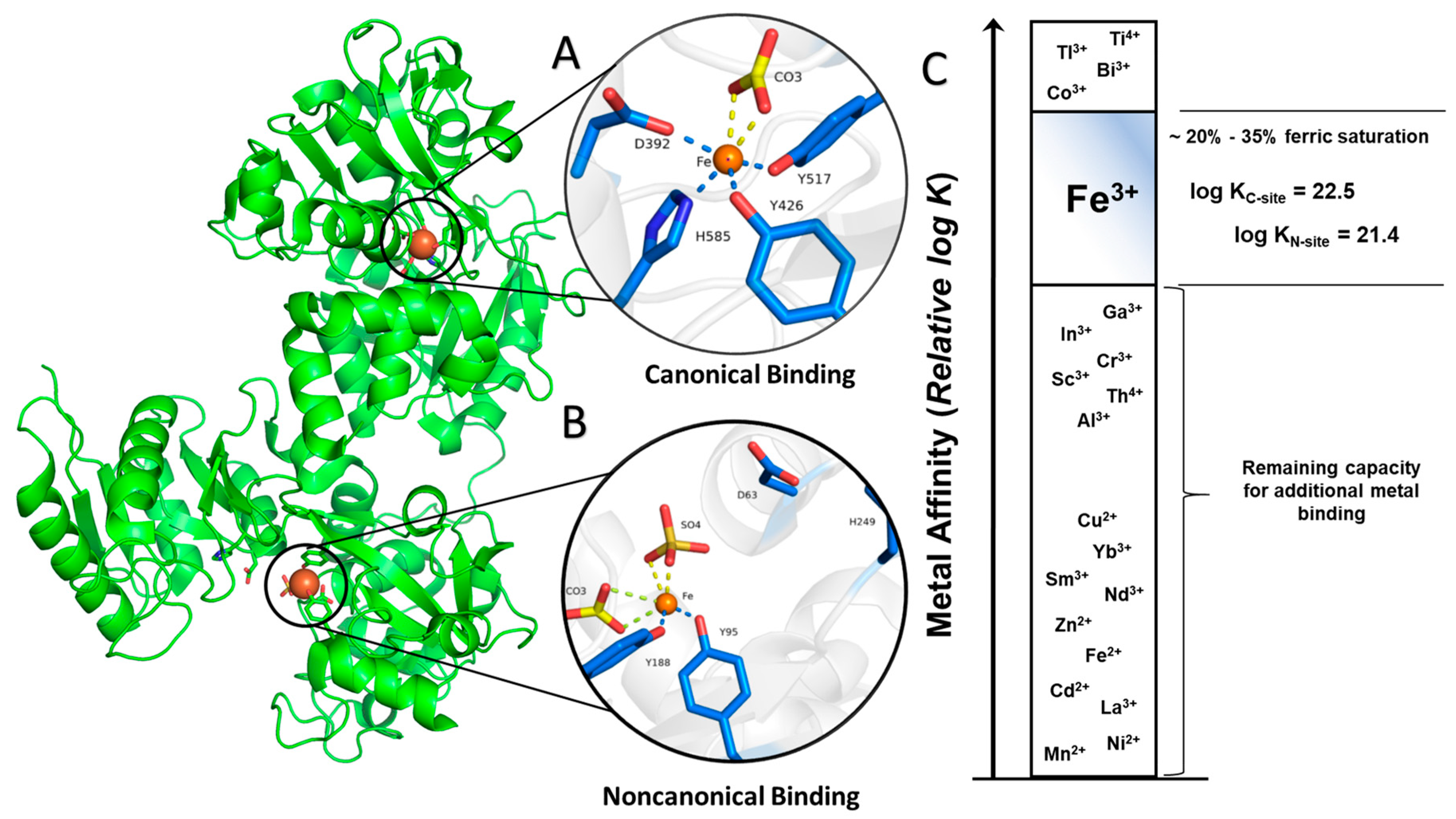
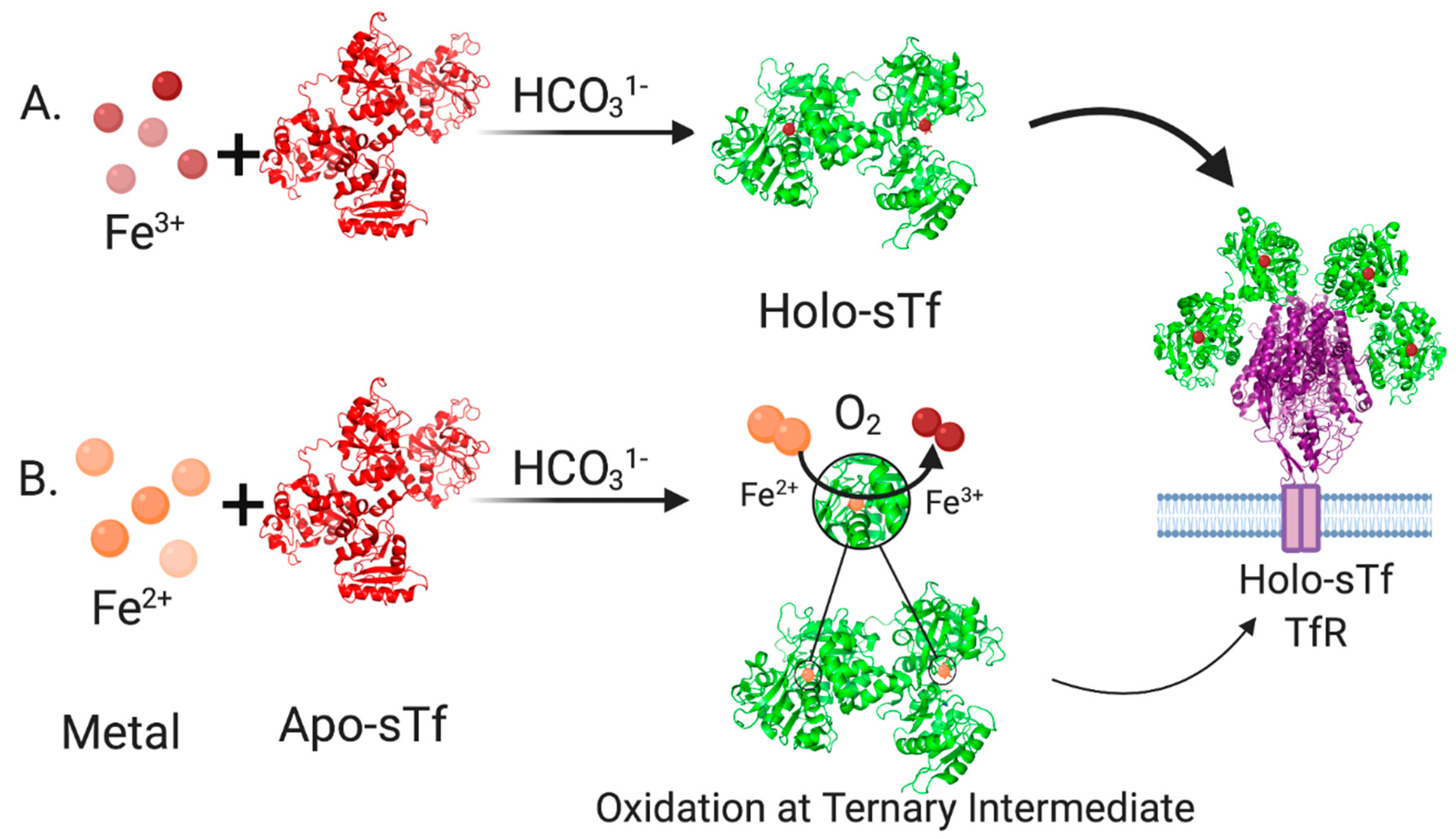
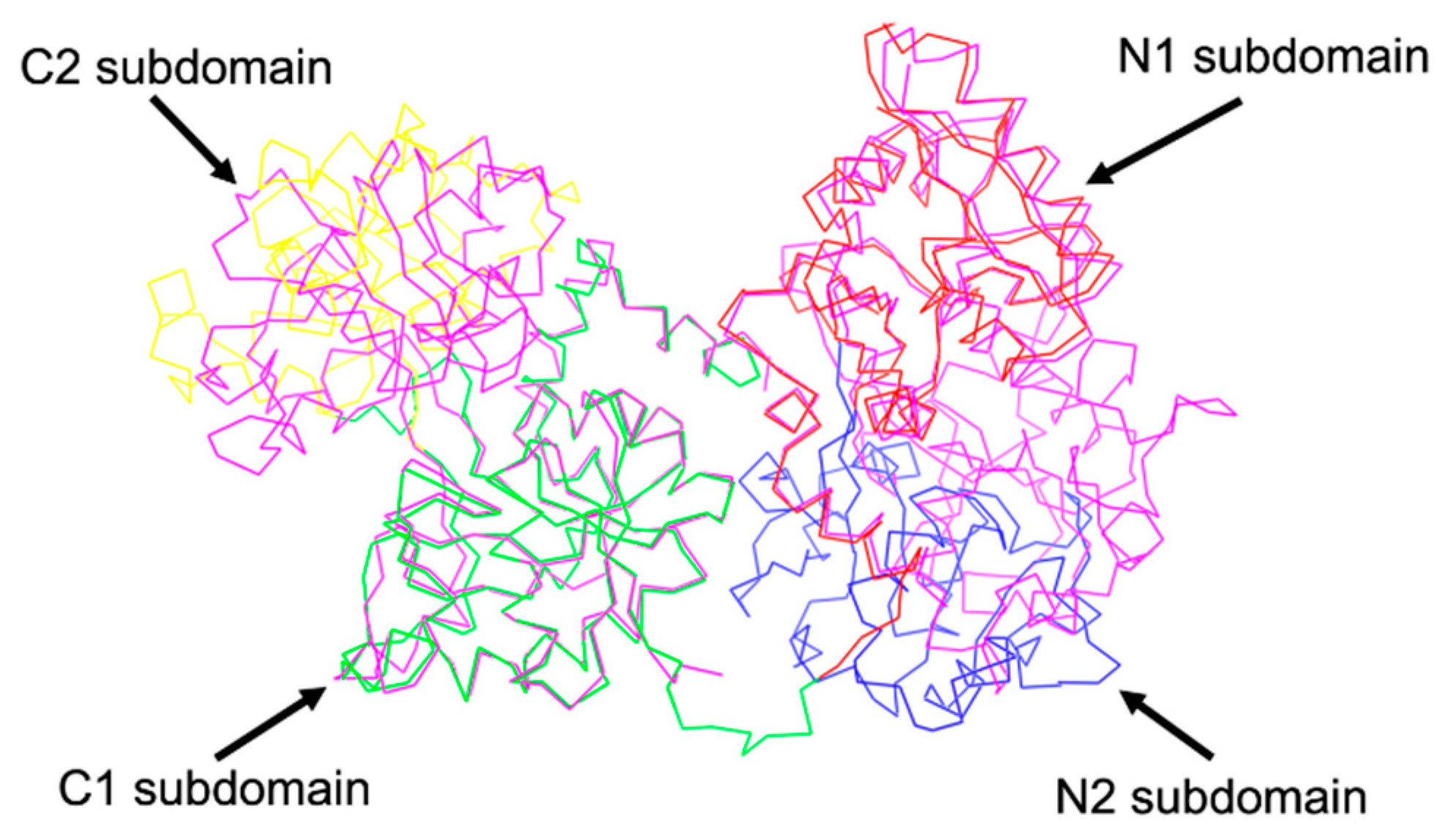
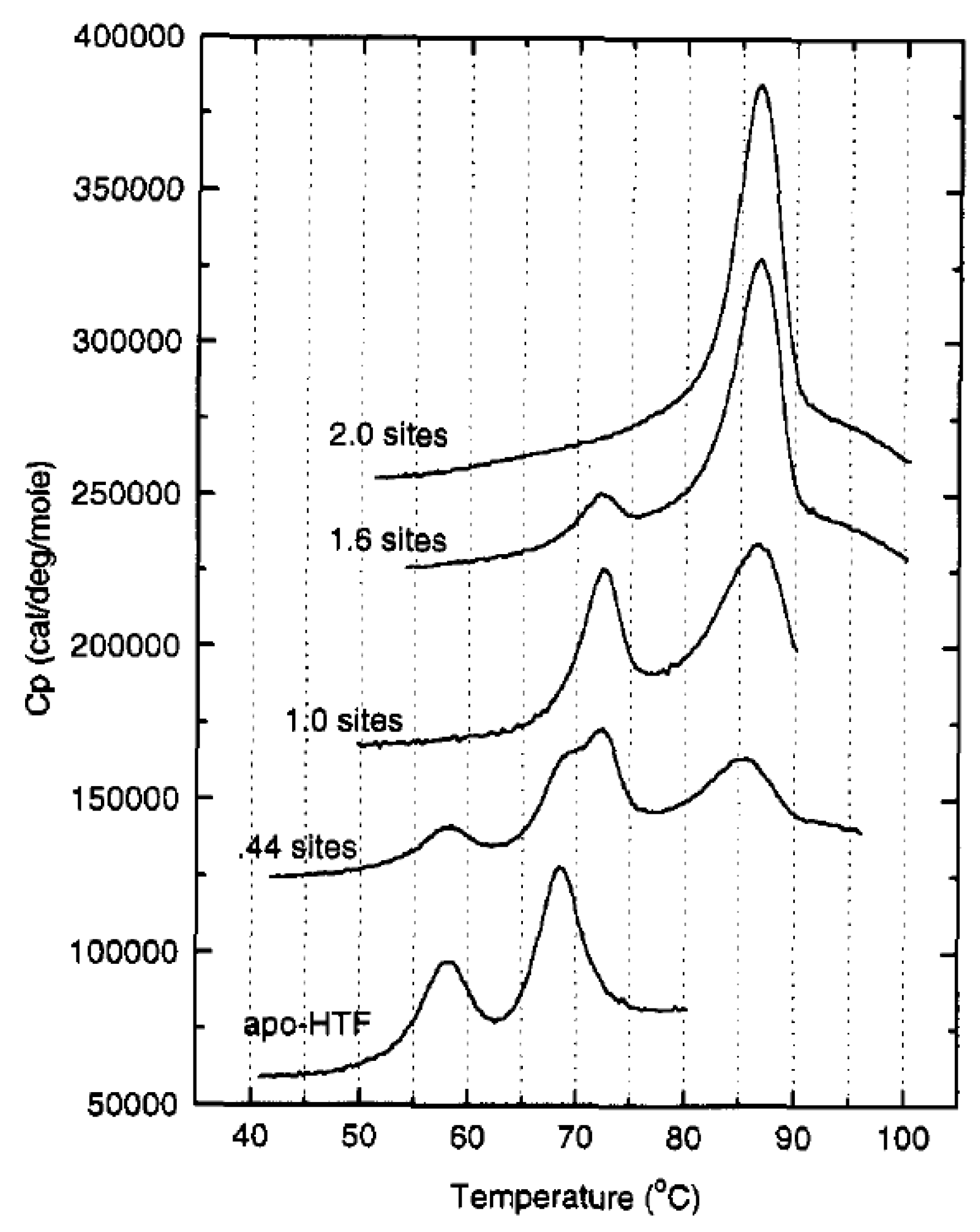
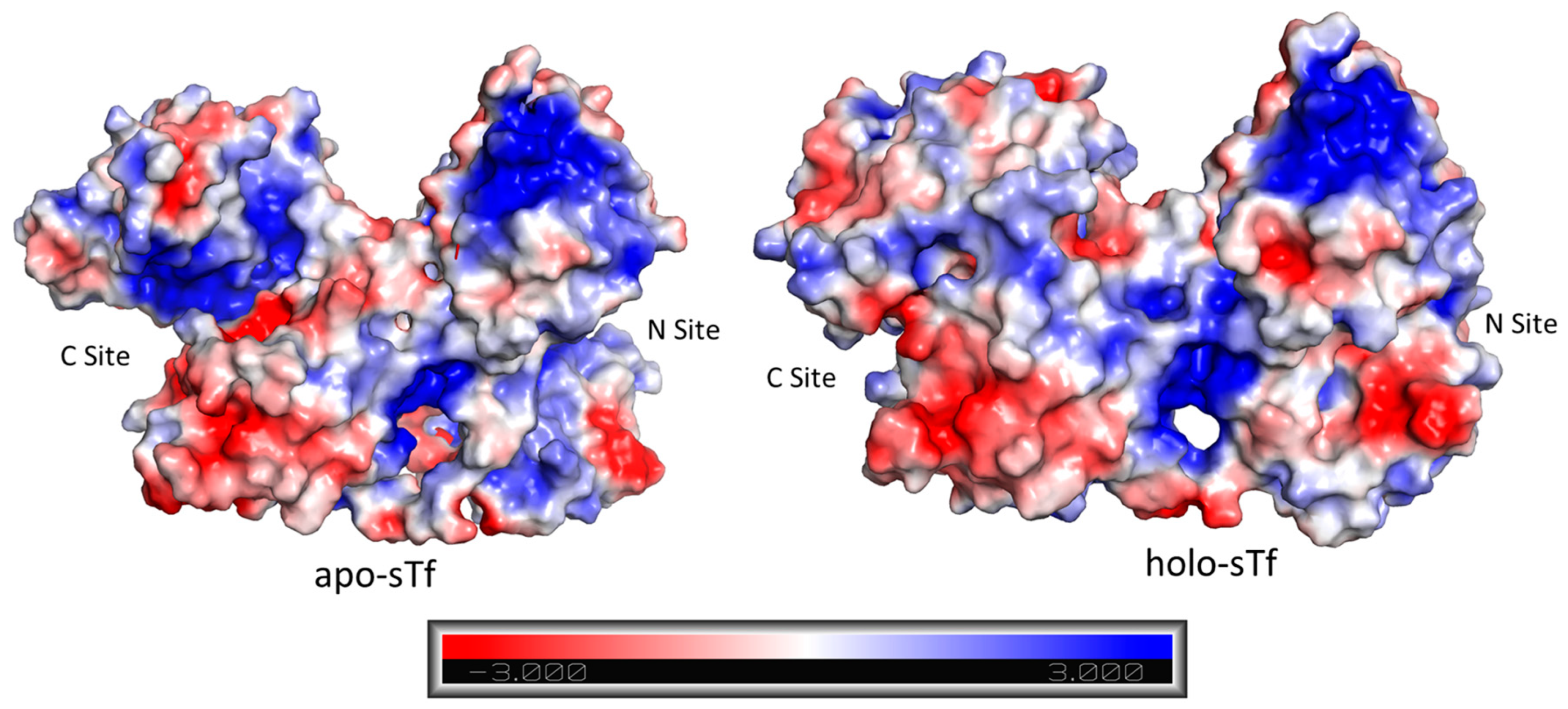
2. Structural Characterization of sTf and Its Fe(III) Binding
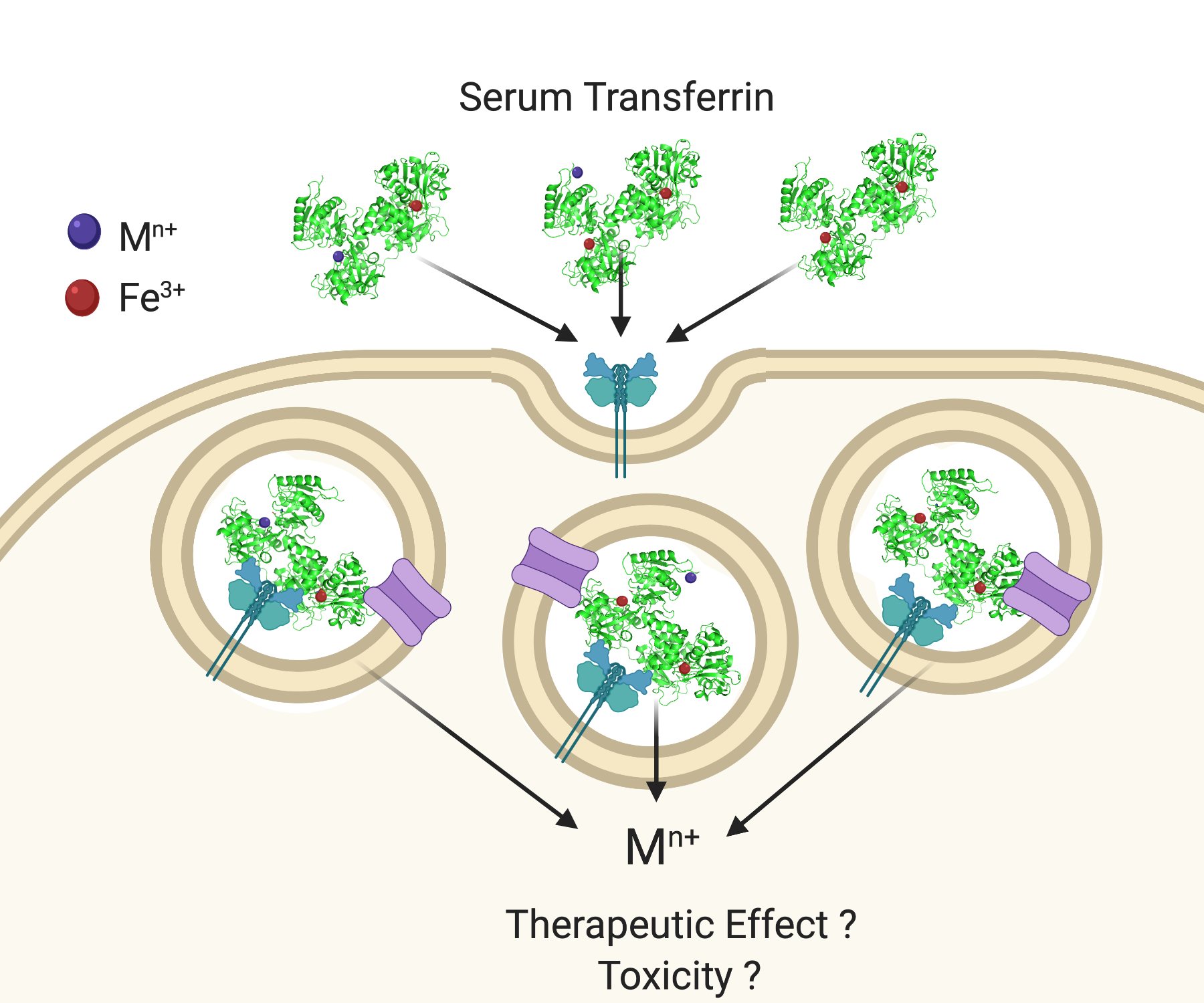
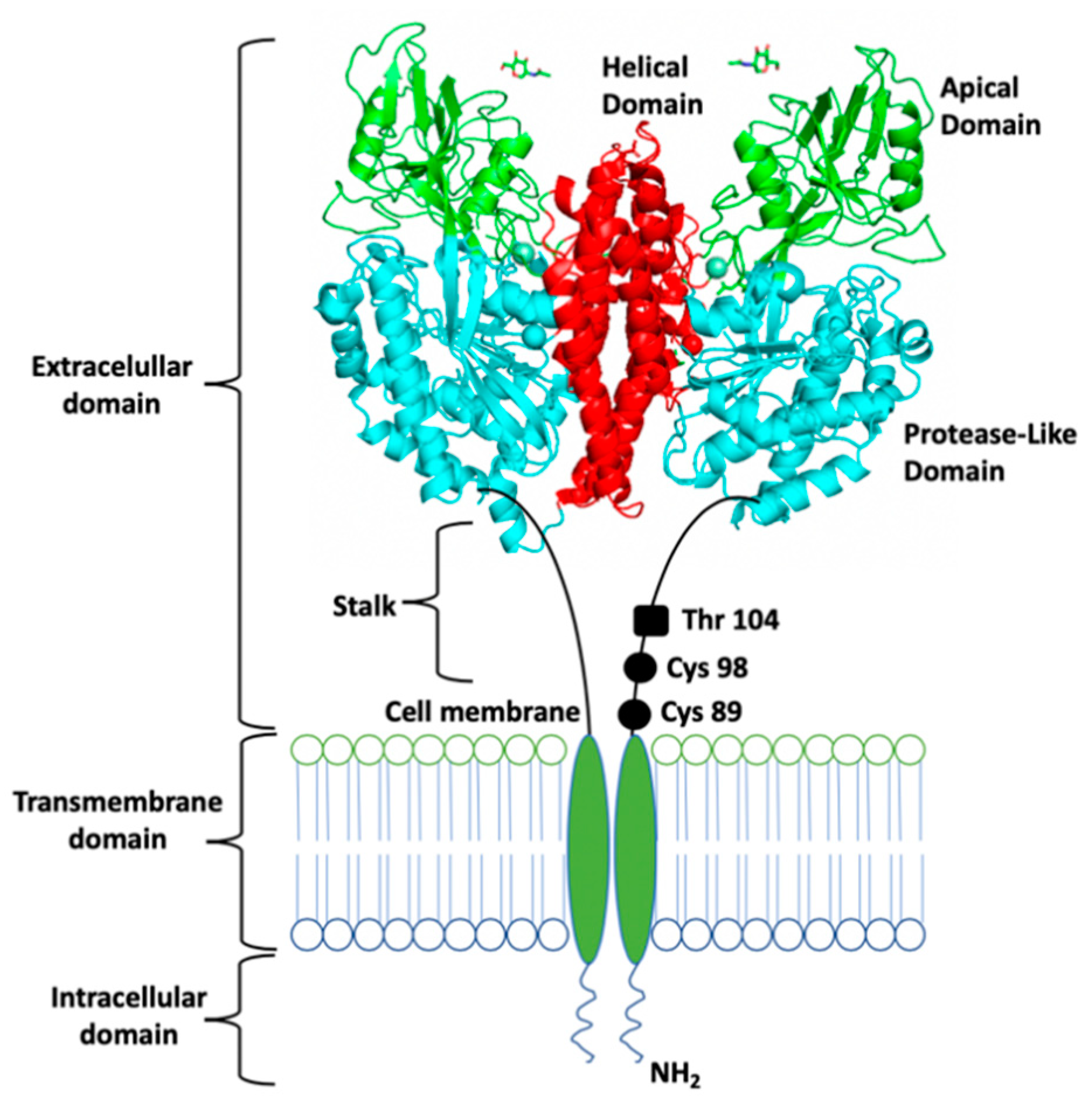
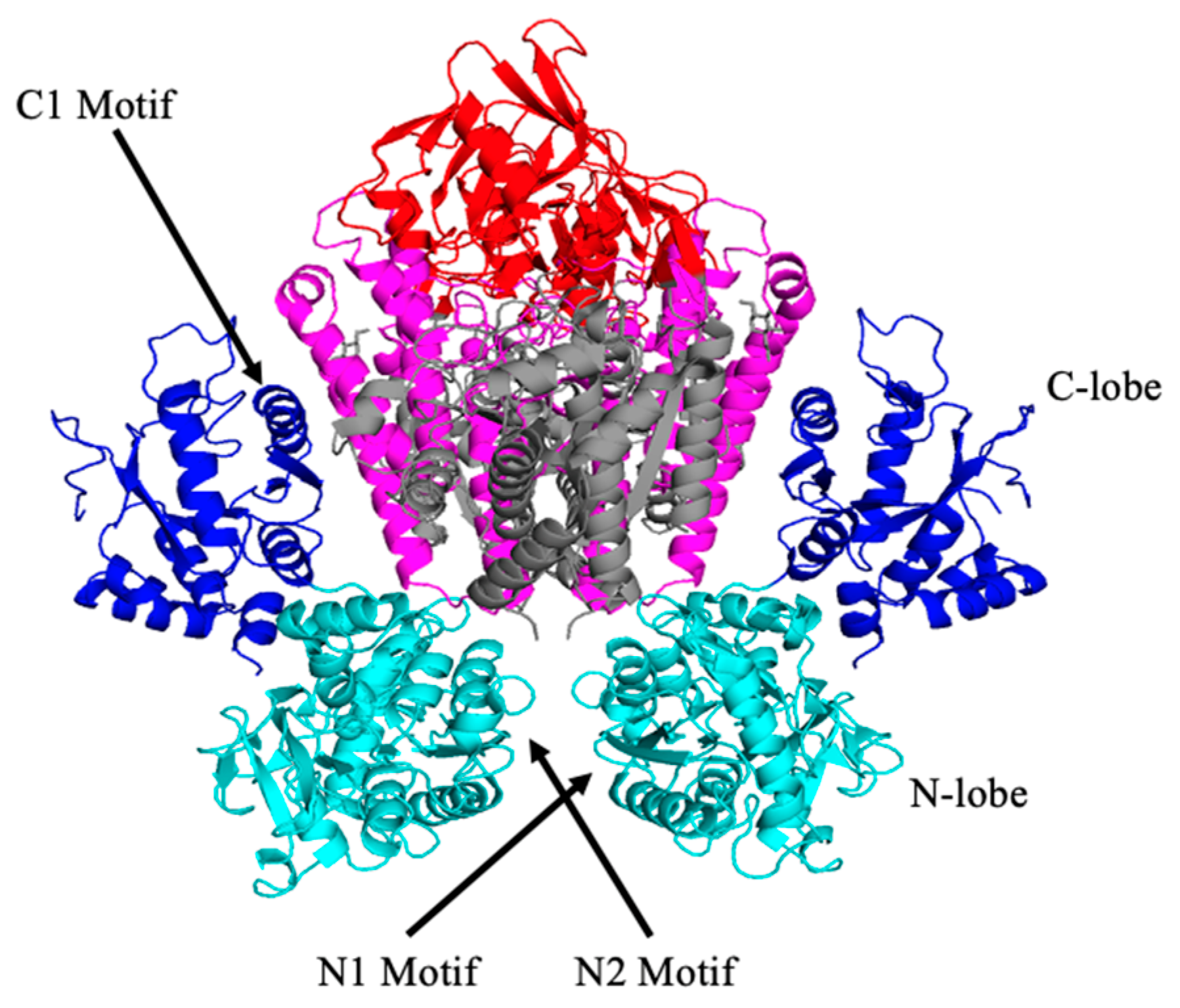
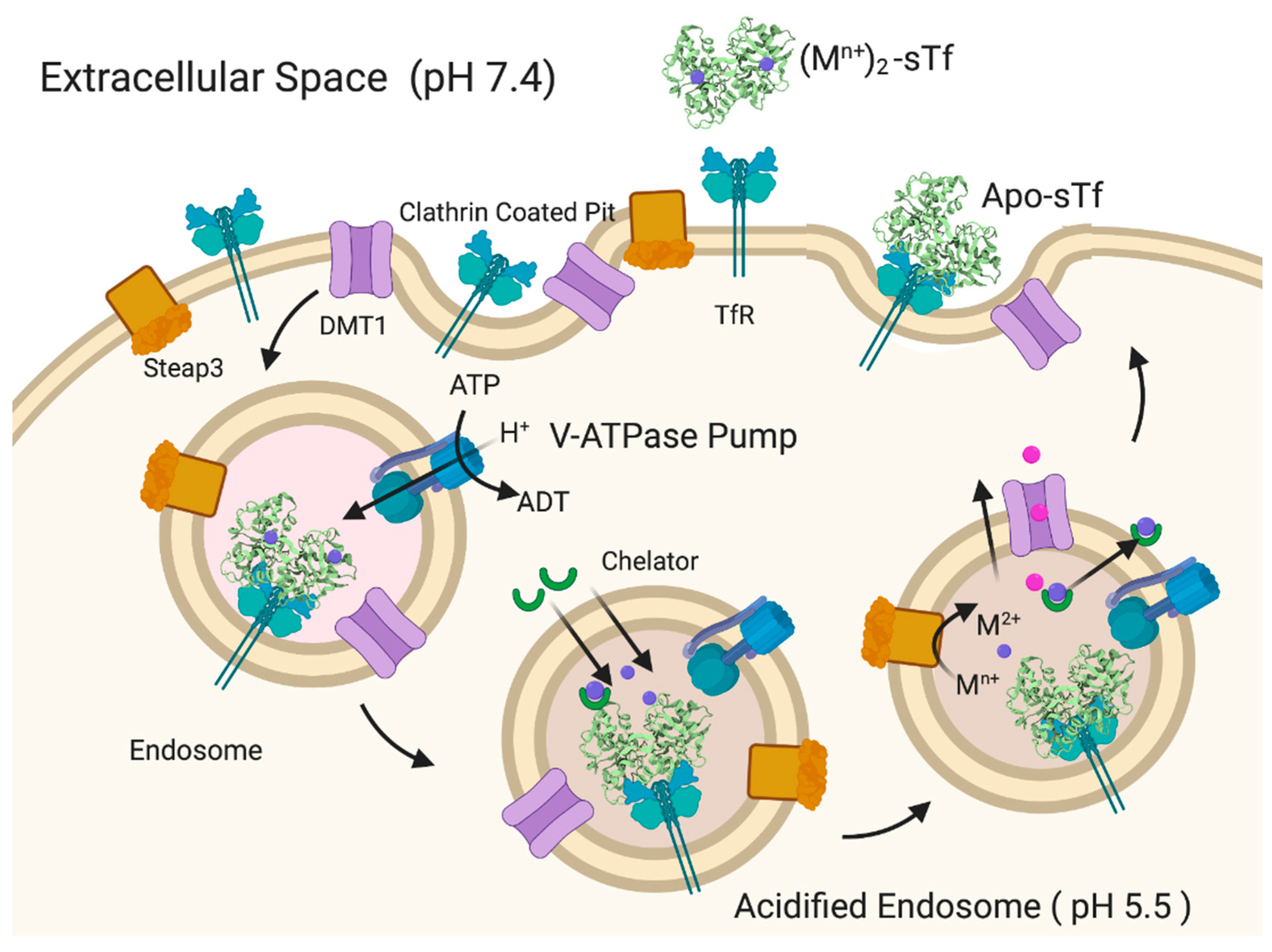
3. STf Interaction with TfR and Cellular Delivery of Fe(III) by Endocytosis
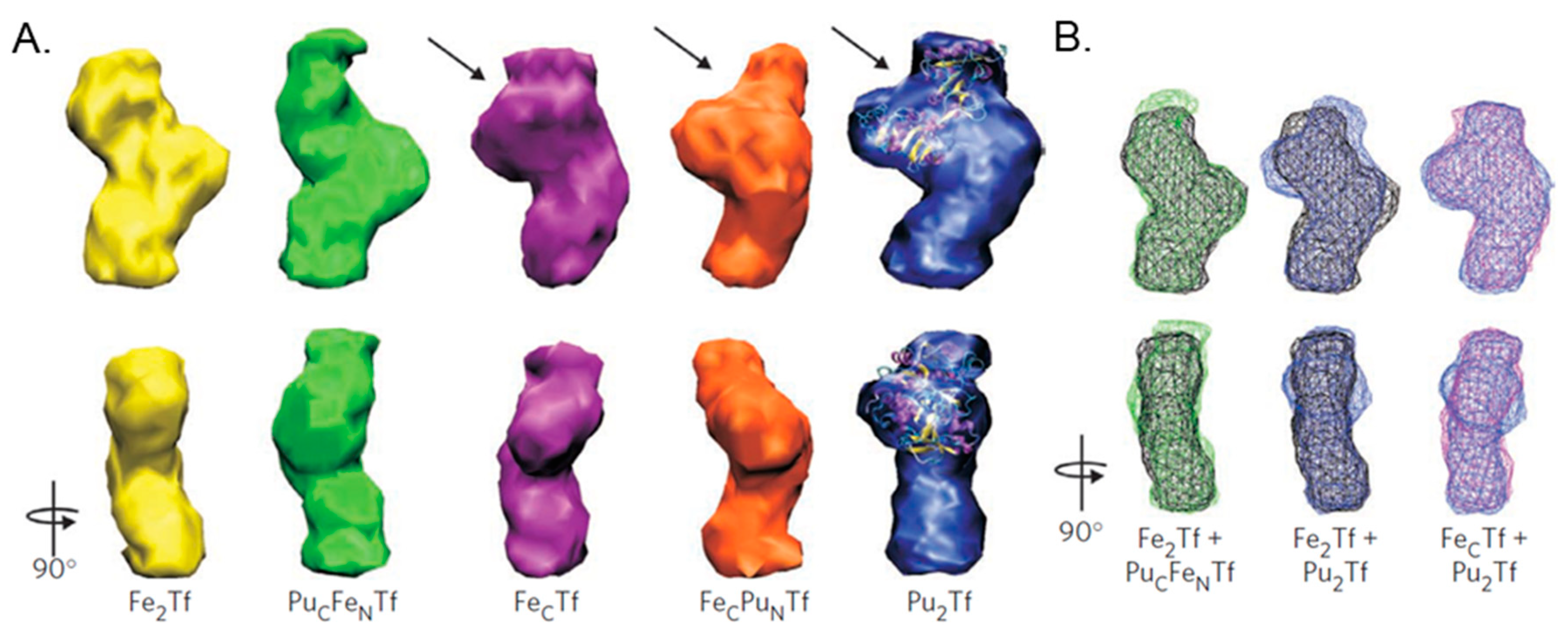
4. Challenging the Perception of Structural Requirements for Metalated-sTf Endocytotic Uptake into Cells
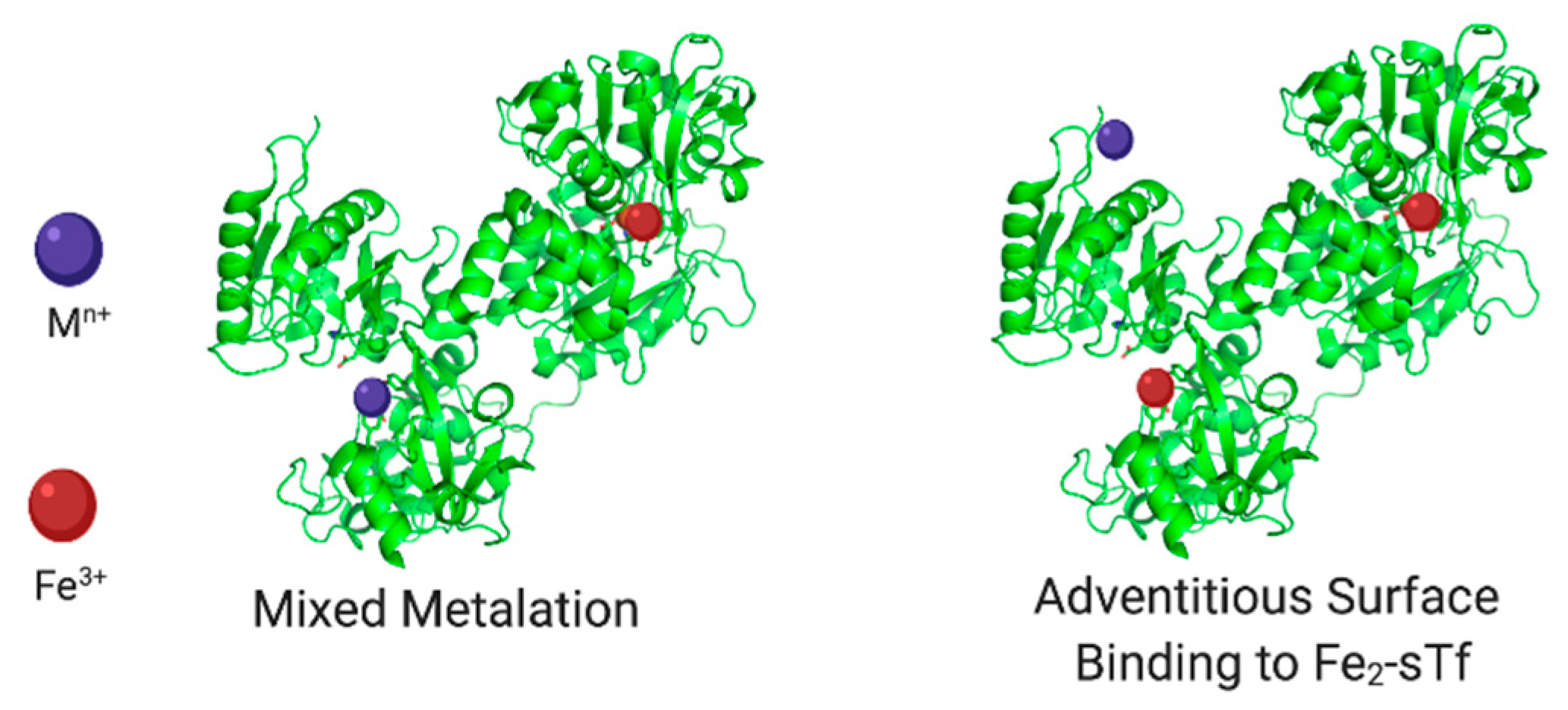
- Binding canonically or noncanonically at the metal binding site;
- Binding in a mixed metalation with Fe(III);
- Adventitious surface binding onto Fe2-sTf.
- Acidification, chelation, and DMT1 transport;
- Acidification, chelation, and ionophoric transport.
5. Examining How sTf May Facilitate the Therapeutic and Toxic Properties of Redox Inert Hard Lewis Metal Ions
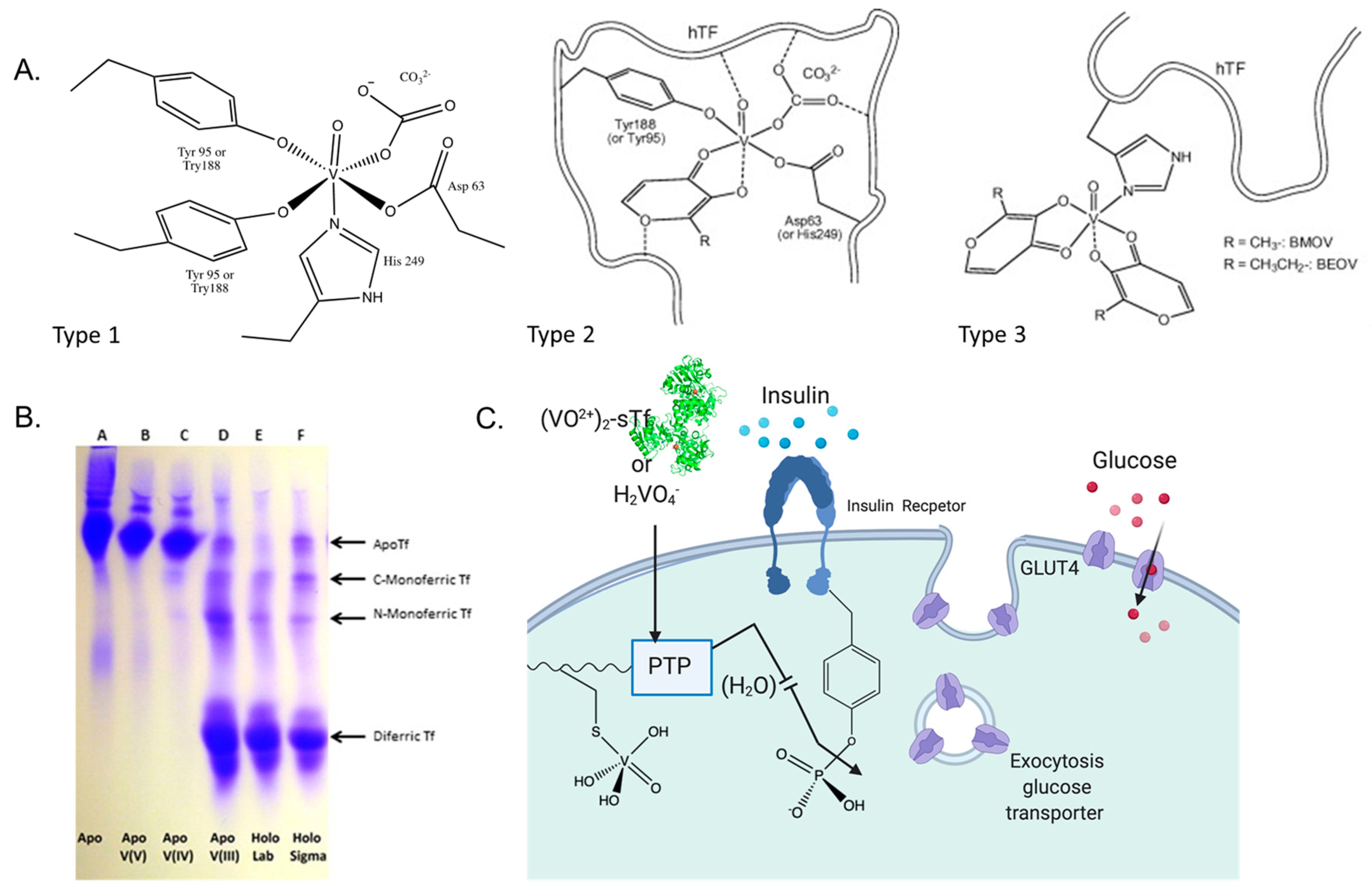
5.1. STf as a Vehicle for the Anti-Type 2 Diabetes V(IV) and Cr(III)
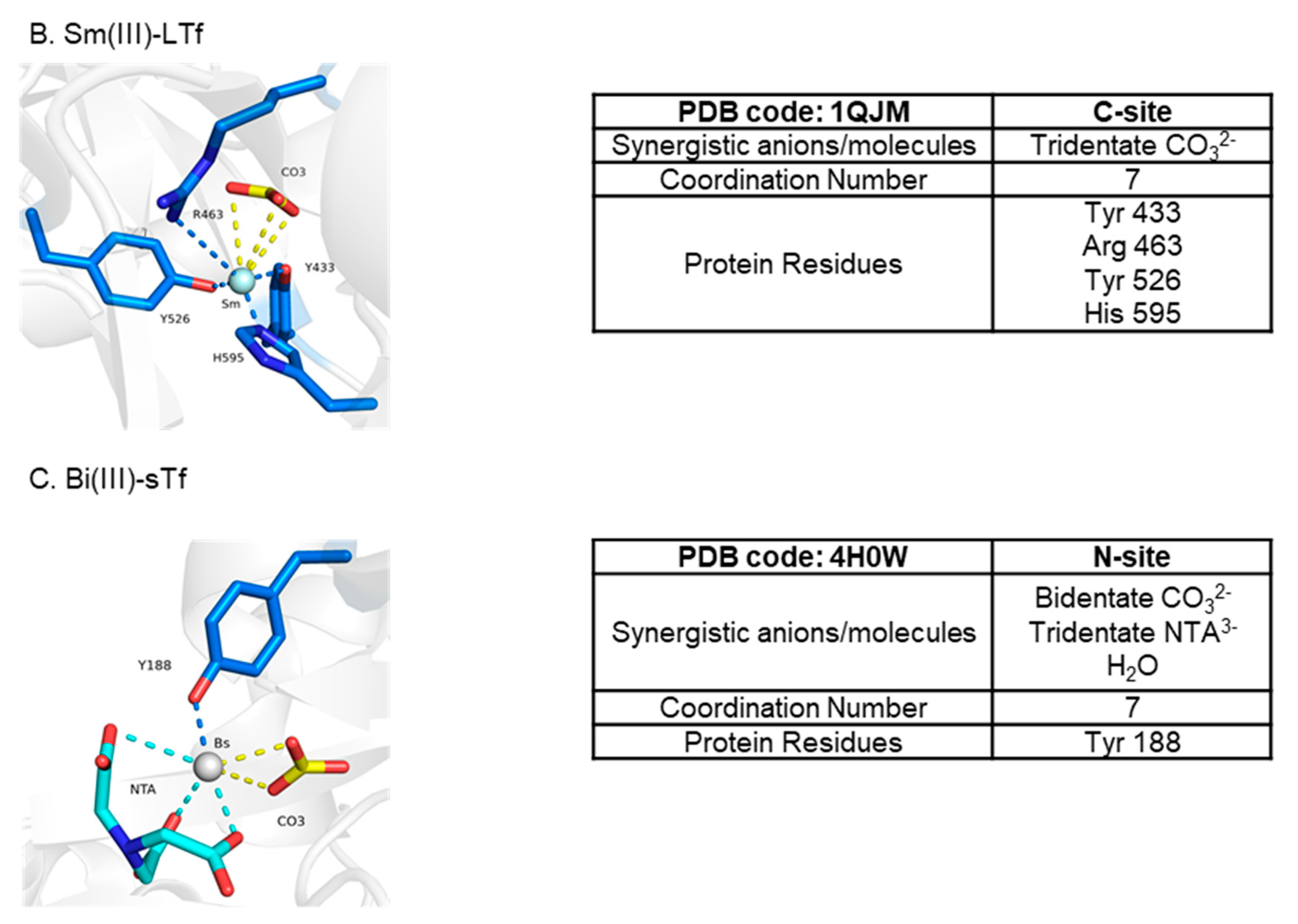
5.1.1. The Anti-Type 2 Diabetes Mechanism of Action of V(IV)
5.1.2. The Anti-Type 2 Diabetes Mechanism of Action of Cr(III)
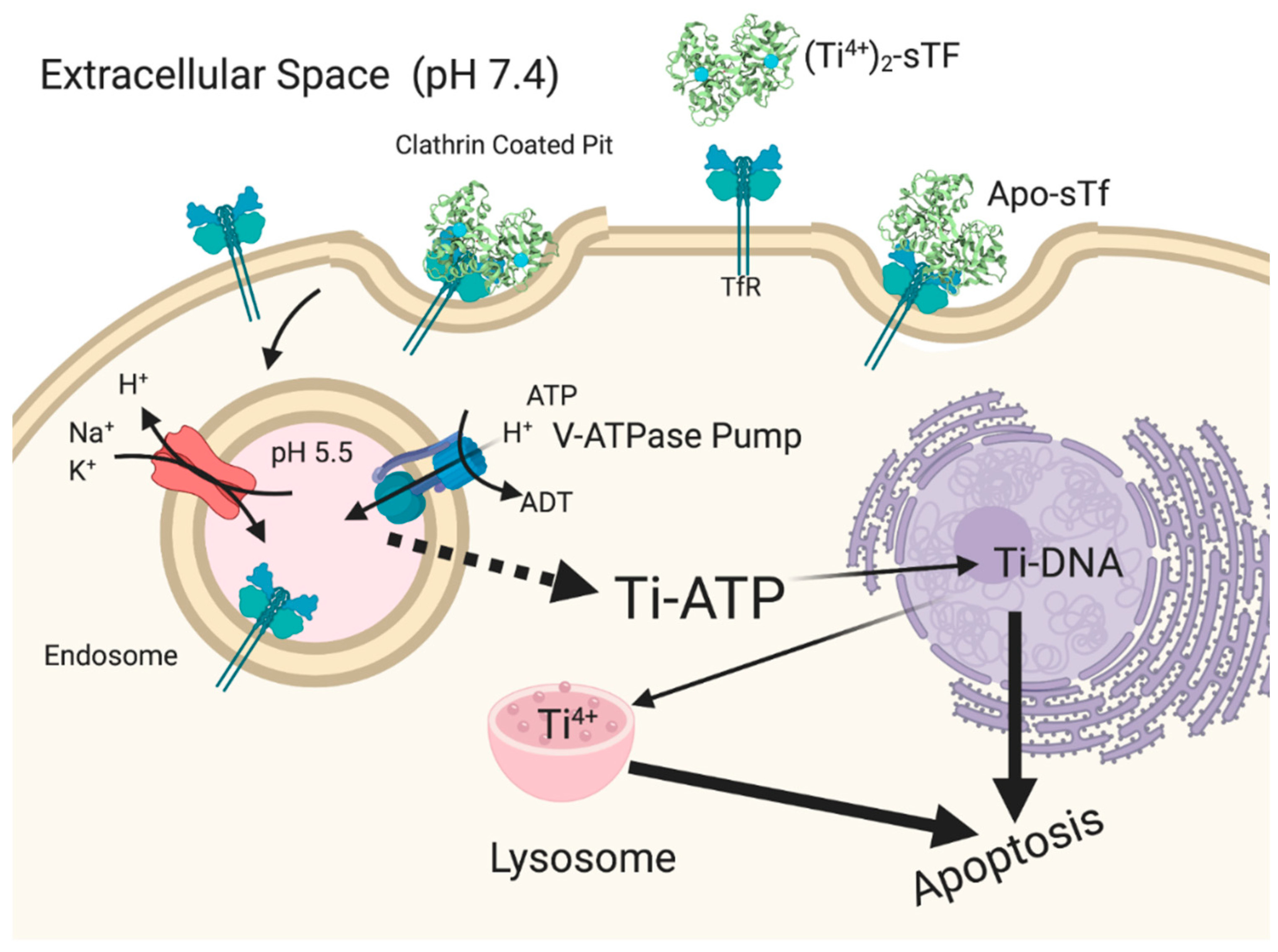
5.2. STf Mediation of the Cytotoxic/Antiproliferative Properties of Ti(IV) and Ga(III)
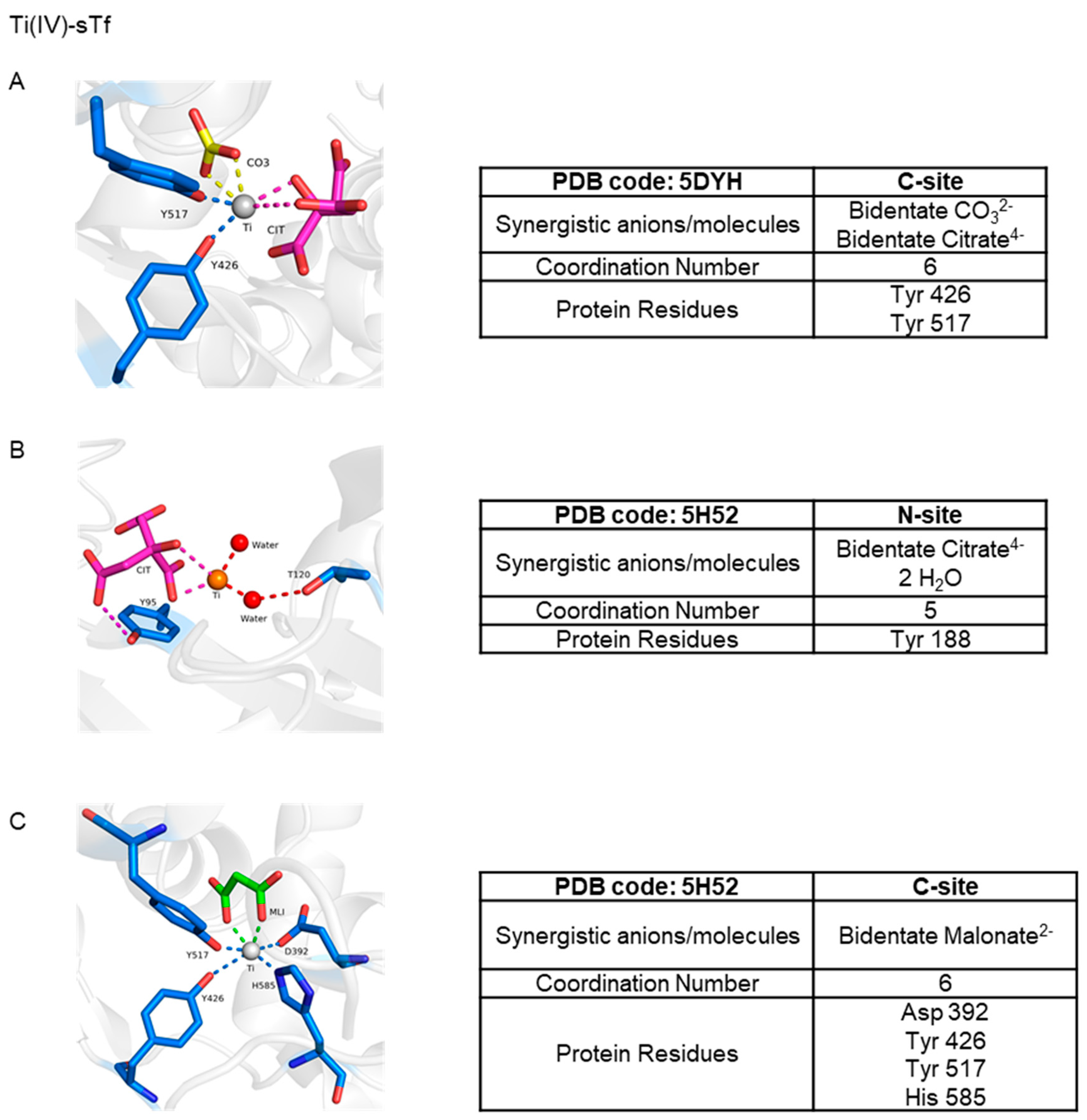
5.2.1. STf May Facilitate the Cytotoxic/Antiproliferative Properties of Ti(IV) at High Metal Concentration
5.2.2. The Cytotoxic/Antiproliferative Properties of Ga(III) Is Owed to Its Biomimicry of Fe(III)
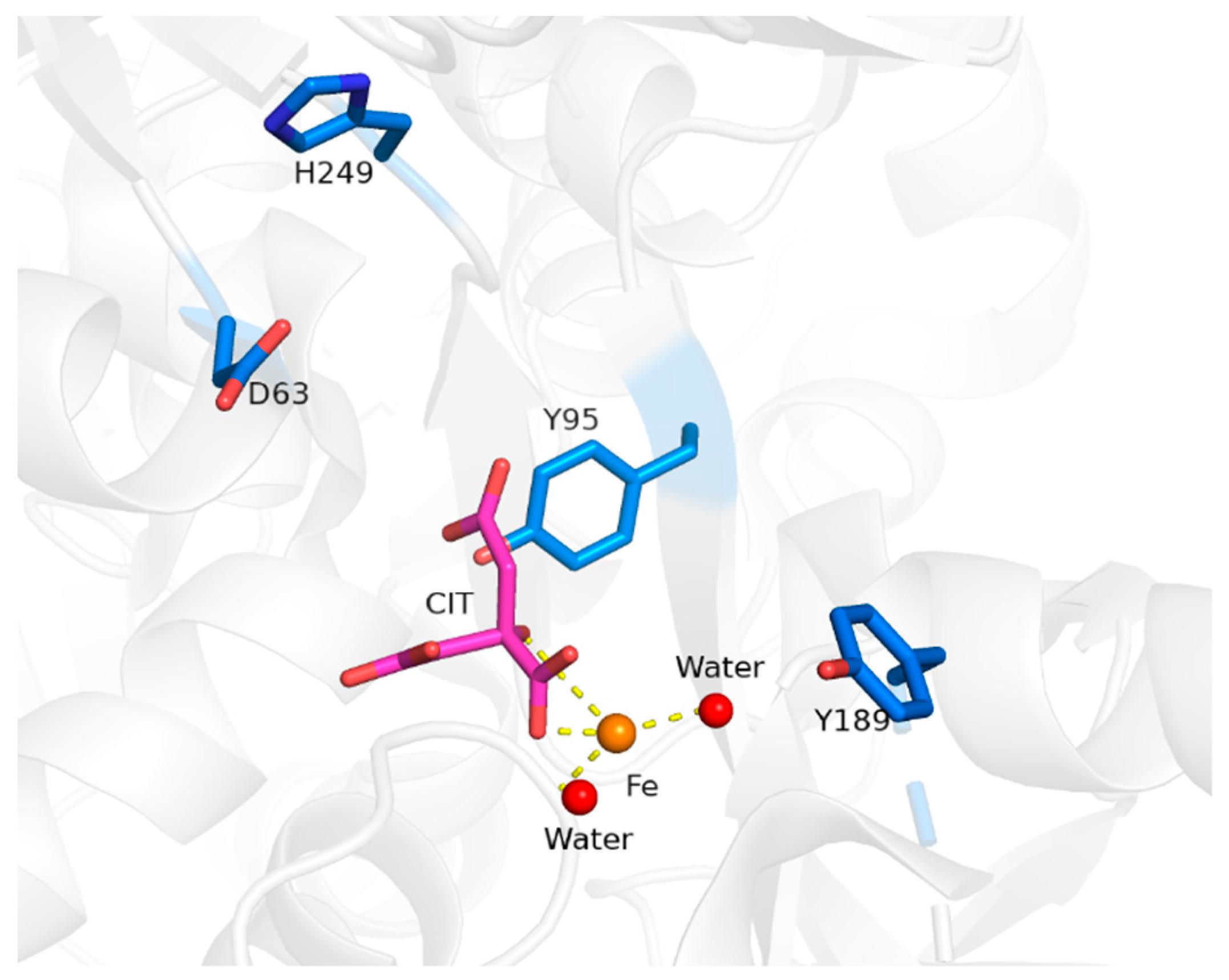
5.3. STf May Facilitate a Lifelong Exposure to Radioactive Pu(IV)
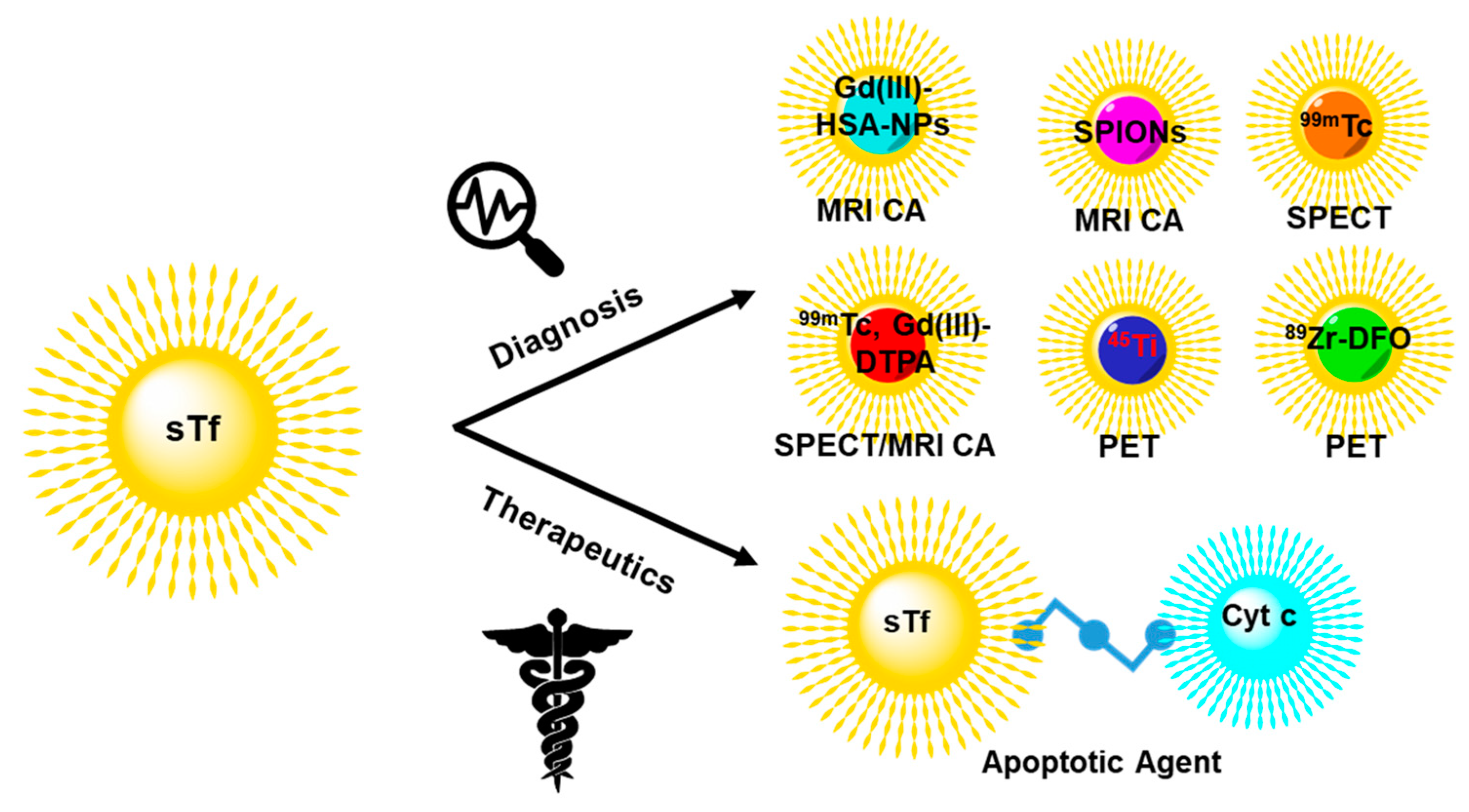
6. Engineering sTf for Delivery of Metal-Based Biomedical Tools for Cancer Applications
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dertz, E.A.; Raymond, K.N. Comprehensive Coordination Chemistry II; McCleverty, J.A., Meyer, T.J., Eds.; Pergamon: Oxford, UK, 2003; pp. 141–168. [Google Scholar]
- Bertini, I.; Gray, H.B.; Stiefel, E.I.; Valentine, J.S. Biological Inorganic Chemistry: Structure and Reactivity; University Science Books: Mill Valley, CA, USA, 2007. [Google Scholar]
- Stefánsson, A. Iron(III) Hydrolysis and Solubility at 25 °C. Environ. Sci. Technol. 2007, 41, 6117–6123. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sadler, P.J.; Sun, H. Rationalization of the strength of metal binding to human serum transferrin. Eur. J. Biochem. 1996, 242, 387–393. [Google Scholar] [CrossRef]
- Housecroft, C.E.; Sharpe, A.G. Inorganic Chemistry, 4th ed.; Pearson: London, UK, 2012. [Google Scholar]
- Schlabach, M.R.; Bates, G.W. The synergistic binding of anions and Fe3+ by transferrin. Implications for the interlocking sites hypothesis. J. Biol. Chem. 1975, 250, 2182–2188. [Google Scholar]
- Yang, N.; Zhang, H.; Wang, M.; Hao, Q.; Sun, H. Iron and bismuth bound human serum transferrin reveals a partially-opened conformation in the N-lobe. Sci. Rep. 2012, 2, 999. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, A.D.; Saxena, M.; Sharma, S.; Noinaj, N.; Delgado, Y.; Quinones Gonzalez, E.P.; Conklin, S.E.; Zambrana, N.; Loza-Rosas, S.A.; Parks, T.B. Unusual synergism of transferrin and citrate in the regulation of Ti(IV) speciation, transport, and toxicity. J. Am. Chem. Soc. 2016, 138, 5659–5665. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.; Moreton, K. The distribution of iron between the metal-binding sites of transferrin human serum. Biochem. J. 1980, 185, 483–488. [Google Scholar] [CrossRef]
- Bates, G.W.; Workman, E.F., Jr.; Schlabach, M.R. Does transferrin exhibit ferroxidase activity? Biochem. Biophys. Res. Commun. 1973, 50, 84–90. [Google Scholar] [CrossRef]
- Kojima, N.; Bates, G.W. The formation of Fe3+-transferrin-CO32- via the binding and oxidation of Fe2+. J. Biol. Chem. 1981, 256, 12034–12039. [Google Scholar]
- Sun, H.; Li, H.; Sadler, P.J. Transferrin as a metal ion mediator. Chem. Rev. 1999, 99, 2817–2842. [Google Scholar] [CrossRef]
- Vincent, J.B.; Love, S. The binding and transport of alternative metals by transferrin. Biochim. Biophys. Acta 2012, 1820, 362–378. [Google Scholar] [CrossRef]
- Lambert, L.A.; Perri, H.; Halbrooks, P.J.; Mason, A.B. Evolution of the transferrin family: Conservation of residues associated with iron and anion binding. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2005, 142, 129–141. [Google Scholar] [CrossRef]
- Lambert, L.A.; Perri, H.; Meehan, T.J. Evolution of duplications in the transferrin family of proteins. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2005, 140, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, A.D.; Peterson, C.W.; Lucchese, B.; Doyle, R.P.; Valentine, A.M. On the evolutionary significance and metal-binding characteristics of a monolobal transferrin from Ciona intestinalis. Proc. Natl. Acad. Sci. USA 2008, 105, 3268–3273. [Google Scholar] [CrossRef] [PubMed]
- Wally, J.; Halbrooks, P.J.; Vonrhein, C.; Rould, M.A.; Everse, S.J.; Mason, A.B.; Buchanan, S.K. The crystal structure of iron-free human serum transferrin provides insight into inter-lobe communication and receptor binding. J. Biol. Chem. 2006, 281, 24934–24944. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, J.G.; Crawley, J.B.; Strange, R.W.; Patel, K.J.; Murphy, L.M.; Neu, M.; Evans, R.W.; Hasnain, S.S. The nature of ligand-induced conformational change in transferrin in solution. An investigation using X-ray scattering, XAFS and site-directed mutants. J. Mol. Biol. 1998, 279, 461–472. [Google Scholar] [CrossRef]
- Lin, L.N.; Mason, A.B.; Woodworth, R.C.; Brandts, J.F. Calorimetric studies of serum transferrin and ovotransferrin-Estimates of domain interactions, and study of the kinetic complexities of ferric ion-binding. Biochemistry 1994, 33, 1881–1888. [Google Scholar] [CrossRef]
- Evans, R.W.; Williams, J. The electrophoresis of transferrins in urea/polyacrylamide gels. Biochem. J. 1980, 189, 541–546. [Google Scholar] [CrossRef]
- Byrne, S.L.; Mason, A.B. Human serum transferrin: A tale of two lobes. Urea gel and steady state fluorescence analysis of recombinant transferrins as a function of pH, time, and the soluble portion of the transferrin receptor. J. Biol. Inorg. Chem. 2009, 14, 771–781. [Google Scholar] [CrossRef]
- Yajima, H.; Sakajiri, T.; Kikuchi, T.; Morita, M.; Ishii, T. Molecular modeling of human serum transferrin for rationalizing the changes in its physicochemical properties induced by iron binding. Implication of the mechanism of binding to its receptor. J. Protein Chem. 2000, 19, 215–223. [Google Scholar] [CrossRef]
- Dewan, J.C.; Mikami, B.; Hirose, M.; Sacchettini, J.C. Structural evidence for a pH-sensitive dilysine trigger in the hen ovotransferrin N-lobe: Implications for transferrin iron release. Biochemistry 1993, 32, 11963–11968. [Google Scholar] [CrossRef]
- Peterson, N.A.; Arcus, V.L.; Anderson, B.F.; Tweedie, J.W.; Jameson, G.B.; Baker, E.N. “Dilysine trigger” in transferrins probed by mutagenesis of lactoferrin: Crystal structures of the R210G, R210E, and R210L mutants of human lactoferrin. Biochemistry 2002, 41, 14167–14175. [Google Scholar] [CrossRef] [PubMed]
- Halbrooks, P.J.; Giannetti, A.M.; Klein, J.S.; Björkman, P.J.; Larouche, J.R.; Smith, V.C.; MacGillivray, R.T.A.; Everse, S.J.; Mason, A.B. Composition of pH-Sensitive triad in C-Lobe of human serum transferrin. Comparison to sequences of ovotransferrin and lactoferrin provides insight into functional differences in iron release. Biochemistry 2005, 44, 15451–15460. [Google Scholar] [CrossRef]
- Aisen, P. Transferrin receptor 1. Int. J. Biochem. Cell Biol. 2004, 36, 2137–2143. [Google Scholar] [CrossRef]
- Cheng, Y.; Zak, O.; Aisen, P.; Harrison, S.C.; Walz, T. Structure of the human transferrin receptor-transferrin complex. Cell 2004, 116, 565–576. [Google Scholar] [CrossRef]
- Daniels, T.R.; Delgado, T.; Rodriguez, J.A.; Helguera, G.; Penichet, M.L. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin. Immunol. 2006, 121, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, C.M.; Ray, S.; Babyonyshev, M.; Galluser, R.; Borhani, D.W.; Harrison, S.C. Crystal structure of the ectodomain of human transferrin receptor. Science 1999, 286, 779–782. [Google Scholar] [CrossRef]
- Eckenroth, B.E.; Steere, A.N.; Chasteen, N.D.; Everse, S.J.; Mason, A.B. How the binding of human transferrin primes the transferrin receptor potentiating iron release at endosomal pH. Proc. Natl. Acad. Sci. USA 2011, 108, 13089–13094. [Google Scholar] [CrossRef]
- Leverence, R.; Mason, A.B.; Kaltashov, I.A. Noncanonical interactions between serum transferrin and transferrin receptor evaluated with electrospray ionization mass spectrometry. Proc. Natl. Acad. Sci. USA 2010, 107, 8123–8128. [Google Scholar] [CrossRef]
- Conner, S.D.; Schmid, S.L. Differential requirements for AP-2 in clathrin-mediated endocytosis. J. Cell Biol. 2003, 162, 773–779. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef]
- Lafourcade, C.; Sobo, K.; Kieffer-Jaquinod, S.; Garin, J.; van der Goot, F.G. Regulation of the V-ATPase along the endocytic pathway occurs through reversible subunit association and membrane localization. PLoS ONE 2008, 3, e2758. [Google Scholar] [CrossRef]
- Steere, A.N.; Byrne, S.L.; Chasteen, N.D.; Mason, A.B. Kinetics of iron release from transferrin bound to the transferrin receptor at endosomal pH. Biochim. Biophys. Acta 2012, 1820, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Cabantchik, Z.I.; Kakhlon, O.; Epsztejn, S.; Zanninelli, G.; Breuer, W. Iron Chelation Therapy; Hershko, C., Ed.; Springer: Boston, MA, USA, 2002; pp. 55–75. [Google Scholar]
- Kakhlon, O.; Cabantchik, Z.I. The labile iron pool: Characterization, measurement, and participation in cellular processes. Free Radic. Biol. Med. 2002, 33, 1037–1046. [Google Scholar] [CrossRef]
- Kruszewski, M. Labile iron pool: The main determinant of cellular response to oxidative stress. Mutat. Res. Fundam. Mol. Mech. Mutag. 2003, 531, 81–92. [Google Scholar] [CrossRef]
- Breuer, W.; Shvartsman, M.; Cabantchik, Z.I. Intracellular labile iron. Int. J. Biochem. Cell Biol. 2008, 40, 350–354. [Google Scholar] [CrossRef]
- Cabantchik, Z.I. Labile iron in cells and body fluids: Physiology, pathology, and pharmacology. Front. Pharmacol. 2014, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Kraiter, D.C.; Zak, O.; Aisen, P.; Crumbliss, A.L. A determination of the reduction potentials for diferric and C- and N-lobe monoferric transferrins at endosomal pH (5.8). Inorg. Chem. 1998, 37, 964–968. [Google Scholar] [CrossRef]
- Dhungana, S.; Taboy, C.H.; Zak, O.; Larvie, M.; Crumbliss, A.L.; Aisen, P. Redox properties of human transferrin bound to its receptor. Biochemistry 2004, 43, 205–209. [Google Scholar] [CrossRef]
- Bou-Abdallah, F. Does Iron Release from Transferrin Involve a Reductive Process? Bioenerg. Open Access 2012, 1, 1000e111. [Google Scholar] [CrossRef]
- Harris, W.R. Thermodynamic binding constants of the zinc-human serum transferrin complex. Biochemistry 1983, 22, 3920–3926. [Google Scholar] [CrossRef]
- Levina, A.; Lay, P.A. Transferrin cycle and clinical roles of citrate and ascorbate in improved iron metabolism. ACS Chem. Biol. 2019, 14, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.P.; Gorman-Lewis, D.; Aryal, B.; Paunesku, T.; Vogt, S.; Rickert, P.G.; Seifert, S.; Lai, B.; Woloschak, G.E.; Soderholm, L. An iron-dependent and transferrin-mediated cellular uptake pathway for plutonium. Nat. Chem. Biol. 2011, 7, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.P.; Wang, M.; Cheng, T.; Jin, L.; Sun, H. The role of citrate, lactate and transferrin in determining titanium release from surgical devices into human serum. J. Biol. Inorg. Chem. 2018, 23, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, A.D.; Eames, E.V.; Valentine, A.M. Reconsideration of serum Ti(IV) transport: Albumin and transferrin trafficking of Ti(IV) and its complexes. J. Am. Chem. Soc. 2008, 130, 2262–2270. [Google Scholar] [CrossRef]
- Bonvin, G.; Bobst, C.E.; Kaltashov, I.A. Interaction of transferrin with non-cognate metals studied by native electrospray ionization mass spectrometry. Int. J. Mass Spectrom. 2017, 420, 74–82. [Google Scholar] [CrossRef]
- Smith, C.A.; Anderson, B.F.; Baker, H.M.; Baker, E.N. Metal substitution in transferrins: The crystal structure of human copper-lactoferrin at 2.1-A resolution. Biochemistry 1992, 31, 4527–4533. [Google Scholar] [CrossRef]
- Sharma, A.K.; Singh, T.P. Lactoferrin–metal interactions: First crystal structure of a complex of lactoferrin with a lanthanide ion (Sm3+) at 3.4 Å resolution. Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 1799–1804. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Zhang, M.; Gumerov, D.R.; Kaltashov, I.A.; Mason, A.B. Indirect detection of protein-Metal binding: Interaction of serum transferrin with In3+ and Bi3+. J. Am. Soc. Mass Spectrom. 2004, 15, 1658–1664. [Google Scholar] [CrossRef]
- Mishra, L.; Sawant, P.D.; Sundararajan, M.; Bandyopadhyay, T. Binding of Cm(III) and Th(IV) with Human Transferrin at Serum pH: Combined QM and MD Investigations. J. Phys. Chem. B 2019, 123, 2729–2744. [Google Scholar] [CrossRef]
- Mishra, L.; Sundararajan, M.; Bandyopadhyay, T. Molecular dynamics simulations of plutonium binding and its decorporation from the binding-cleft of human serum transferrin. J. Biol. Inorg. Chem. 2020, 25, 213–231. [Google Scholar] [CrossRef] [PubMed]
- Chitambar, C.R. Gallium and its competing roles with iron in biological systems. Biochim. Biophys. Acta 2016, 1863, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Hacht, B. Gallium(III) Ion Hydrolysis under Physiological Conditions. Bull. Korean Chem. Soc. 2008, 29, 372. [Google Scholar]
- Harris, W.R.; Pecoraro, V.L. Thermodynamic binding constants for gallium transferrin. Biochemistry 1983, 22, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Brulfert, F.; Aupiais, J. Topological speciation of actinide-transferrin complexes by capillary isoelectric focusing coupled with inductively coupled plasma mass spectrometry: Evidence of the non-closure of the lobes. Dalton Trans. 2018, 47, 9994–10001. [Google Scholar] [CrossRef]
- Deblonde, G.J.; Sturzbecher-Hoehne, M.; Mason, A.B.; Abergel, R.J. Receptor recognition of transferrin bound to lanthanides and actinides: A discriminating step in cellular acquisition of f-block metals. Metallomics 2013, 5, 619–626. [Google Scholar] [CrossRef]
- El Hage Chahine, J.M.; Hemadi, M.; Ha-Duong, N.T. Uptake and release of metal ions by transferrin and interaction with receptor 1. Biochim. Biophys. Acta 2012, 1820, 334–347. [Google Scholar] [CrossRef]
- Wang, M.; Ding, W.; Wang, D. Binding mechanism of uranyl to transferrin implicated by density functional theory study. RSC Adv. 2017, 7, 3667–3675. [Google Scholar] [CrossRef]
- Wolff, N.A.; Garrick, M.D.; Zhao, L.; Garrick, L.M.; Ghio, A.J.; Thévenod, F. A role for divalent metal transporter (DMT1) in mitochondrial uptake of iron and manganese. Sci. Rep. 2018, 8, 211. [Google Scholar] [CrossRef]
- Levina, A.; Pham, T.H.; Lay, P.A. Binding of Chromium(III) to Transferrin Could Be Involved in Detoxification of Dietary Chromium(III) Rather than Transport of an Essential Trace Element. Angew. Chem. 2016, 55, 8104–8107. [Google Scholar] [CrossRef]
- Levina, A.; Lay, P.A. Influence of an anti-metastatic ruthenium(III) prodrug on extracellular protein–protein interactions: Studies by bio-layer interferometry. Inorg. Chem. 2014, 1, 44–48. [Google Scholar] [CrossRef]
- Pongratz, M.; Schluga, P.; Jakupec, M.A.; Arion, V.B.; Hartinger, C.G.; Allmaier, G.; Keppler, B.K. Transferrin binding and transferrin-mediated cellular uptake of the ruthenium coordination compound KP1019, studied by means of AAS, ESI-MS and CD spectroscopy. J. Anal. Atomic Spectrom. 2004, 19, 46–51. [Google Scholar] [CrossRef]
- Costa Pessoa, J.; Gonçalves, G.; Roy, S.; Correia, I.; Mehtab, S.; Santos, M.F.A.; Santos-Silva, T. New insights on vanadium binding to human serum transferrin. Inorg. Chim. Acta 2014, 420, 60–68. [Google Scholar] [CrossRef]
- Levina, A.; Lay, P.A. Vanadium(V/IV)–Transferrin Binding Disrupts the Transferrin Cycle and Reduces Vanadium Uptake and Antiproliferative Activity in Human Lung Cancer Cells. Inorg. Chem. 2020. [Google Scholar] [CrossRef]
- Lutsenko, S. Copper trafficking to the secretory pathway. Metallomics 2016, 8, 840–852. [Google Scholar] [CrossRef]
- Linder, M.C. Ceruloplasmin and other copper binding components of blood plasma and their functions: An update. Metallomics 2016, 8, 887–905. [Google Scholar] [CrossRef]
- Ramos, D.; Mar, D.; Ishida, M.; Vargas, R.; Gaite, M.; Montgomery, A.; Linder, M.C. Mechanism of copper uptake from blood plasma ceruloplasmin by mammalian cells. PLoS ONE 2016, 11, e0149516. [Google Scholar] [CrossRef]
- Crans, D.C.; Woll, K.A.; Prusinskas, K.; Johnson, M.D.; Norkus, E. Metal speciation in health and medicine represented by iron and vanadium. Inorg. Chem. 2013, 52, 12262–12275. [Google Scholar] [CrossRef]
- Templeton, D.M. Speciation in metal toxicity and metal-based therapeutics. Toxics 2015, 3, 170–186. [Google Scholar] [CrossRef]
- Levina, A.; Crans, D.C.; Lay, P.A. Speciation of metal drugs, supplements and toxins in media and bodily fluids controls in vitro activities. Coord. Chem. Rev. 2017, 352, 473–498. [Google Scholar] [CrossRef]
- Gaur, K.; Cruz, Y.M.; Santiago Espinoza, J.A.; Morales Rueda, C.A.; Loza-Rosas, S.A.; Fernández-Vega, L.; Benjamín-Rivera, J.A.; Álvarez, A.; Tinoco, A.D. Exploring the pH dependent aqueous speciation of metal complexes through UV–Vis spectroscopy. J. Chem. Educ. 2020, 97, 1970–1975. [Google Scholar] [CrossRef]
- Manfredi, M.; McCullough, M.J.; Vescovi, P.; Al-Kaarawi, Z.M.; Porter, S.R. Update on diabetes mellitus and related oral diseases. Oral Dis. 2004, 10, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, E.; Storm, P.; Käräjämäki, A.; Martinell, M.; Dorkhan, M.; Carlsson, A.; Vikman, P.; Prasad, R.B.; Aly, D.M.; Almgren, P.; et al. Novel subgroups of adult-onset diabetes and their association with outcomes: A data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018, 6, 361–369. [Google Scholar] [CrossRef]
- Rother, K.I. Diabetes treatment—Bridging the divide. N. Engl. J. Med. 2007, 356, 1499–1501. [Google Scholar] [CrossRef]
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of insulin synthesis and secretion and pancreatic β-cell dysfunction in diabetes. Curr. Diabetes Rev. 2013, 9, 25–53. [Google Scholar] [CrossRef]
- Virkamäki, A.; Ueki, K.; Kahn, C.R. Protein–protein interaction in insulin signaling and the molecular mechanisms of insulin resistance. J. Clin. Investig. 1999, 103, 931–943. [Google Scholar] [CrossRef]
- Le Roith, D.; Zick, Y. Recent advances in our understanding of insulin action and insulin resistance. Diabetes Care 2001, 24, 588. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Goldwaser, I.; Gefel, D.; Gershonov, E.; Fridkin, M.; Shechter, Y. Insulin-like effects of vanadium: Basic and clinical implications. J. Inorg. Biochem. 2000, 80, 21–25. [Google Scholar] [CrossRef]
- Perseghin, G.; Ghosh, S.; Gerow, K.; Shulman, G.I. Metabolic defects in lean nondiabetic offspring of NIDDM Parents: A cross-sectional study. Diabetes 1997, 46, 1001. [Google Scholar] [CrossRef]
- Crans, D.C.; Smee, J.J.; Gaidamauskas, E.; Yang, L. The chemistry and biochemistry of vanadium and the biological activities exerted by vanadium compounds. Chem. Rev. 2004, 104, 849–902. [Google Scholar] [CrossRef] [PubMed]
- Di Bona, K.R.; Love, S.; Rhodes, N.R.; McAdory, D.; Sinha, S.H.; Kern, N.; Kent, J.; Strickland, J.; Wilson, A.; Beaird, J.; et al. Chromium is not an essential trace element for mammals: Effects of a “low-chromium” diet. J. Biol. Inorg. Chem. 2011, 16, 381–390. [Google Scholar] [CrossRef]
- Thompson, K.H.; Orvig, C. Coordination chemistry of vanadium in metallopharmaceutical candidate compounds. Coord. Chem. Rev. 2001, 219–221, 1033–1053. [Google Scholar] [CrossRef]
- Thompson, K.H.; Orvig, C. Vanadium in diabetes: 100 years from Phase 0 to Phase I. J. Inorg. Biochem. 2006, 100, 1925–1935. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.H.; Lichter, J.; LeBel, C.; Scaife, M.C.; McNeill, J.H.; Orvig, C. Vanadium treatment of type 2 diabetes: A view to the future. J. Inorg. Biochem. 2009, 103, 554–558. [Google Scholar] [CrossRef]
- Hoffman, N.J.; Penque, B.A.; Habegger, K.M.; Sealls, W.; Tackett, L.; Elmendorf, J.S. Chromium enhances insulin responsiveness via AMPK. J. Nutr. Biochem. 2014, 25, 565–572. [Google Scholar] [CrossRef]
- Deng, G.; Wu, K.; Cruce, A.A.; Bowman, M.K.; Vincent, J.B. Binding of trivalent chromium to serum transferrin is sufficiently rapid to be physiologically relevant. J. Inorg. Biochem. 2015, 143, 48–55. [Google Scholar] [CrossRef]
- Edwards, K.C.; Kim, H.; Vincent, J.B. Release of trivalent chromium from serum transferrin is sufficiently rapid to be physiologically relevant. J. Inorg. Biochem. 2020, 202, 110901. [Google Scholar] [CrossRef]
- Melchior, M.; Rettig, S.J.; Liboiron, B.D.; Thompson, K.H.; Yuen, V.G.; McNeill, J.H.; Orvig, C. Insulin-enhancing vanadium(III) complexes. Inorg. Chem. 2001, 40, 4686–4690. [Google Scholar] [CrossRef]
- Thompson, K.H.; Orvig, C. Design of vanadium compounds as insulin enhancing agents. Dalton Trans. 2000, 17, 2885–2892. [Google Scholar] [CrossRef]
- Yuen, V.G.; McNeill, J.H.; Orvig, C. Comparison of the glucose-lowering properties of vanadyl sulfate and bis(maltolato)oxovanadium(IV) following acute and chronic administration. Can. J. Physiol. Pharmacol. 1995, 73, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Wang, S.; Zhu, M.; Liu, Z.; Guo, M.; Xing, S.; Fu, X. Inhibition protein tyrosine phosphatases by an oxovanadium glutamate complex, Na2[VO(Glu)2(CH3OH)](Glu = glutamate). BioMetals 2010, 23, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Lu, L.; Gao, X.; Wu, Y.; Guo, M.; Li, Y.; Fu, X.; Zhu, M. Ternary oxovanadium(IV) complexes of ONO-donor Schiff base and polypyridyl derivatives as protein tyrosine phosphatase inhibitors: Synthesis, characterization, and biological activities. J. Biol. Inorg. Chem. 2009, 14, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Brandão, T.A.S.; Hengge, A.C.; Johnson, S.J. Insights into the reaction of protein-tyrosine phosphatase 1B: Crystal structures for transition state analogs of both catalytic steps. J. Biol. Chem. 2010, 285, 15874–15883. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ding, W.; Baruah, B.; Crans, D.C.; Wang, R. Inhibition of protein tyrosine phosphatase 1B and alkaline phosphatase by bis(maltolato)oxovanadium(IV). J. Inorg. Biochem. 2008, 102, 1846–1853. [Google Scholar] [CrossRef]
- Crans, D.C.; Tarlton, M.L.; McLauchlan, C.C. Trigonal bipyramidal or square pyramidal coordination geometry? Investigating the most potent geometry for vanadium phosphatase inhibitors. Eur. J. Inorg. Chem. 2014, 2014, 4450–4468. [Google Scholar] [CrossRef]
- Sánchez-Lombardo, I.; Alvarez, S.; McLauchlan, C.C.; Crans, D.C. Evaluating transition state structures of vanadium-phosphatase protein complexes using shape analysis. J. Inorg. Biochem. 2015, 147, 153–164. [Google Scholar] [CrossRef]
- Peters, K.G.; Davis, M.G.; Howard, B.W.; Pokross, M.; Rastogi, V.; Diven, C.; Greis, K.D.; Eby-Wilkens, E.; Maier, M.; Evdokimov, A.; et al. Mechanism of insulin sensitization by BMOV (bis maltolato oxo vanadium); unliganded vanadium (VO4) as the active component. J. Inorg. Biochem. 2003, 96, 321–330. [Google Scholar] [CrossRef]
- Costa Pessoa, J.; Garribba, E.; Santos, M.F.A.; Santos-Silva, T. Vanadium and proteins: Uptake, transport, structure, activity and function. Coord. Chem. Rev. 2015, 301–302, 49–86. [Google Scholar] [CrossRef]
- Kiss, T.; Jakusch, T.; Hollender, D.; Dörnyei, Á.; Enyedy, É.A.; Pessoa, J.C.; Sakurai, H.; Sanz-Medel, A. Biospeciation of antidiabetic VO(IV) complexes. Coord. Chem. Rev. 2008, 252, 1153–1162. [Google Scholar] [CrossRef]
- Costa Pessoa, J.; Tomaz, I. Transport of therapeutic vanadium and ruthenium complexes by blood plasma components. Curr. Med. Chem. 2010, 17, 3701–3738. [Google Scholar] [CrossRef] [PubMed]
- Jakusch, T.; Costa Pessoa, J.; Kiss, T. The speciation of vanadium in human serum. Coord. Chem. Rev. 2011, 255, 2218–2226. [Google Scholar] [CrossRef]
- Justino, G.C.; Garribba, E.; Pessoa, J.C. Binding of VIVO2+ to the Fe binding sites of human serum transferrin. A theoretical study. J. Biol. Inorg. Chem. 2013, 18, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Sanna, D.; Micera, G.; Garribba, E. Interaction of insulin-enhancing vanadium compounds with human serum holo-transferrin. Inorg. Chem. 2013, 52, 11975–11985. [Google Scholar] [CrossRef]
- Tsaramyrsi, M.; Kaliva, M.; Salifoglou, A.; Raptopoulou, C.P.; Terzis, A.; Tangoulis, V.; Giapintzakis, J. Vanadium(IV)–citrate complex interconversions in aqueous solutions. A pH-Dependent synthetic, structural, spectroscopic, and magnetic study. Inorg. Chem. 2001, 40, 5772–5779. [Google Scholar] [CrossRef]
- Kiss, T.; Jakusch, T. Metallotherapeutic Drugs and Metal-Based Diagnostic Agents; John Wiley & Sons: Hoboken, NJ, USA, 2005; pp. 143–158. [Google Scholar]
- Sakurai, H.; Goda, T.; Shimomura, S.; Yoshimura, T. ATP(adenosine triphosphate)-vanadyl complex. Biochem. Biophys. Res. Commun. 1982, 104, 1421–1426. [Google Scholar] [CrossRef]
- Illing, A.C.; Shawki, A.; Cunningham, C.L.; Mackenzie, B. Substrate profile and metal-ion selectivity of human divalent metal-ion transporter-1. J. Biol. Chem. 2012, 287, 30485–30496. [Google Scholar] [CrossRef]
- Striffler, J.S.; Polansky, M.M.; Anderson, R.A. Dietary chromium decreases insulin resistance in rats fed a high-fat, mineral-imbalanced diet. Metabolism 1998, 47, 396–400. [Google Scholar] [CrossRef]
- Sun, Y.; Ramirez, J.; Woski, S.A.; Vincent, J.B. The binding of trivalent chromium to low-molecular-weight chromium-binding substance (LMWCr) and the transfer of chromium from transferrin and chromium picolinate to LMWCr. J. Biol. Inorg. Chem. 2000, 5, 129–136. [Google Scholar] [CrossRef]
- Petersen, C.M.; Edwards, K.C.; Gilbert, N.C.; Vincent, J.B.; Thompson, M.K. X-ray structure of chromium(III)-containing transferrin: First structure of a physiological Cr(III)-binding protein. J. Inorg. Biochem. 2020, 210, 111101. [Google Scholar] [CrossRef]
- Yamamoto, A.; Wada, O.; Ono, T. Isolation of a biologically active low-molecular-mass chromium compound from rabbit liver. Eur. J. Biochem. 1987, 165, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Kandror, K.V. Insulin regulation of protein traffic in rat adipose cells. J. Biol. Chem. 1999, 274, 25210–25217. [Google Scholar] [CrossRef] [PubMed]
- Institute for Health Metrics and Evaluation. Global Burden of Disease. Available online: https://vizhub.healthdata.org/cod/ (accessed on 31 July 2020).
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, H.; Fan, D.; Ozawa, S.; Yano, S.; Van Arsdell, M.; Viner, J.L.; Beers, R.; Pastan, I.; Fidler, I.J. Site-specific expression of transferrin receptor by human colon cancer cells directly correlates with eradication by antitransferrin recombinant immunotoxin. Int. J. Oncol. 2000, 17, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Shindelman, J.E.; Ortmeyer, A.E.; Sussman, H.H. Demonstration of the transferrin receptor in human breast cancer tissue. Potential marker for identifying dividing cells. Int. J. Cancer. 1981, 27, 329–334. [Google Scholar] [CrossRef]
- Philpott, C.C.; Ryu, M.-S. Special delivery: Distributing iron in the cytosol of mammalian cells. Front. Pharmacol. 2014, 5, 173. [Google Scholar] [CrossRef]
- Sutherland, R.; Delia, D.; Schneider, C.; Newman, R.; Kemshead, J.; Greaves, M. Ubiquitous cell-surface glycoprotein on tumor cells is proliferation-associated receptor for transferrin. Proc. Natl. Acad. Sci. USA 1981, 78, 4515. [Google Scholar] [CrossRef]
- Muggia, F.; Farrell, N. Platinum coordination compounds in cancer chemotherapy. Crit. Rev. Oncol. Hematol. 2005, 1, 1–2. [Google Scholar] [CrossRef]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef]
- Fernández-Vega, L.; Ruiz Silva, V.A.; Domínguez-González, T.M.; Claudio-Betancourt, S.; Toro-Maldonado, R.E.; Capre Maso, L.C.; Sanabria Ortiz, K.; Pérez-Verdejo, J.A.; Román González, J.; Rosado-Fraticelli, G.T.; et al. Evaluating ligand modifications of the titanocene and auranofin moieties for the development of more potent anticancer drugs. Inorganics 2020, 8, 10. [Google Scholar] [CrossRef]
- Heim, M.E.; Flechtner, H.; Keppler, B.K. Clinical studies with budotitane—A new non-platinum metal complex for cancer therapy. In Ruthenium and Other Non-Platinum Metal Complexes in Cancer Chemotherapy; Baulieu, E., Forman, D.T., Ingelman-Sundberg, M., Jaenicke, L., Kellen, J.A., Nagai, Y., Springer, G.F., Träger, L., Will-Shahab, L., Wittliff, J.L., Eds.; Springer: Berlin/Heidelberg, Germany, 1989; pp. 217–223. [Google Scholar]
- Schilling, T.; Keppler, K.B.; Heim, M.E.; Niebch, G.; Dietzfelbinger, H.; Rastetter, J.; Hanauske, A.-R. Clinical phase I and pharmacokinetic trial of the new titanium complex budotitane. Invest. New Drugs 1996, 13, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, C.V.; Ferry, D.R.; Fyfe, D.W.; Young, A.; Doran, J.; Sheehan, T.M.; Eliopoulos, A.; Hale, K.; Baumgart, J.; Sass, G.; et al. Phase I trial of weekly scheduling and pharmacokinetics of titanocene dichloride in patients with advanced cancer. J. Clin. Oncol. 1998, 16, 2761–2769. [Google Scholar] [CrossRef]
- Korfel, A.; Scheulen, M.; Schmoll, H.-J.; Gründel, O.; Harstrick, A.; Knoche, M.; Fels, L.M.; Skorzec, M.; Bach, F.; Baumgart, J.; et al. Phase I clinical and pharmacokinetic study of titanocene dichloride in adults with advanced solid tumors. Clin. Cancer Res. 1998, 4, 2701–2708. [Google Scholar] [PubMed]
- Mross, K.; Robben-Bathe, P.; Edler, L.; Baumgart, J.; Berdel, W.E.; Fiebig, H.; Unger, C. Phase I clinical trial of a day-1,-3,-5 every 3 weeks schedule with titanocene dichloride (MKT 5) in patients with advanced cancer—A study of the phase I study group of the Association for Medical Oncology (AIO) of the German Cancer Society. Onkologie 2000, 23, 576–579. [Google Scholar] [PubMed]
- Mokdsi, G.; Harding, M.M. Inhibition of human topoisomerase II by the antitumor metallocenes. J. Inorg. Biochem. 2001, 83, 205–209. [Google Scholar] [CrossRef]
- Christodoulou, C.V.; Eliopoulos, A.G.; Young, L.S.; Hodgkins, L.; Ferry, D.R.; Kerr, D.J. Anti-proliferative activity and mechanism of action of titanocene dichloride. Br. J. Cancer. 1998, 77, 2088–2097. [Google Scholar] [CrossRef]
- Toney, J.H.; Marks, T.J. Hydrolysis chemistry of the metallocene dichlorides M(η5-C5H5)2Cl2, M = titanium, vanadium, or zirconium. Aqueous kinetics, equilibria, and mechanistic implications for a new class of antitumor agents. J. Am. Chem. Soc. 1985, 107, 947–953. [Google Scholar] [CrossRef]
- Sun, H.; Li, H.; Weir, R.A.; Sadler, P.J. The First Specific Ti(IV)-Protein Complex: Potential Relevance to Anticancer Activity of Titanocenes. Angew. Chem. 1998, 37, 1577–1579. [Google Scholar] [CrossRef]
- Messori, L.; Orioli, P.; Banholzer, V.; Pais, I.; Zatta, P. Formation of titanium(IV) transferrin by reaction of human serum apotransferrin with titanium complexes. FEBS Lett. 1999, 442, 157–161. [Google Scholar] [CrossRef]
- Guo, M.; Sun, H.; McArdle, H.J.; Gambling, L.; Sadler, P.J. Ti(IV) Uptake and Release by Human Serum Transferrin and Recognition of Ti(IV)-Transferrin by Cancer Cells: Understanding the Mechanism of Action of the Anticancer Drug Titanocene Dichloride. Biochemistry 2000, 39, 10023–10033. [Google Scholar] [CrossRef]
- Guo, M.L.; Sadler, P.J. Competitive binding of the anticancer drug titanocene dichloride to N,N-ethylenebis(o-hydroxyphenylglycine) and adenosine triphosphate: A model for Ti-IV uptake and release by transferrin. Dalton Trans. 2000, 1, 2885–2892. [Google Scholar] [CrossRef]
- Guo, M.L.; Sun, H.Z.; Bihari, S.; Parkinson, J.A.; Gould, R.O.; Parsons, S.; Sadler, P.J. Stereoselective formation of seven-coordinate titanium(IV) monomer and dimer complexes of ethylenebis(o-hydroxyphenyl)glycine. Inorg. Chem. 2000, 39, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, A.D.; Valentine, A.M. Ti(IV) binds to human serum transferrin more tightly than Does Fe(III). J. Am. Chem. Soc. 2005, 127, 11218–11219. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, A.D.; Incarvito, C.D.; Valentine, A.M. Calorimetric, Spectroscopic, and Model Studies Provide Insight into the Transport of Ti(IV) by Human Serum Transferrin. J. Am. Chem. Soc. 2007, 129, 3444–3454. [Google Scholar] [CrossRef]
- Saxena, M.; Loza Rosas, S.; Gaur, K.; Sharma, S.; Perez Otero, S.C.; Tinoco, A.D. Exploring titanium(IV) chemical proximity to iron(III) to elucidate a function for Ti(IV) in the human body. Coord. Chem. Rev. 2018, 363, 109–125. [Google Scholar] [CrossRef]
- Guo, M.L.; Harvey, I.; Campopiano, D.J.; Sadler, P.J. Short oxo-titanium(IV) bond in bacterial transferrin: A protein target for metalloantibiotics. Angew. Chem. 2006, 45, 2758–2761. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, A.; Kulesha, E.; Gough, J.; Murzin, A.G. The SCOP database in 2020: Expanded classification of representative family and superfamily domains of known protein structures. Nucleic Acids Res. 2019, 48, D376–D382. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.M.; Uppal, R.; Incarvito, C.D.; Valentine, A.M. Titanium(IV) citrate speciation and structure under environmentally and biologically relevant conditions. Inorg. Chem. 2005, 44, 3431–3440. [Google Scholar] [CrossRef]
- Guo, M.; Guo, Z.; Sadler, P.J. Titanium(IV) targets phosphoesters on nucleotides: Implications for the mechanism of action of the anticancer drug titanocene dichloride. J. Biol. Inorg. Chem. 2001, 6, 698–707. [Google Scholar] [CrossRef]
- Gurgueira, S.A.; Meneghini, R. An ATP-dependent iron transport system in isolated rat liver nuclei. J. Biol. Chem. 1996, 271, 13616–13620. [Google Scholar] [CrossRef]
- Köpf-Maier, P. Electron-spectroscopic imaging—A method for analysing the distribution of light elements in mammalian cells and tissues. Acta Histochem. 1991, 91, 25–37. [Google Scholar] [CrossRef]
- Erxleben, A.; Claffey, J.; Tacke, M. Binding and hydrolysis studies of antitumoural titanocene dichloride and Titanocene Y with phosphate diesters. J. Inorg. Biochem. 2010, 104, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhong, J.; Xiao, K.; Tian, Z. Enrichment of intact phosphoproteins using immobilized titanium(IV) affinity chromatography microspheres. Sep. Sci. Plus 2018, 1, 93–99. [Google Scholar] [CrossRef]
- Chimento, A.; Saturnino, C.; Iacopetta, D.; Mazzotta, R.; Caruso, A.; Plutino, M.R.; Mariconda, A.; Ramunno, A.; Sinicropi, M.S.; Pezzi, V.; et al. Inhibition of human topoisomerase I and II and anti-proliferative effects on MCF-7 cells by new titanocene complexes. Biorg. Med. Chem. 2015, 23, 7302–7312. [Google Scholar] [CrossRef] [PubMed]
- Olszewski, U.; Hamilton, G. Mechanisms of cytotoxicity of anticancer titanocenes. Anticancer Agents Med. Chem. 2010, 10, 302–311. [Google Scholar] [CrossRef]
- Tinoco, A.D.; Thomas, H.R.; Incarvito, C.D.; Saghatelian, A.; Valentine, A.M. Cytotoxicity of a Ti(IV) compound is independent of serum proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 5016–5021. [Google Scholar] [CrossRef]
- Soto-Alvaredo, J.; Blanco, E.; Bettmer, J.; Hevia, D.; Sainz, R.M.; Chaves, C.L.; Sanchez, C.; Llopis, J.; Sanz-Medel, A.; Montes-Bayon, M. Evaluation of the biological effect of Ti generated debris from metal implants: Ions and nanoparticles. Metallomics 2014, 6, 1702–1708. [Google Scholar] [CrossRef]
- Loza-Rosas, S.A.; Saxena, M.; Delgado, Y.; Gaur, K.; Pandrala, M.; Tinoco, A.D. A ubiquitous metal, difficult to track: Towards an understanding of the regulation of titanium(IV) in humans. Metallomics 2017, 9, 346–356. [Google Scholar] [CrossRef]
- Nuevo-Ordonez, Y.; Montes-Bayon, M.; Blanco-Gonzalez, E.; Paz-Aparicio, J.; Raimundez, J.D.; Tejerina, J.M.; Pena, M.A.; Sanz-Medel, A. Titanium release in serum of patients with different bone fixation implants and its interaction with serum biomolecules at physiological levels. Anal. Bioanal. Chem. 2011, 401, 2747–2754. [Google Scholar] [CrossRef]
- Nuevo-Ordonez, Y.; Montes-Bayon, M.; Gonzalez, E.B.; Sanz-Medel, A. Titanium preferential binding sites in human serum transferrin at physiological concentrations. Metallomics 2011, 3, 1297–1303. [Google Scholar] [CrossRef]
- Jakupec, M.A.; Keppler, B.K. Gallium in cancer treatment. Curr. Top. Med. Chem. 2004, 4, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Rasey, J.S.; Nelson, N.J.; Larson, S.M. Tumor cell toxicity of stable gallium nitrate: Enhancement by transferrin and protection by iron. Eur. J. Cancer Clin. Oncol. 1982, 18, 661–668. [Google Scholar] [CrossRef]
- Chitambar, C.R.; Zivkovic, Z. Uptake of gallium-67 by human leukemic cells: Demonstration of transferrin receptor-dependent and transferrin-independent mechanisms. Cancer Res. 1987, 47, 3929–3934. [Google Scholar] [PubMed]
- Straus, D.J. Gallium nitrate in the treatment of lymphoma. Semin. Oncol. 2003, 30, 25–33. [Google Scholar] [CrossRef]
- Seligman, P.A.; Moran, P.L.; Schleicher, R.B.; Crawford, E.D. Treatment with gallium nitrate: Evidence for interference with iron metabolism in vivo. Am. J. Hematol. 1992, 41, 232–240. [Google Scholar] [CrossRef]
- Gaur, K.; Vázquez-Salgado, A.M.; Duran-Camacho, G.; Dominguez-Martinez, I.; Benjamín-Rivera, J.A.; Fernández-Vega, L.; Carmona Sarabia, L.; Cruz García, A.; Pérez-Deliz, F.; Méndez Román, J.A.; et al. Iron and Copper Intracellular Chelation as an Anticancer Drug Strategy. Inorganics 2018, 6, 126. [Google Scholar] [CrossRef]
- Weiner, R.E. Role of phosphate-containing compounds in the transfer of indium-111 and gallium-67 from transferrin to ferritin. J. Nucl. Med. 1989, 30, 70–79. [Google Scholar]
- Weiner, R.E.; Schreiber, G.J.; Hoffer, P.B. In vitro transfer of Ga-67 from transferrin to ferritin. J. Nucl. Med. 1983, 24, 608–614. [Google Scholar]
- Chitambar, C.R.; Narasimhan, J.; Guy, J.; Sem, D.S.; O’Brien, W.J. Inhibition of ribonucleotide reductase by gallium in murine leukemic L1210 cells. Cancer Res. 1991, 51, 6199–6201. [Google Scholar]
- Aoyagi, T.; Bander, N.H. Cytotoxicity of gallium nitrate in vitro using bladder cancer cells. Int. J. Urol. 1995, 2, 288–294. [Google Scholar]
- Seligman, P.A.; Crawford, E.D. Treatment of advanced transitional cell carcinoma of the bladder with continuous-infusion gallium nitrate. J. Natl. Cancer Inst. 1991, 83, 1582–1584. [Google Scholar] [CrossRef] [PubMed]
- Chua, M.S.; Bernstein, L.R.; Li, R.; So, S.K. Gallium maltolate is a promising chemotherapeutic agent for the treatment of hepatocellular carcinoma. Anticancer Res. 2006, 26, 1739–1743. [Google Scholar] [PubMed]
- Weiner, R.E. The mechanism of 67Ga localization in malignant disease. Nucl. Med. Biol. 1996, 23, 745–751. [Google Scholar] [CrossRef]
- Allardyce, C.S.; Dorcier, A.; Scolaro, C.; Dyson, P.J. Development of organometallic (organo-transition metal) pharmaceuticals. Appl. Organomet. Chem. 2005, 19, 1–10. [Google Scholar] [CrossRef]
- Mazuryk, O.; Kurpiewska, K.; Lewinski, K.; Stochel, G.; Brindell, M. Interaction of apo-transferrin with anticancer ruthenium complexes NAMI-A and its reduced form. J. Inorg. Biochem. 2012, 116, 11–18. [Google Scholar] [CrossRef]
- Dyson, P.J.; Sava, G. Metal-based antitumour drugs in the post genomic era. Dalton Trans. 2006, 16, 1929–1933. [Google Scholar] [CrossRef]
- Sava, G.; Bergamo, A.; Dyson, P.J. Metal-based antitumour drugs in the post-genomic era: What comes next? Dalton Trans. 2011, 40, 9069–9075. [Google Scholar] [CrossRef]
- Harris, W.R.; Messori, L. A comparative study of aluminum(III), gallium(III), indium(III), and thallium(III) binding to human serum transferrin. Coord. Chem. Rev. 2002, 228, 237–262. [Google Scholar] [CrossRef]
- Kratz, F.; Hartmann, M.; Keppler, B.; Messori, L. The binding properties of two antitumor ruthenium(III) complexes to apotransferrin. J. Biol. Chem. 1994, 269, 2581–2588. [Google Scholar]
- Alessio, E.; Messori, L. NAMI-A and KP1019/1339, Two iconic ruthenium anticancer drug candidates face-to-face: A case story in medicinal inorganic chemistry. Molecules 2019, 24, 1995. [Google Scholar] [CrossRef]
- Rademaker-Lakhai, J.M.; van den Bongard, D.; Pluim, D.; Beijnen, J.H.; Schellens, J.H. A Phase I and pharmacological study with imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a novel ruthenium anticancer agent. Clin. Cancer Res. 2004, 10, 3717–3727. [Google Scholar] [CrossRef] [PubMed]
- Henke, M.M.; Richly, H.; Drescher, A.; Grubert, M.; Alex, D.; Thyssen, D.; Jaehde, U.; Scheulen, M.E.; Hilger, R.A. Pharmacokinetic study of sodium trans[tetrachlorobis(1H-indazole)-ruthenate (III)]/-indazole hydrochloride (1:1.1) (FFC14A) in patients with solid tumors. Int. J. Clin. Pharmacol. Ther. 2009, 47, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Aramini, J.M.; Germann, M.W.; Vogel, H.J. Field-dependent aluminum-27 NMR studies of the transferrins: An approach for the study of metal ion binding sites in larger proteins. J. Am. Chem. Soc. 1993, 115, 9750–9753. [Google Scholar] [CrossRef]
- Aramini, J.M.; Saponja, J.A.; Vogel, H.J. Spectroscopic studies of the interaction of aluminum(III) with transferrins. Coord. Chem. Rev. 1996, 149, 193–229. [Google Scholar] [CrossRef]
- Mujika, J.I.; Escribano, B.; Akhmatskaya, E.; Ugalde, J.M.; Lopez, X. Molecular dynamics simulations of iron- and aluminum-loaded serum transferrin: Protonation of Tyr188 is necessary to prompt metal release. Biochemistry 2012, 51, 7017–7027. [Google Scholar] [CrossRef]
- Campbell, A.; Becaria, A.; Lahiri, D.K.; Sharman, K.; Bondy, S.C. Chronic exposure to aluminum in drinking water increases inflammatory parameters selectively in the brain. J. Neurosci. Res. 2004, 75, 565–572. [Google Scholar] [CrossRef]
- Roskams, A.J.; Connor, J.R. Aluminum access to the brain: A role for transferrin and its receptor. Proc. Natl. Acad. Sci. USA 1990, 87, 9024–9027. [Google Scholar] [CrossRef]
- Shaw, C.A.; Tomljenovic, L. Aluminum in the central nervous system (CNS): Toxicity in humans and animals, vaccine adjuvants, and autoimmunity. Immunol. Res. 2013, 56, 304–316. [Google Scholar] [CrossRef]
- Klein, G.L. Aluminum toxicity to bone: A multisystem effect? Osteoporos. Sarcopenia 2019, 5, 2–5. [Google Scholar] [CrossRef]
- Jeanson, A.; Ferrand, M.; Funke, H.; Hennig, C.; Moisy, P.; Solari, P.L.; Vidaud, C.; Den Auwer, C. The role of transferrin in actinide(IV) uptake: Comparison with iron(III). Chemistry 2010, 16, 1378–1387. [Google Scholar] [CrossRef]
- Taylor, D.M. The bioinorganic chemistry of actinides in blood. J. Alloys Compd. 1998, 271–273, 6–10. [Google Scholar] [CrossRef]
- Vidaud, C.; Gourion-Arsiquaud, S.; Rollin-Genetet, F.; Torne-Celer, C.; Plantevin, S.; Pible, O.; Berthomieu, C.; Quemeneur, E. Structural consequences of binding of UO22+ to apotransferrin: Can this protein account for entry of uranium into human cells? Biochemistry 2007, 46, 2215–2226. [Google Scholar] [CrossRef] [PubMed]
- Kosman, D.J. Plutonium’s Trojan horse. Nat. Chem. Biol. 2011, 7, 498–499. [Google Scholar] [CrossRef] [PubMed]
- Boocock, G.; Popplewell, D.S. Distribution of plutonium in serum proteins following intravenous injection into rats. Nature 1965, 208, 282–283. [Google Scholar] [CrossRef]
- Mostapha, S.; Fontaine-Vive, F.; Berthon, L.; Boubals, N.; Zorz, N.; Solari, P.L.; Charbonnel, M.C.; Den Auwer, C. On the structure of thorium and americium adenosine triphosphate complexes. Int. J. Radiat. Biol. 2014, 90, 966–974. [Google Scholar] [CrossRef]
- Prutki, M.; Poljak-Blazi, M.; Jakopovic, M.; Tomas, D.; Stipancic, I.; Zarkovic, N. Altered iron metabolism, transferrin receptor 1 and ferritin in patients with colon cancer. Cancer Lett. 2006, 238, 188–196. [Google Scholar] [CrossRef]
- Dabrowiak, J.C. Metals in Medicine, 2nd ed.; Wiley: Hoboken, NJ, USA, 2017. [Google Scholar]
- Zak, O.; Aisen, P. Spectroscopic and thermodynamic studies on the binding of gadolinium (III) to human serum transferrin. Biochemistry 1988, 27, 1075–1080. [Google Scholar] [CrossRef]
- Korkusuz, H.; Ulbrich, K.; Welzel, K.; Koeberle, V.; Watcharin, W.; Bahr, U.; Chernikov, V.; Knobloch, T.; Petersen, S.; Huebner, F.; et al. Transferrin-coated gadolinium nanoparticles as MRI contrast agent. Mol. Imag. Biol. 2013, 15, 148–154. [Google Scholar] [CrossRef]
- Jiang, W.; Xie, H.; Ghoorah, D.; Shang, Y.; Shi, H.; Liu, F.; Yang, X.; Xu, H. Conjugation of functionalized SPIONs with transferrin for targeting and imaging brain glial tumors in rat model. PLoS ONE 2012, 7, e37376. [Google Scholar] [CrossRef]
- Smith, T.A.; Perkins, A.C.; Walton, P.H. 99mTc-labelled human serum transferrin for tumour imaging: An in vitro and in vivo study of the complex. Nucl. Med. Commun. 2004, 25, 387–391. [Google Scholar] [CrossRef]
- Gu, B.; Cai, J.; Zhang, J.; Xu, X.; Luo, J.; Zhou, X.; Zheng, Y.; Zhang, Y. 99m Tc-labeled and gadolinium-chelated transferrin enhances the sensitivity and specificity of dual-modality SPECT/MR imaging of breast cancer. RSC Adv. 2016, 6, 20532–20541. [Google Scholar] [CrossRef]
- Evans, M.J.; Holland, J.P.; Rice, S.L.; Doran, M.G.; Cheal, S.M.; Campos, C.; Carlin, S.D.; Mellinghoff, I.K.; Sawyers, C.L.; Lewis, J.S. Imaging tumor burden in the brain with 89Zr-transferrin. J. Nucl. Med. 2013, 54, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Vävere, A.; Jones, L.; McCarthy, T.; Rowland, D.; Welch, M. Preparation, biodistribution, and micropet imaging of 45Ti-transferrin. J. Labelled Compd. Radiopharmaceut. 2001, 44, S793–S795. [Google Scholar] [CrossRef]
- Vavere, A.L.; Welch, M.J. Preparation, biodistribution, and small animal PET of 45Ti-transferrin. J. Nucl. Med. 2005, 46, 683–690. [Google Scholar] [PubMed]
- Ooi, H.K.; Ma, L. Modeling heterogeneous responsiveness of intrinsic apoptosis pathway. BMC Syst. Biol. 2013, 7, 65. [Google Scholar] [CrossRef]
- Saxena, M.; Delgado, Y.; Sharma, R.K.; Sharma, S.; Guzmán, S.L.P.D.L.; Tinoco, A.D.; Griebenow, K. Inducing cell death in vitro in cancer cells by targeted delivery of cytochrome c via a transferrin conjugate. PLoS ONE 2018, 13, e0195542. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal Ion (Mn+) | Mn+-sTf (log K1) | Mn+-sTf-TfR Complex Detected? |
|---|---|---|
| Ti4+ | 35.8 a | Yes b,c |
| Pu4+ | 25.0 d | Weak e,f |
| Tc4+ | 23.0 b | Yes b |
| Fe3+ | 22.5 c | Yes b,c,e,f |
| Co3+ | 21.5 b | Yes b,g |
| Ga3+ | 20.3 h | Moderate b,e,g |
| Bi3+ | 19.4 b | Yes b,g |
| Th4+ | 18.65 e | Yes e |
| In3+ | 18.5 c,e | Yes c |
| Cr3+ | 17 i | Yes c |
| Al3+ | 13.8 b | Weak b,g |
| UO2+ | 13 j | Yes e,g |
| Species | pI |
|---|---|
| Apo-sTf | 6.07 ± 0.02 |
| Fec-sTf | 5.90 ± 0.02 |
| FeN-sTf | 5.82 ± 0.02 |
| Fe2-sTf | 5.63 ± 0.02 |
| Puc-sTf | 6.02 ± 0.02 |
| PuN-sTf | N.D. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benjamín-Rivera, J.A.; Cardona-Rivera, A.E.; Vázquez-Maldonado, Á.L.; Dones-Lassalle, C.Y.; Pabón-Colon, H.L.; Rodríguez-Rivera, H.M.; Rodríguez, I.; González-Espiet, J.C.; Pazol, J.; Pérez-Ríos, J.D.; et al. Exploring Serum Transferrin Regulation of Nonferric Metal Therapeutic Function and Toxicity. Inorganics 2020, 8, 48. https://doi.org/10.3390/inorganics8090048
Benjamín-Rivera JA, Cardona-Rivera AE, Vázquez-Maldonado ÁL, Dones-Lassalle CY, Pabón-Colon HL, Rodríguez-Rivera HM, Rodríguez I, González-Espiet JC, Pazol J, Pérez-Ríos JD, et al. Exploring Serum Transferrin Regulation of Nonferric Metal Therapeutic Function and Toxicity. Inorganics. 2020; 8(9):48. https://doi.org/10.3390/inorganics8090048
Chicago/Turabian StyleBenjamín-Rivera, Josué A., Andrés E. Cardona-Rivera, Ángel L. Vázquez-Maldonado, Christian Y. Dones-Lassalle, Héctor L. Pabón-Colon, Héctor M. Rodríguez-Rivera, Israel Rodríguez, Jean C. González-Espiet, Jessika Pazol, Jobaniel D. Pérez-Ríos, and et al. 2020. "Exploring Serum Transferrin Regulation of Nonferric Metal Therapeutic Function and Toxicity" Inorganics 8, no. 9: 48. https://doi.org/10.3390/inorganics8090048
APA StyleBenjamín-Rivera, J. A., Cardona-Rivera, A. E., Vázquez-Maldonado, Á. L., Dones-Lassalle, C. Y., Pabón-Colon, H. L., Rodríguez-Rivera, H. M., Rodríguez, I., González-Espiet, J. C., Pazol, J., Pérez-Ríos, J. D., Catala-Torres, J. F., Carrasquillo Rivera, M., De Jesus-Soto, M. G., Cordero-Virella, N. A., Cruz-Maldonado, P. M., González-Pagan, P., Hernández-Ríos, R., Gaur, K., Loza-Rosas, S. A., & Tinoco, A. D. (2020). Exploring Serum Transferrin Regulation of Nonferric Metal Therapeutic Function and Toxicity. Inorganics, 8(9), 48. https://doi.org/10.3390/inorganics8090048