Ferrocenyl Migrations and Molecular Rearrangements: The Significance of Electronic Charge Delocalization

Abstract
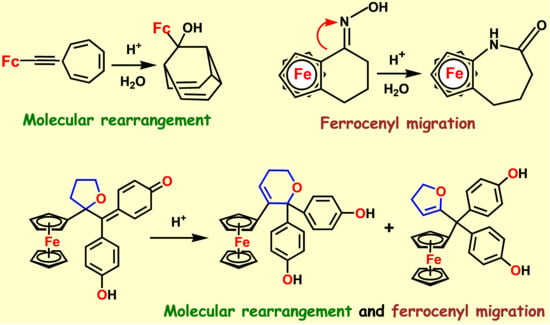
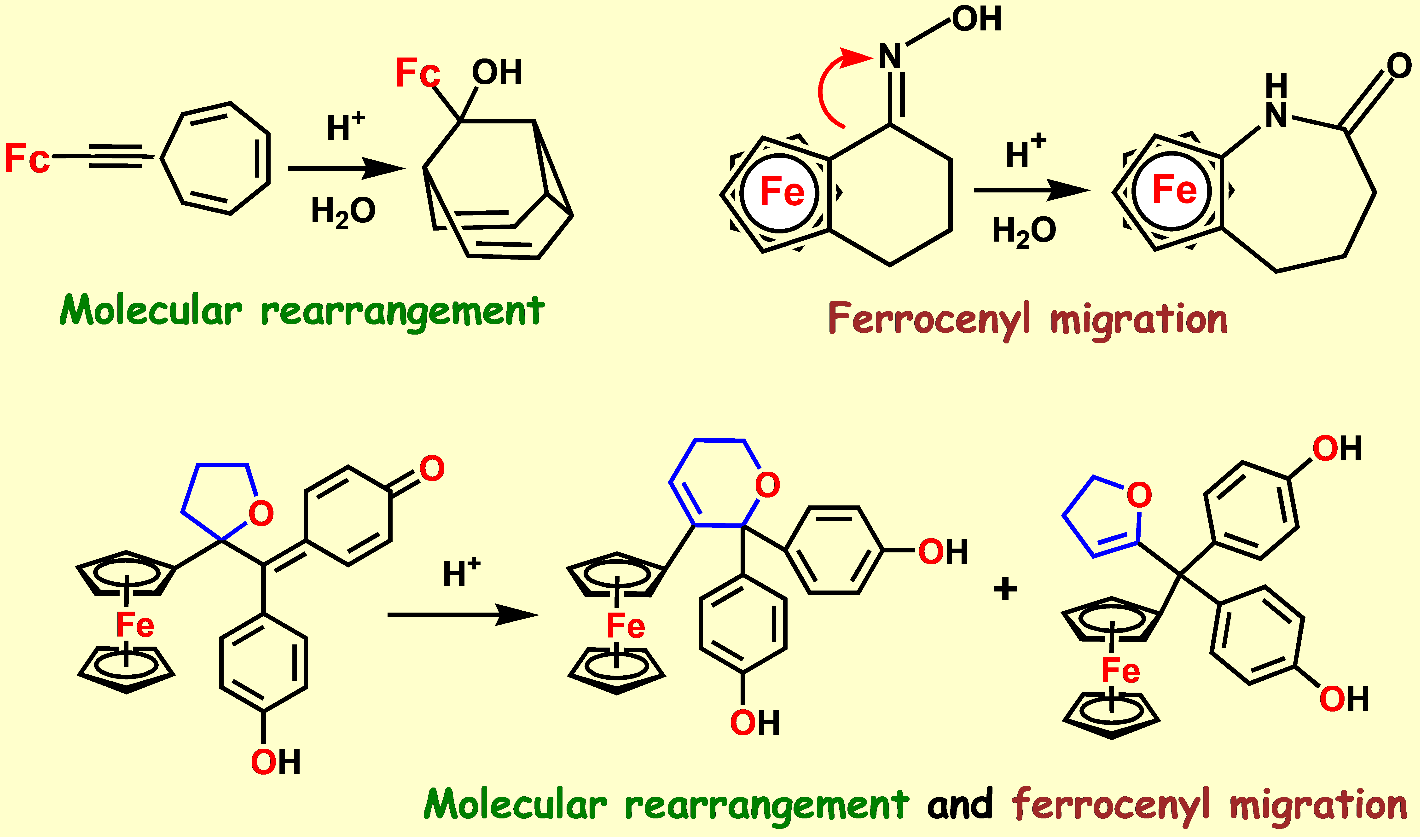{kind=link}
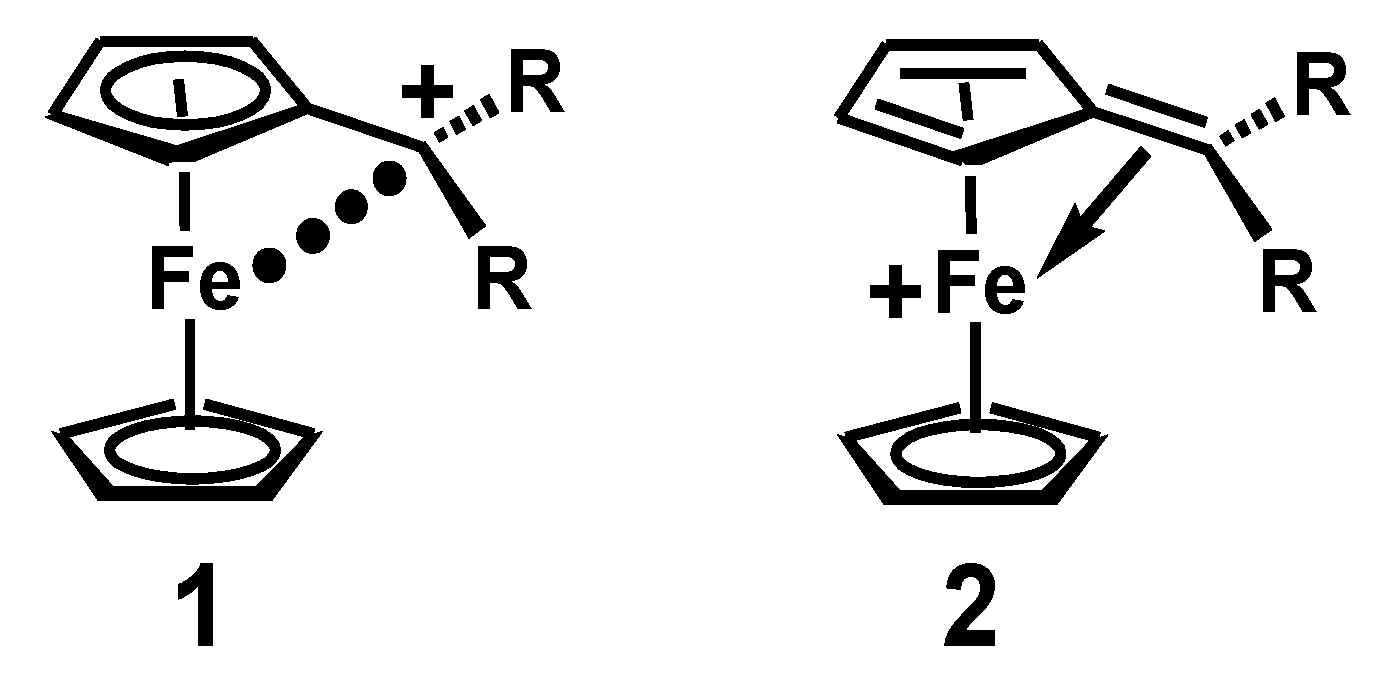{kind=link}
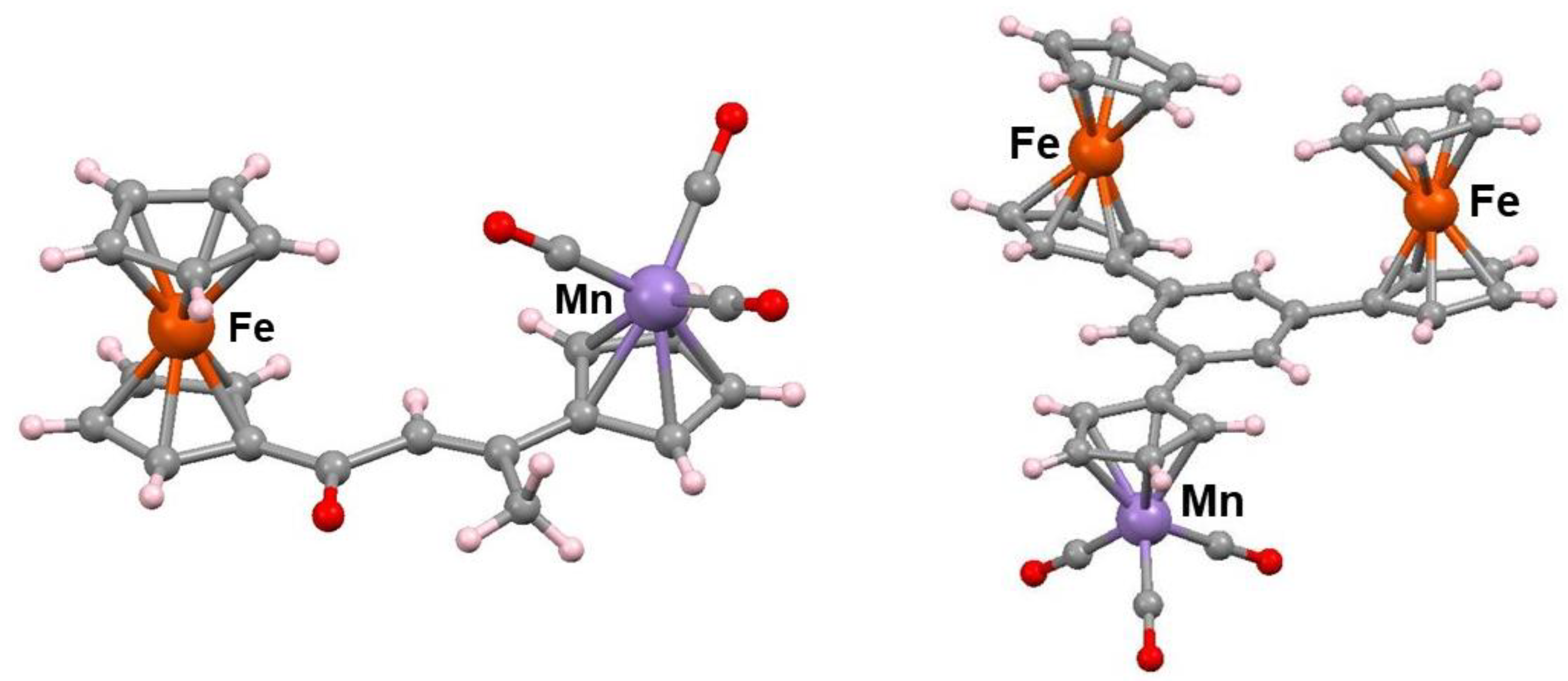{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
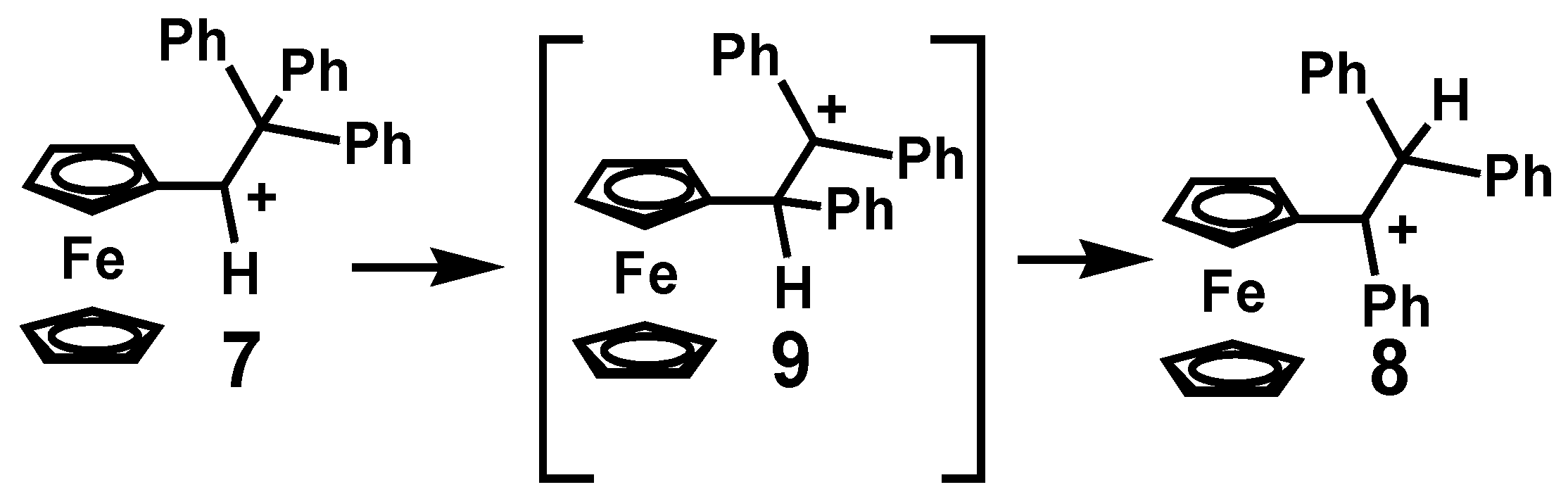{kind=link}
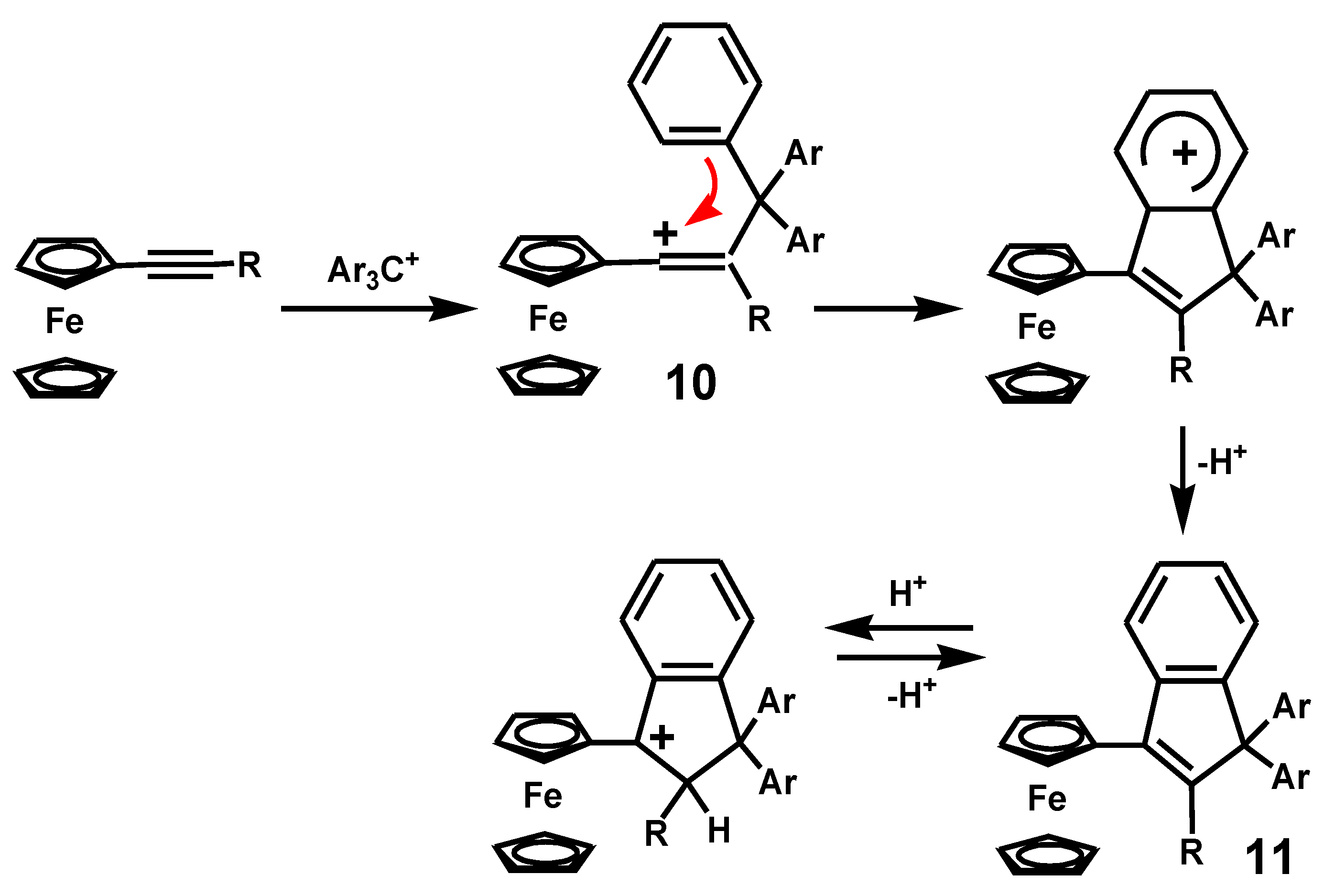{kind=link}
{kind=link}
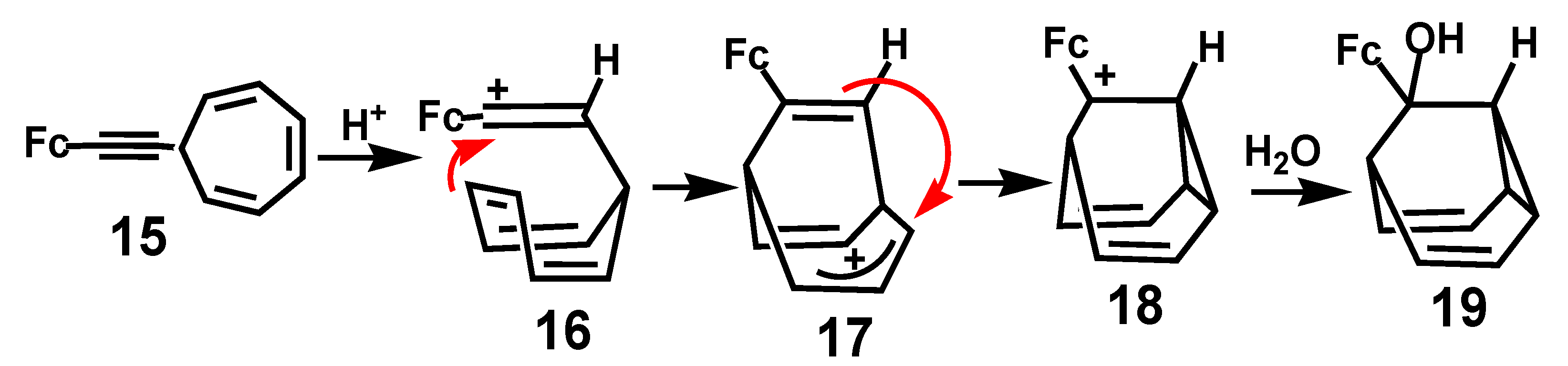{kind=link}
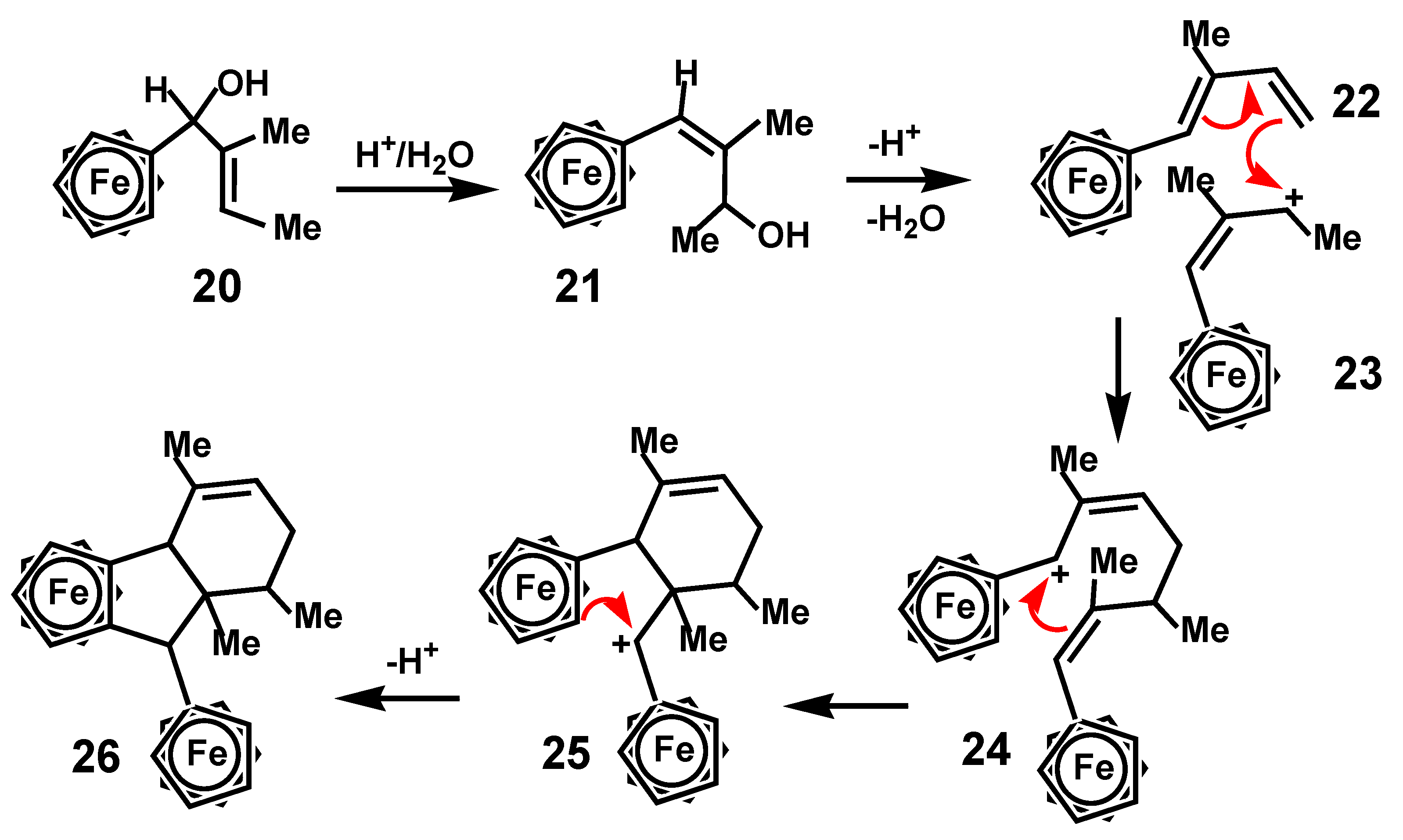{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
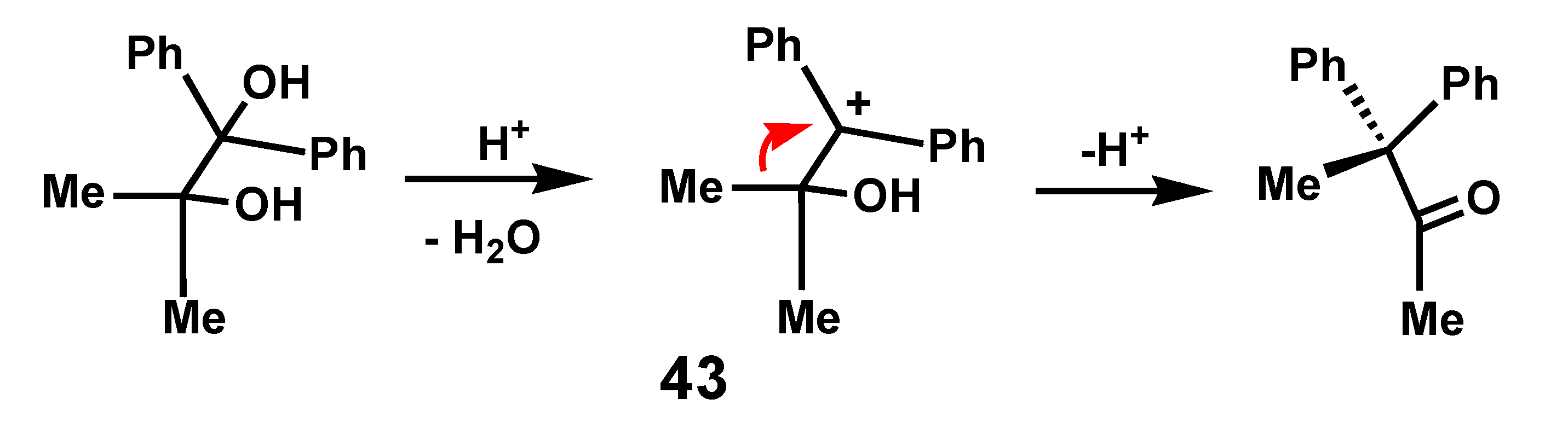{kind=link}
{kind=link}
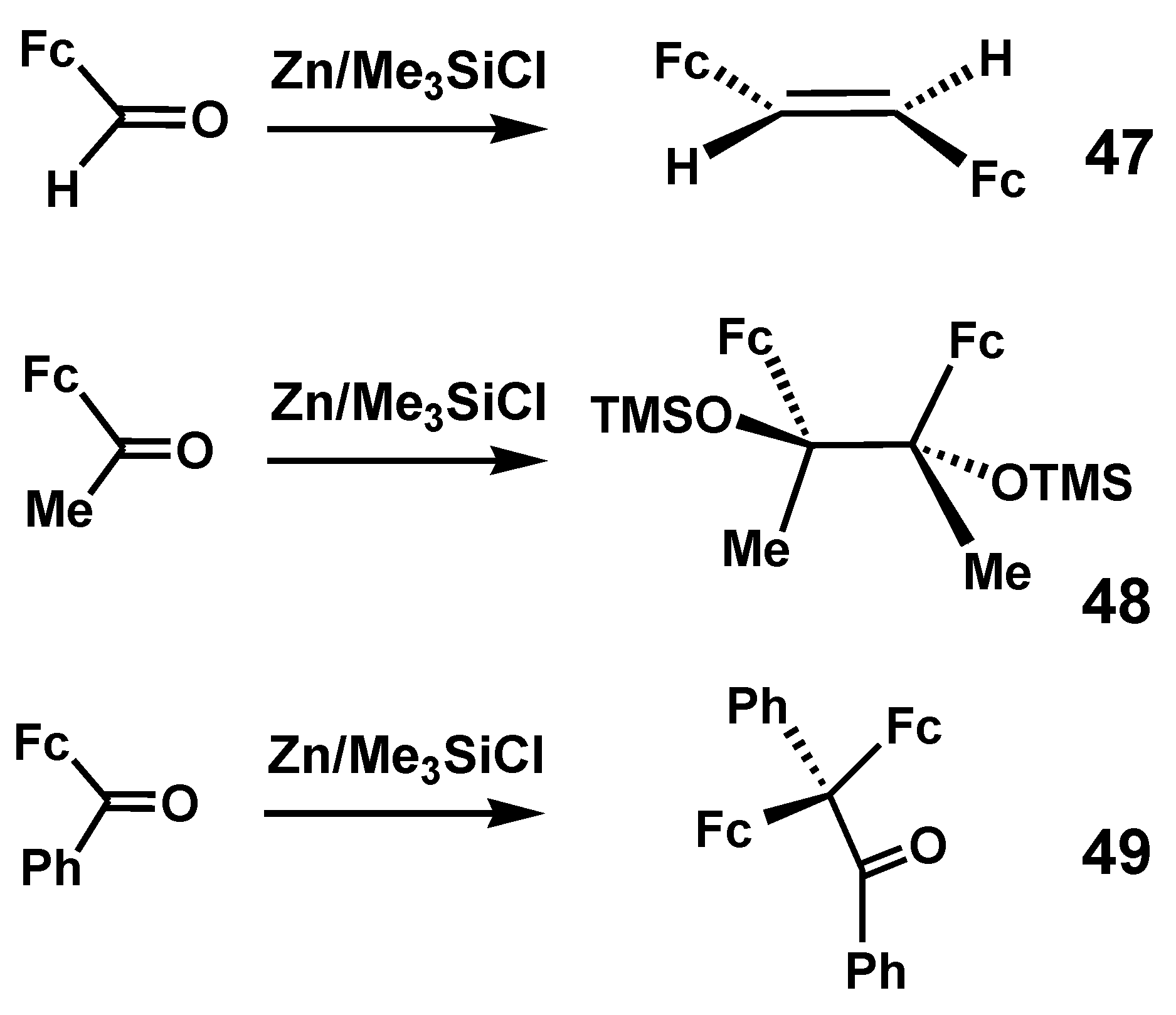{kind=link}
{kind=link}
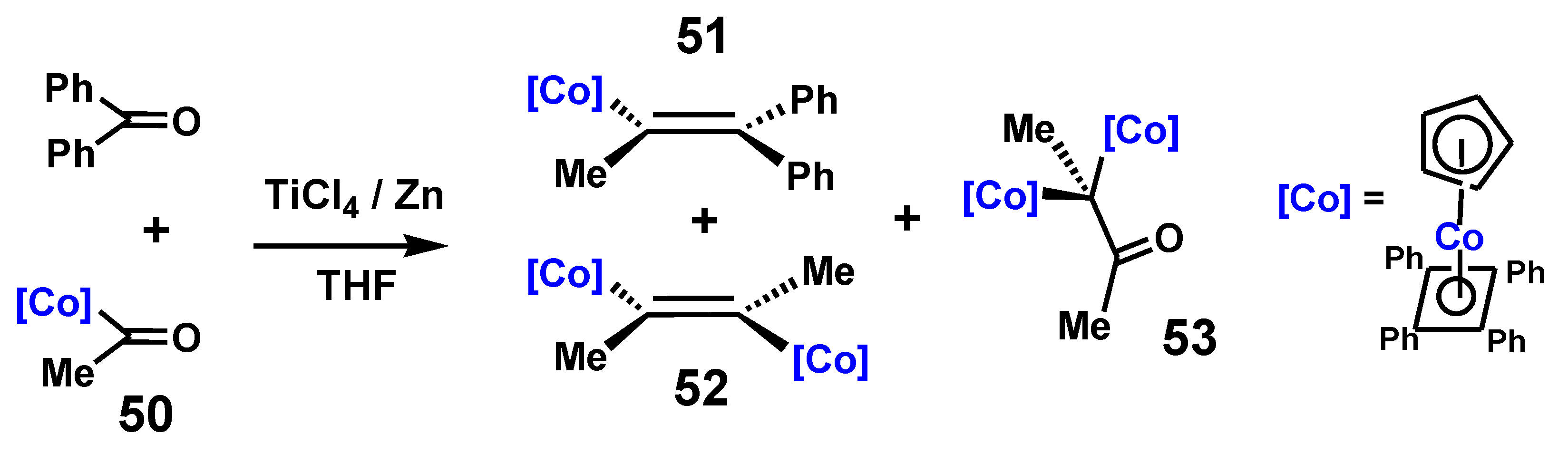
{kind=link}
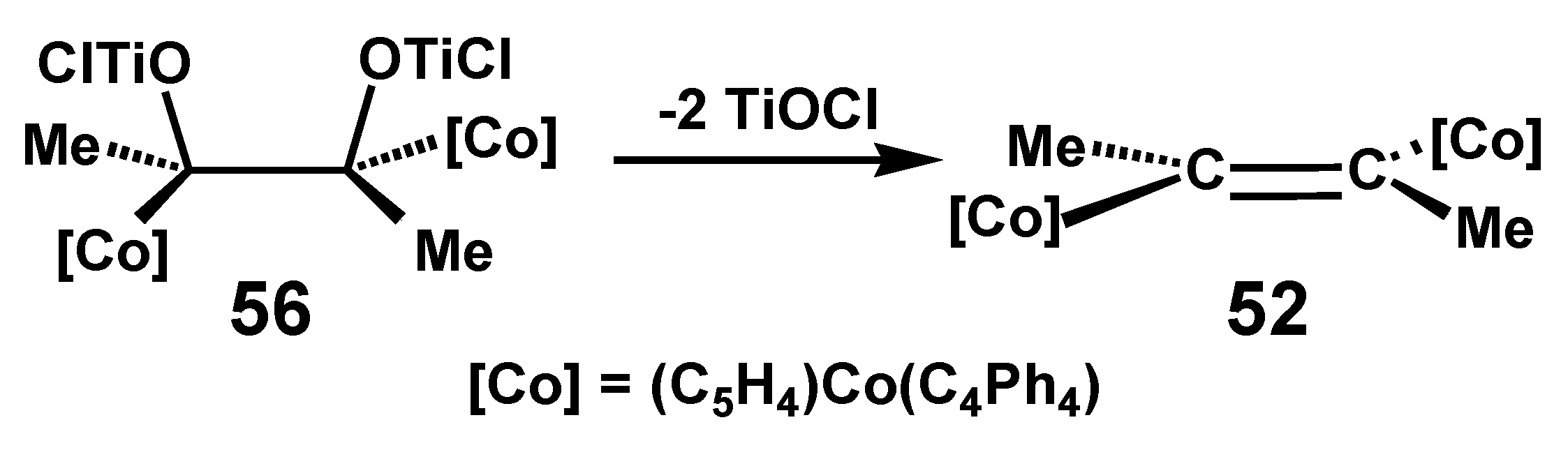{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Ferrocenyl-Mediated Carbocationic Rearrangements
2.1. Isomerizations of Secondary to Tertiary Carbocations
2.2. Rearrangements of Vinyl and Allyl Cations
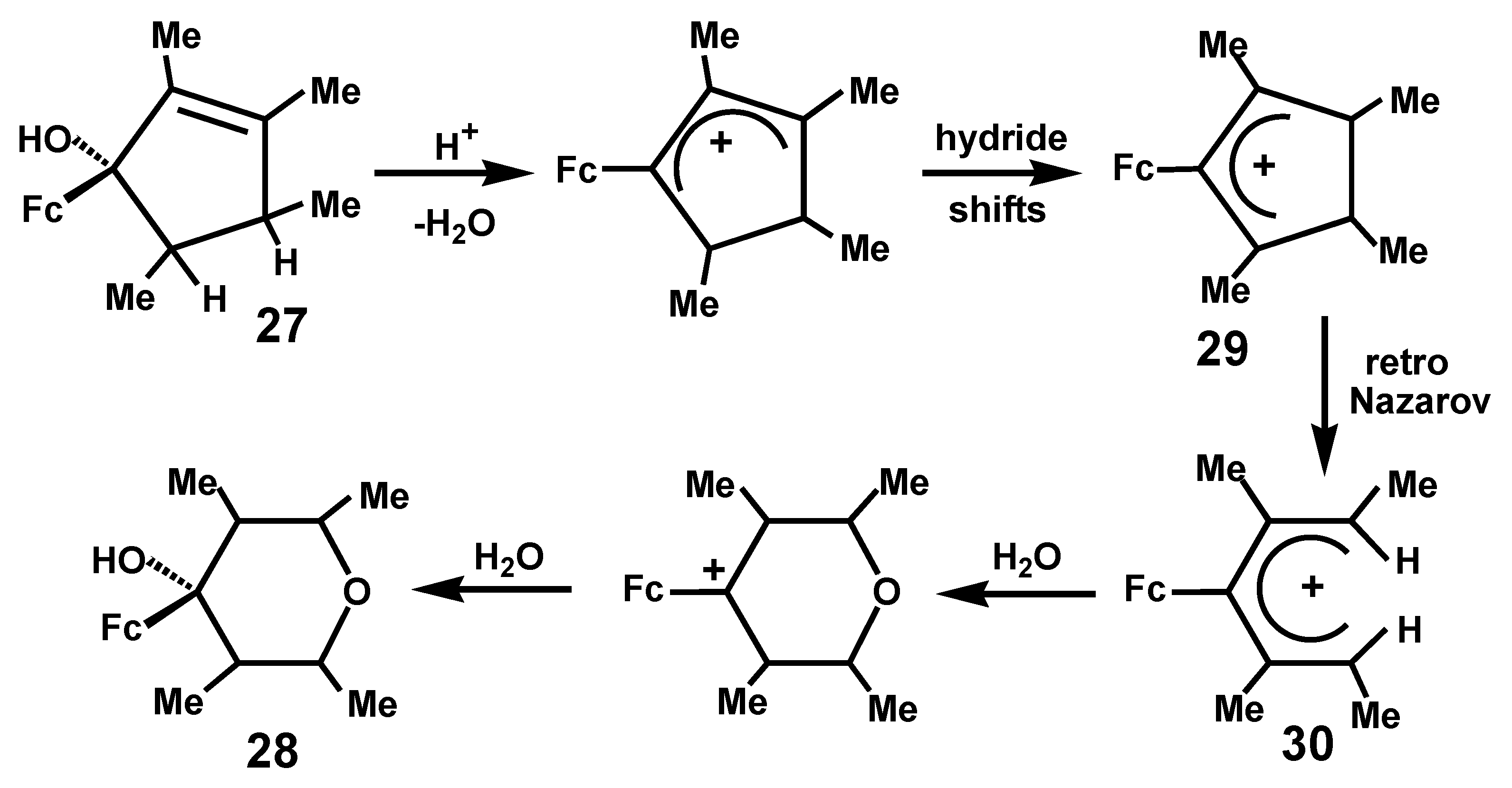
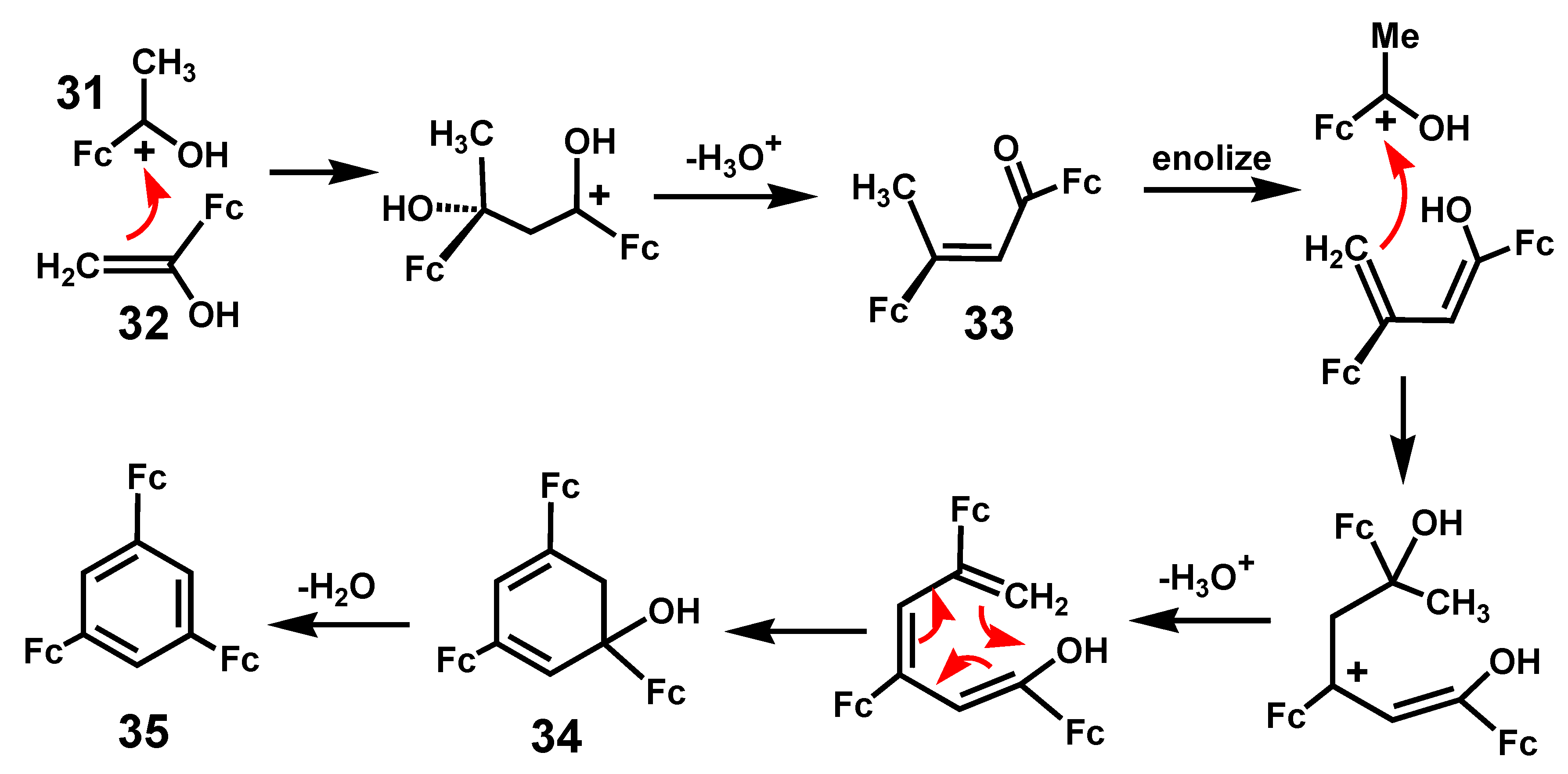
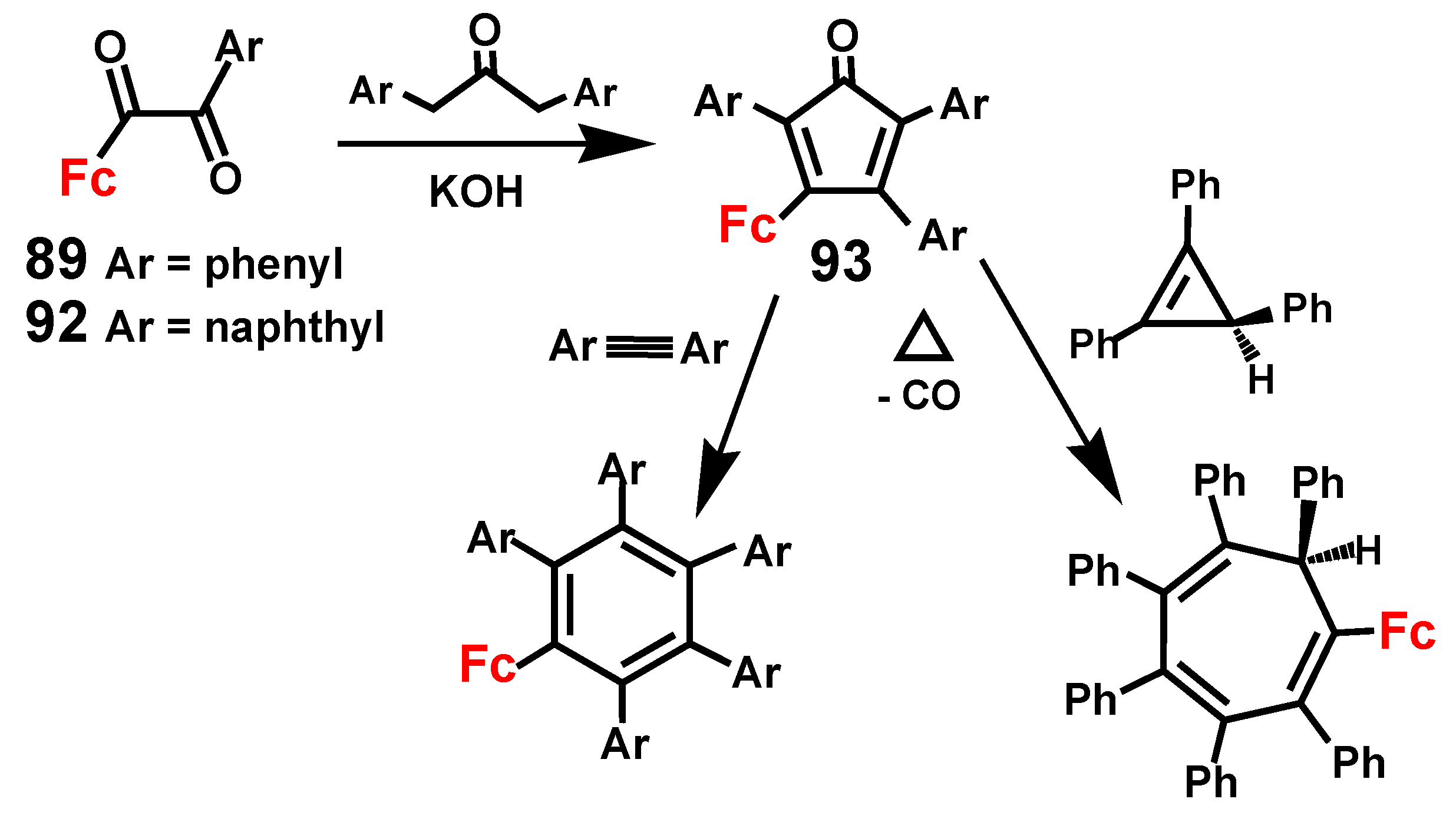
2.3. Ferrocene-Mediated Cyclization to form Trisubstituted Aromatics, 1,3,5-C6H3(MLn)3
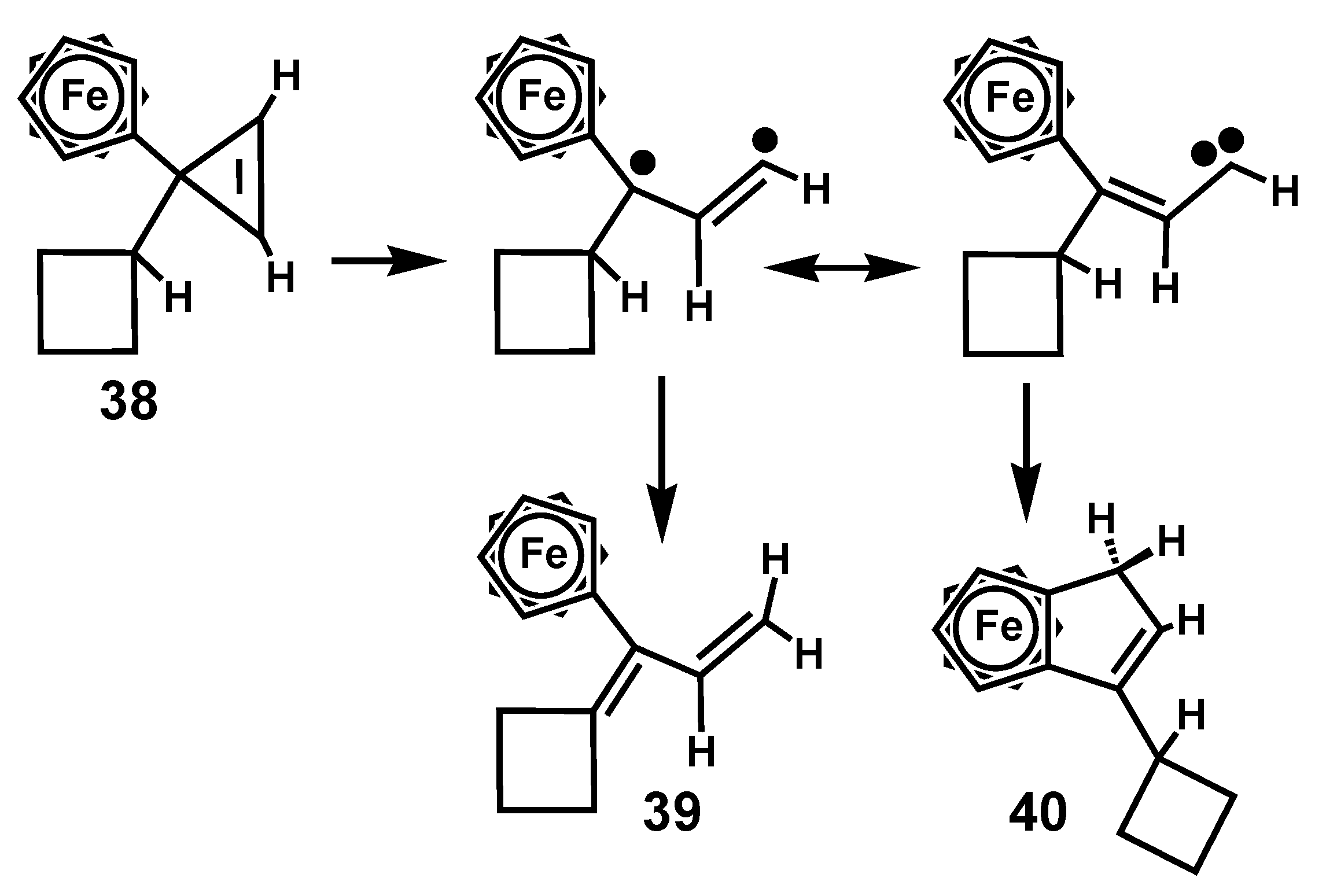
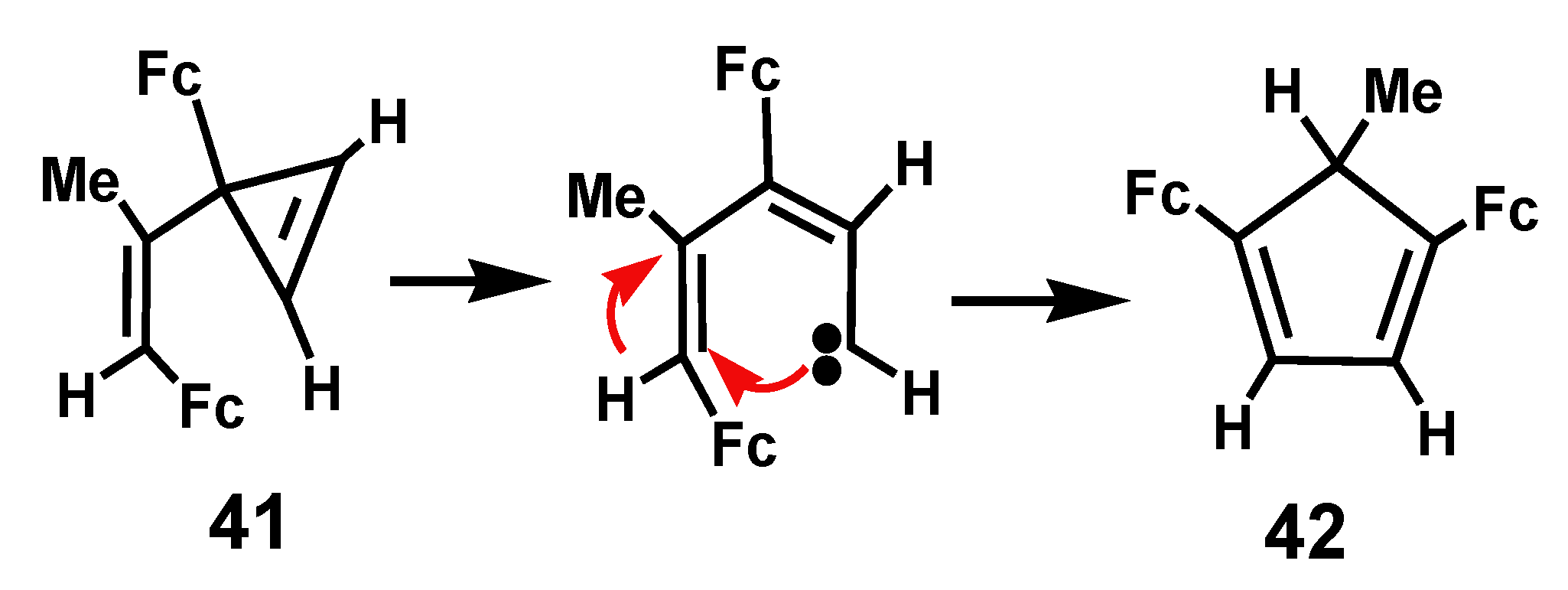
3. Rearrangements of Vinylcyclopropenes
4. Ferrocenyl Migration onto an Electron-Deficient Carbon Center
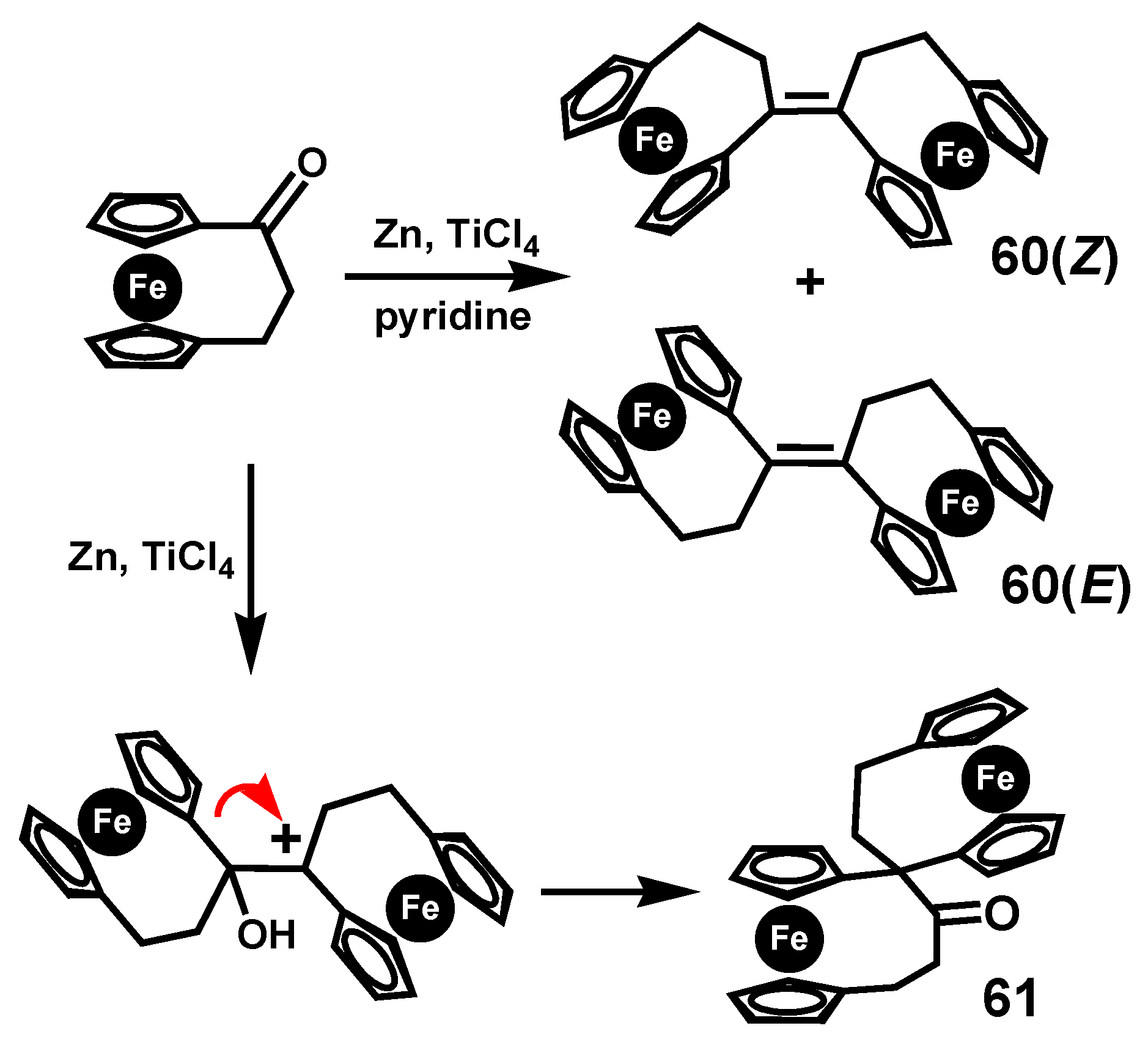
4.1. Pinacol Rearrangements
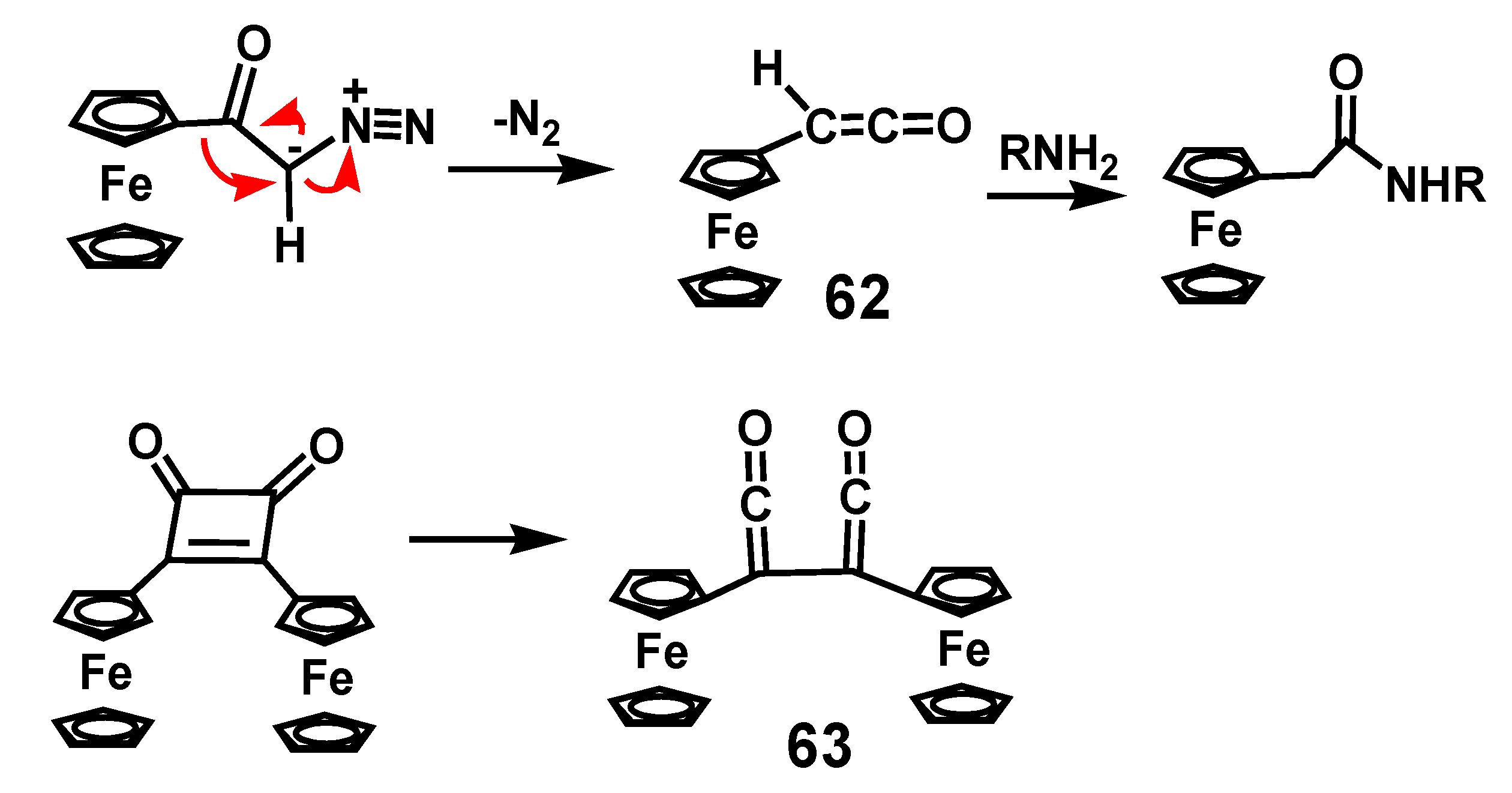
4.2. Wolff Rearrangements
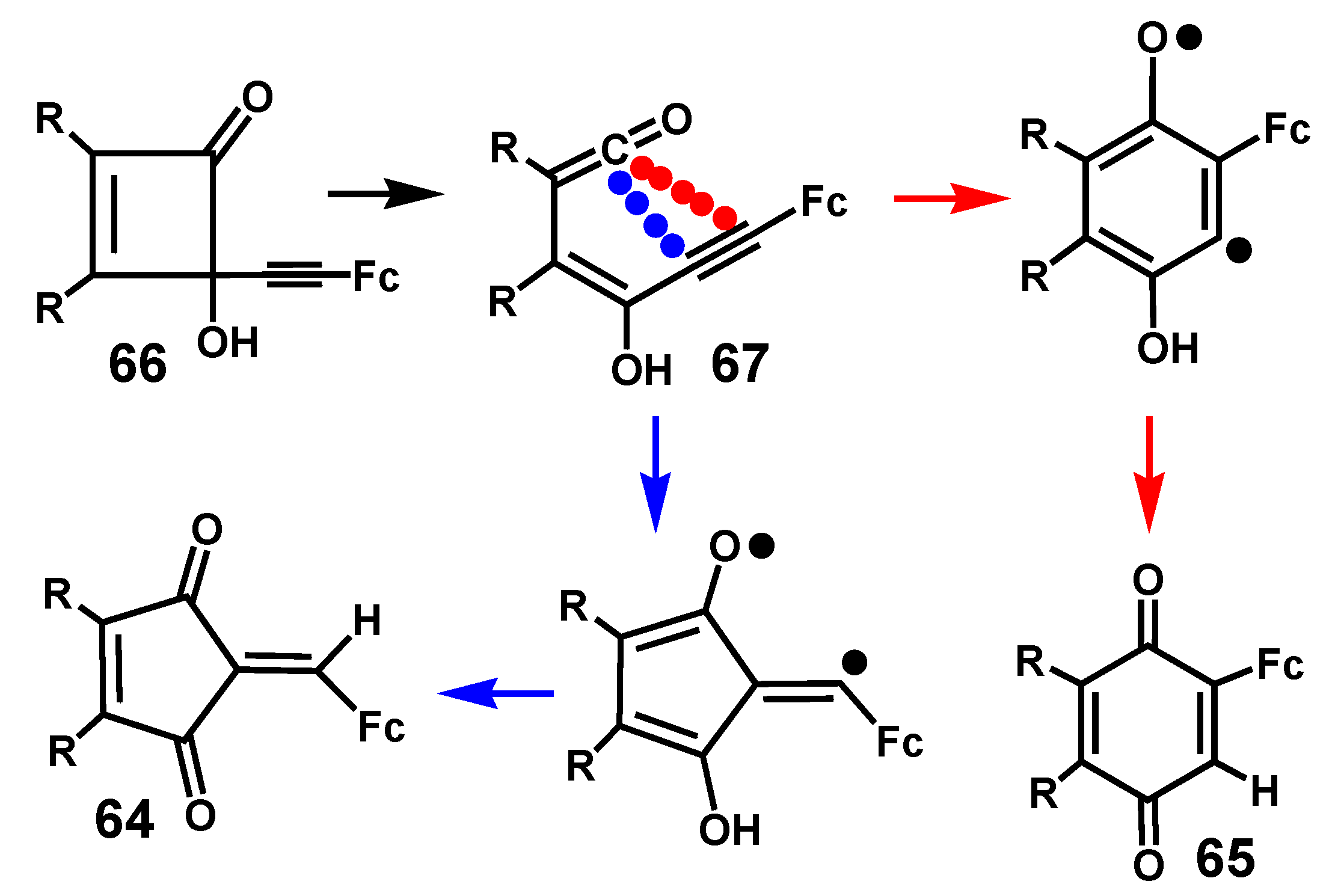
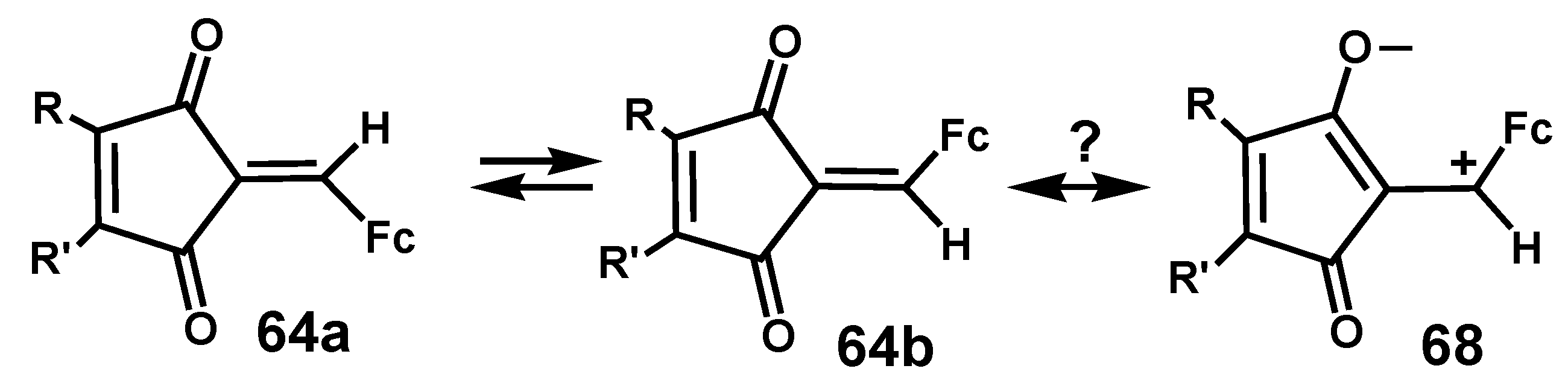
4.3. Rearrangements of Alkynyl-4-hydroxy-2-cyclobutenones
5. Ferrocenyl Migration onto an Electron-Deficient Nitrogen Center
5.1. Beckmann Rearrangements
5.2. Curtius Rearrangements
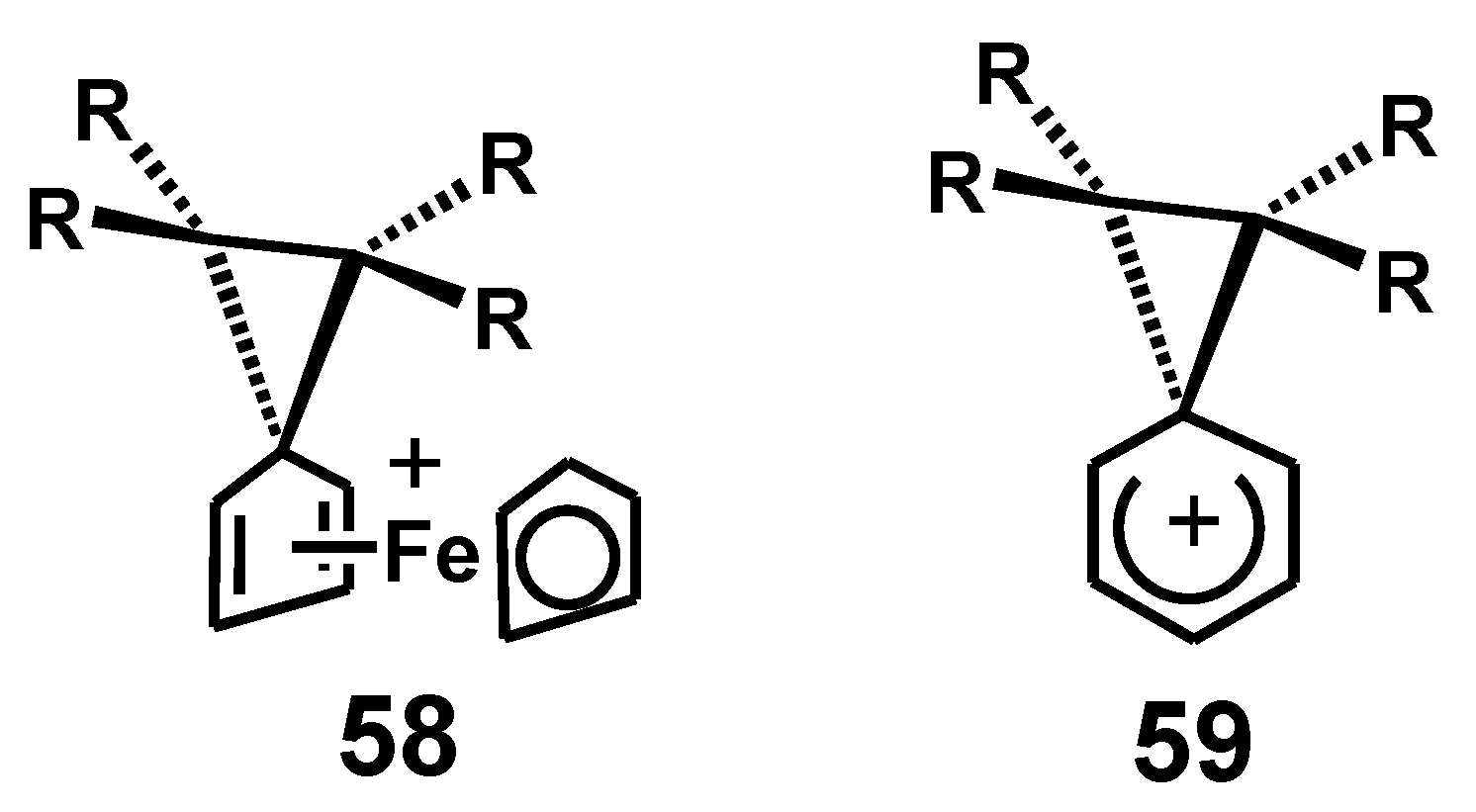
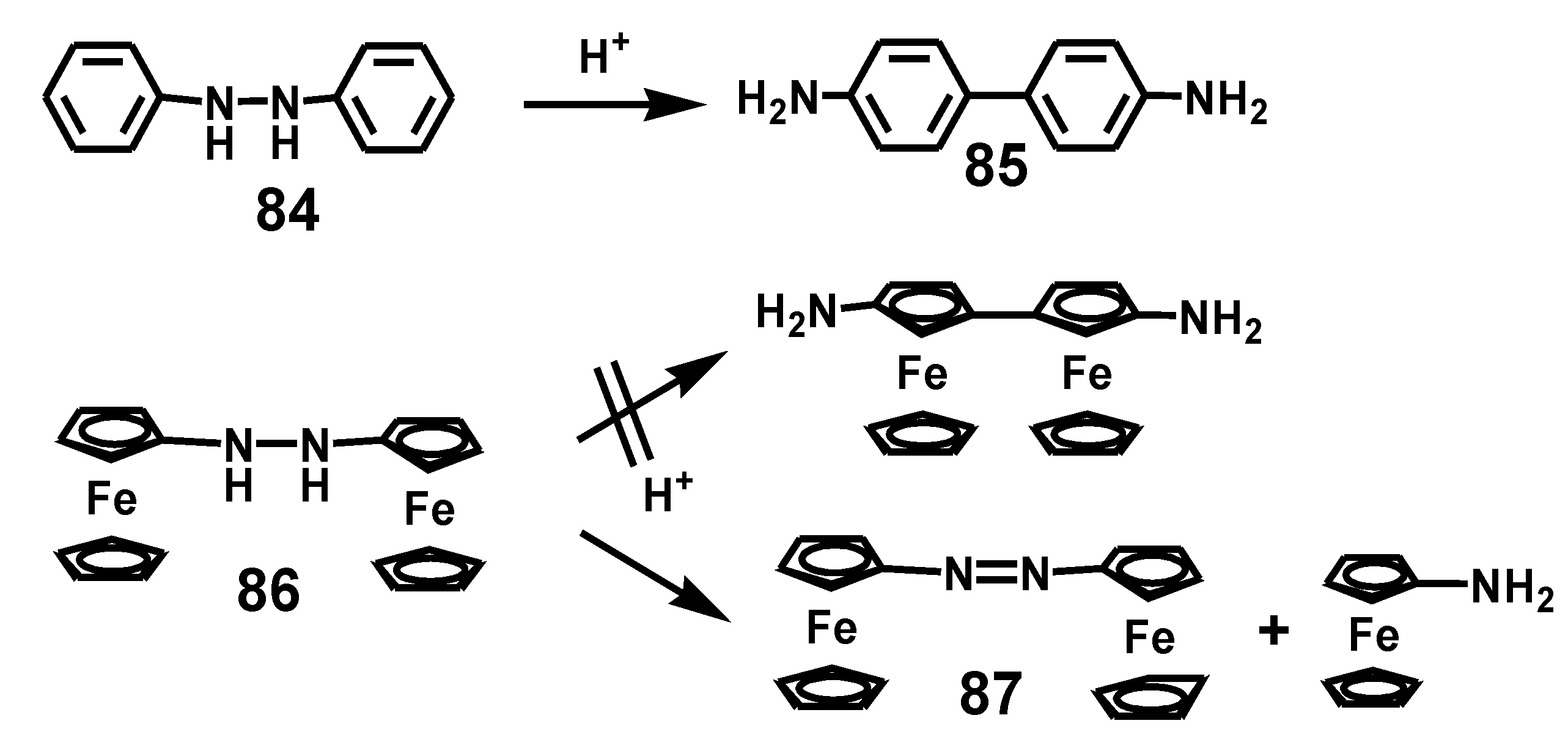
5.3. Benzidine Rearrangement
6. Anionic Rearrangements of Ferrocenyl Compounds
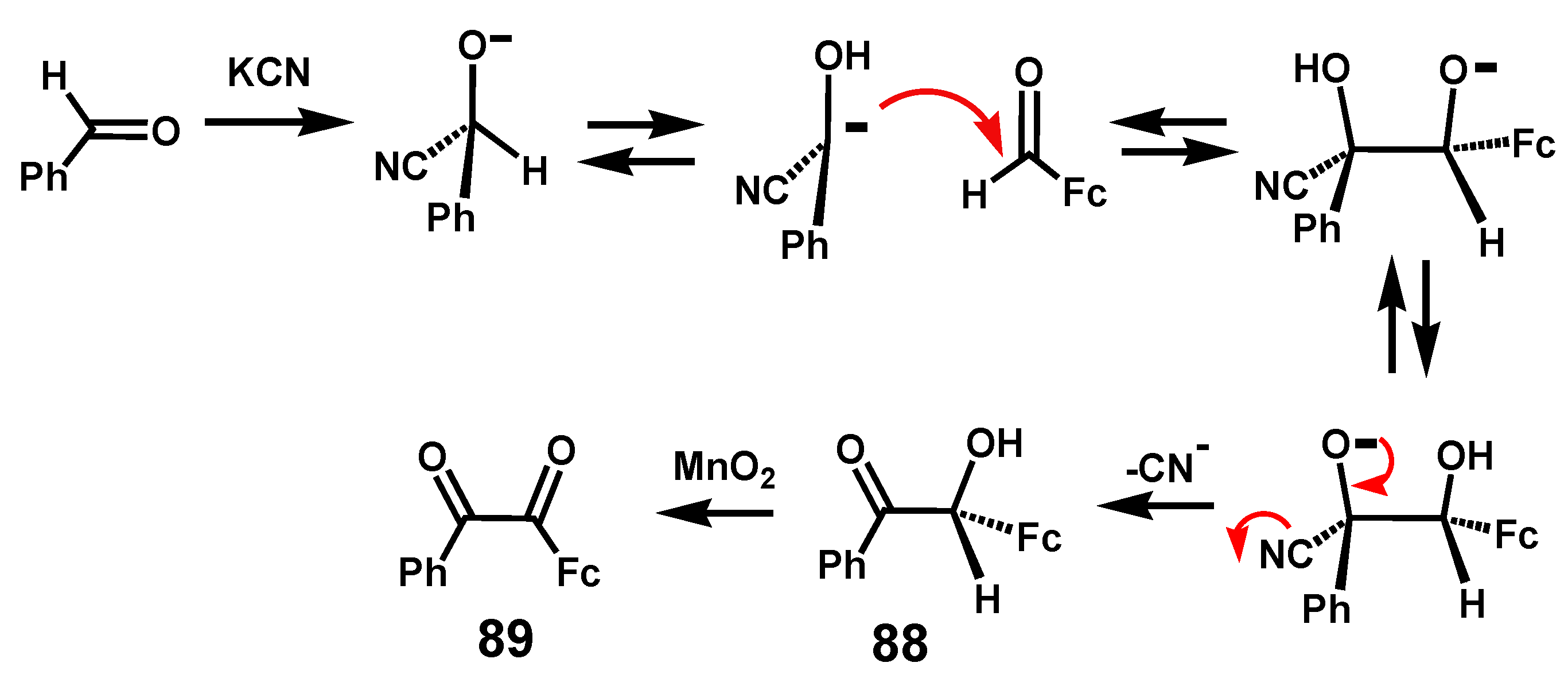
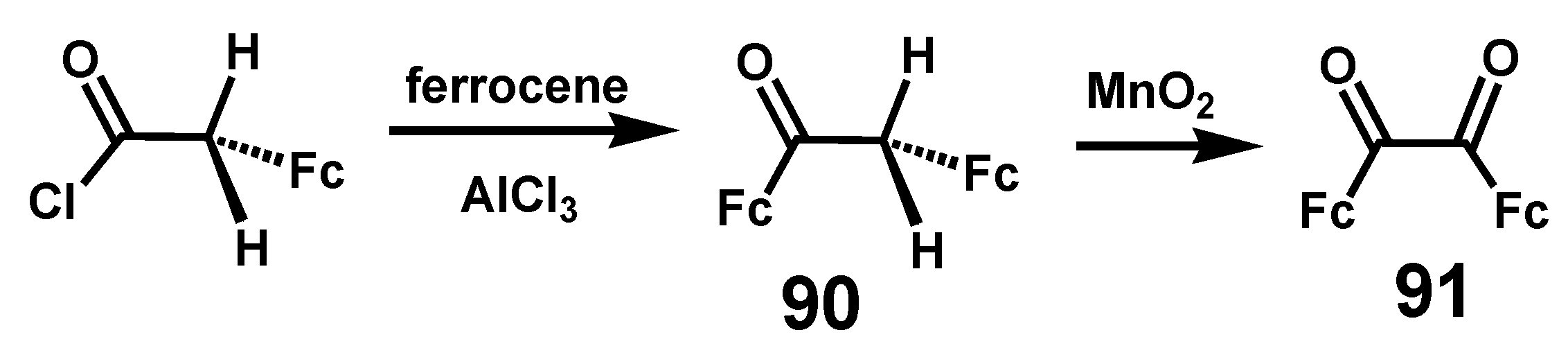
6.1. Benzoins and Benzils
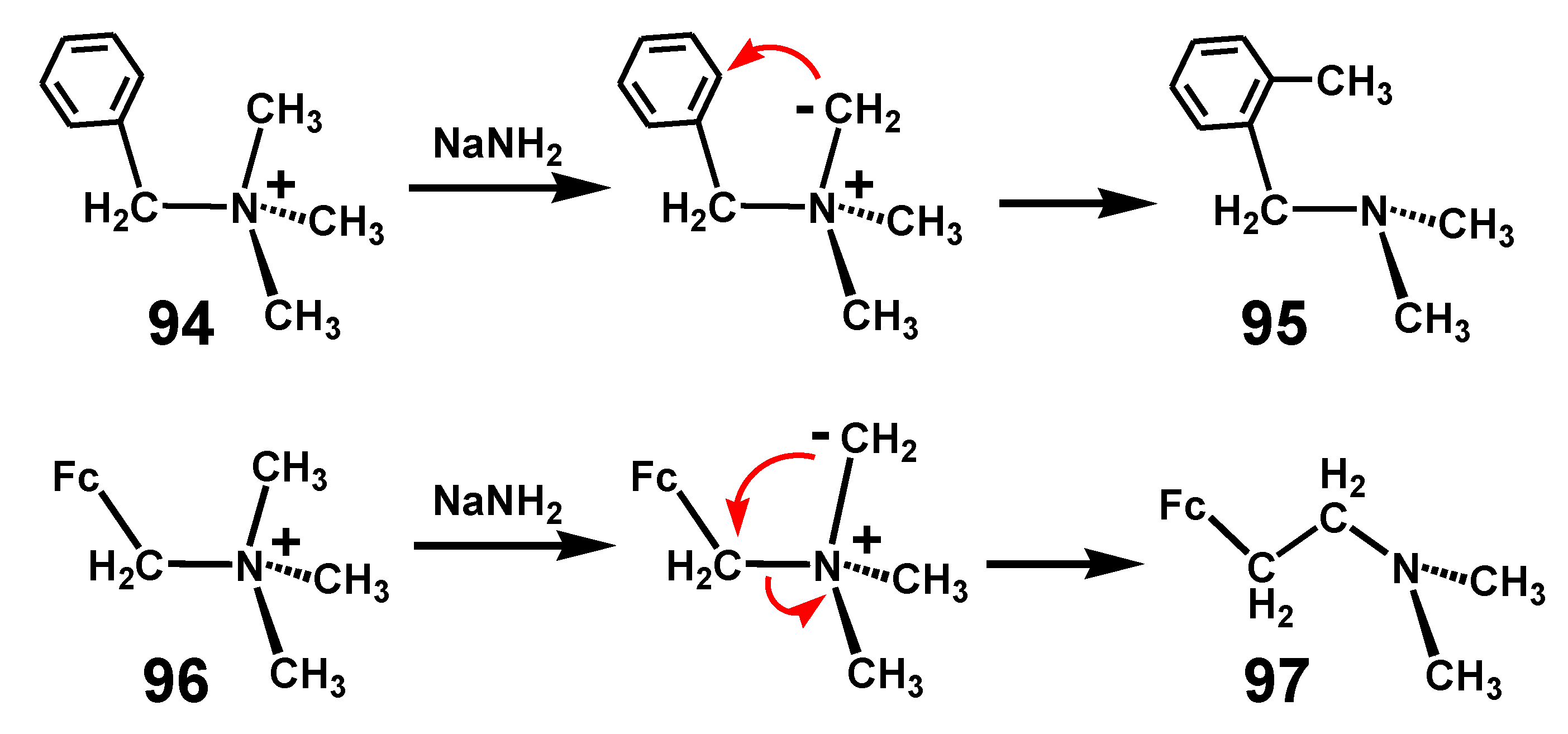
6.2. Stevens Rearrangement
7. Ferrocenes in Bioorganometallic Chemistry
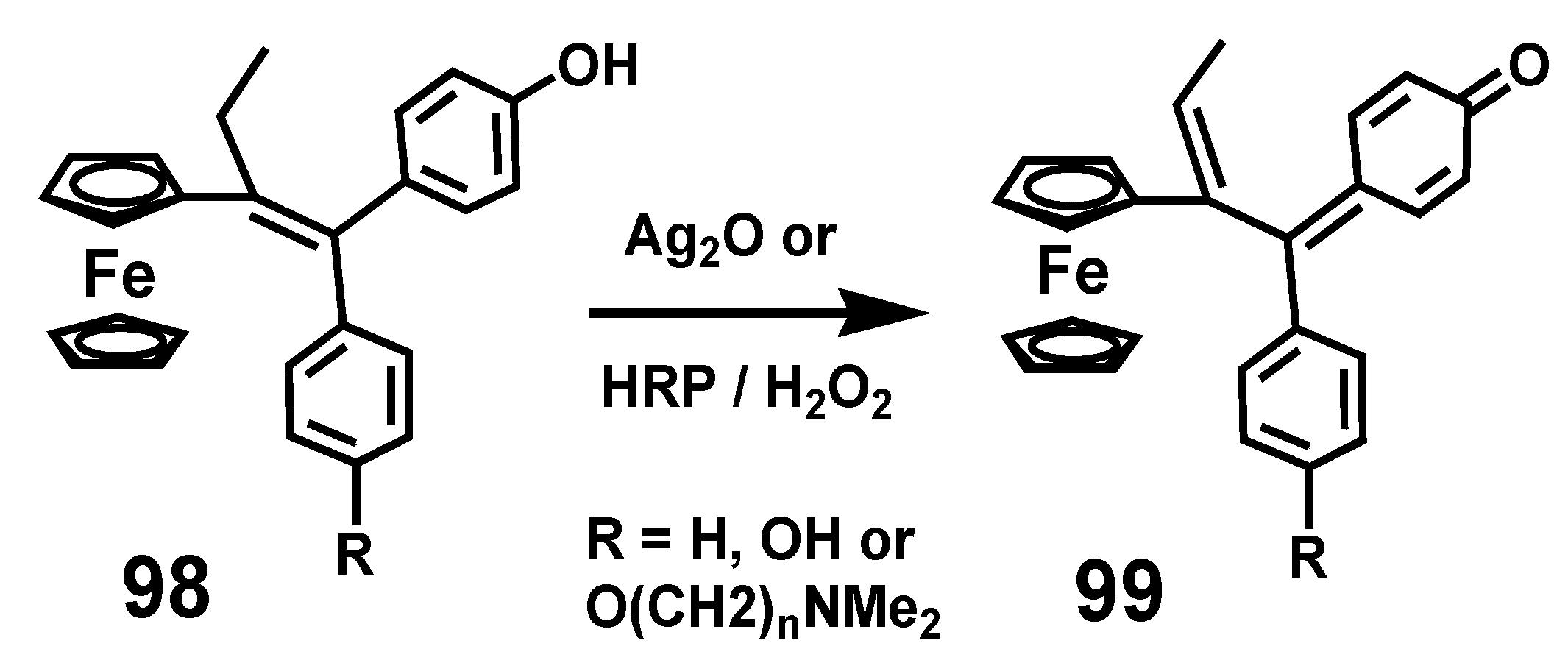
7.1. Quinone Methides
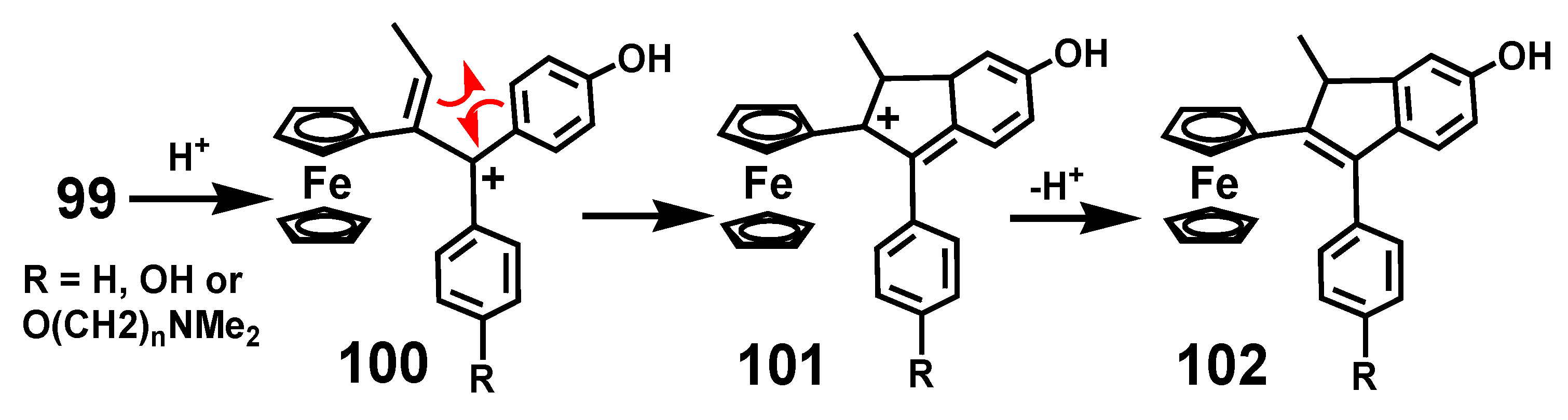
7.2. Formation of Indenes
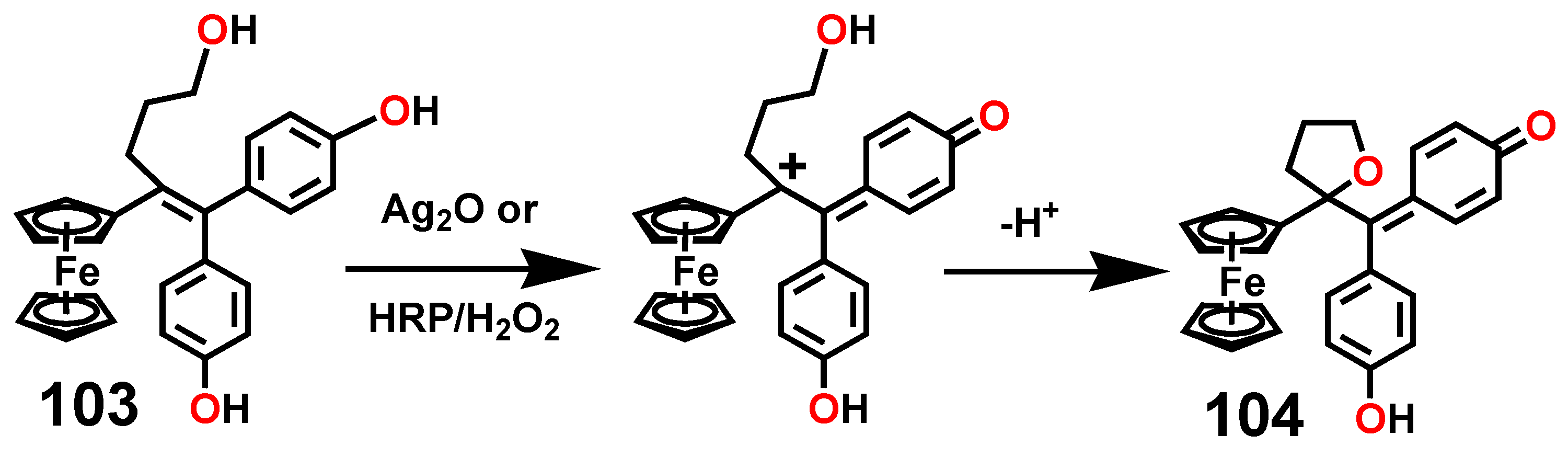
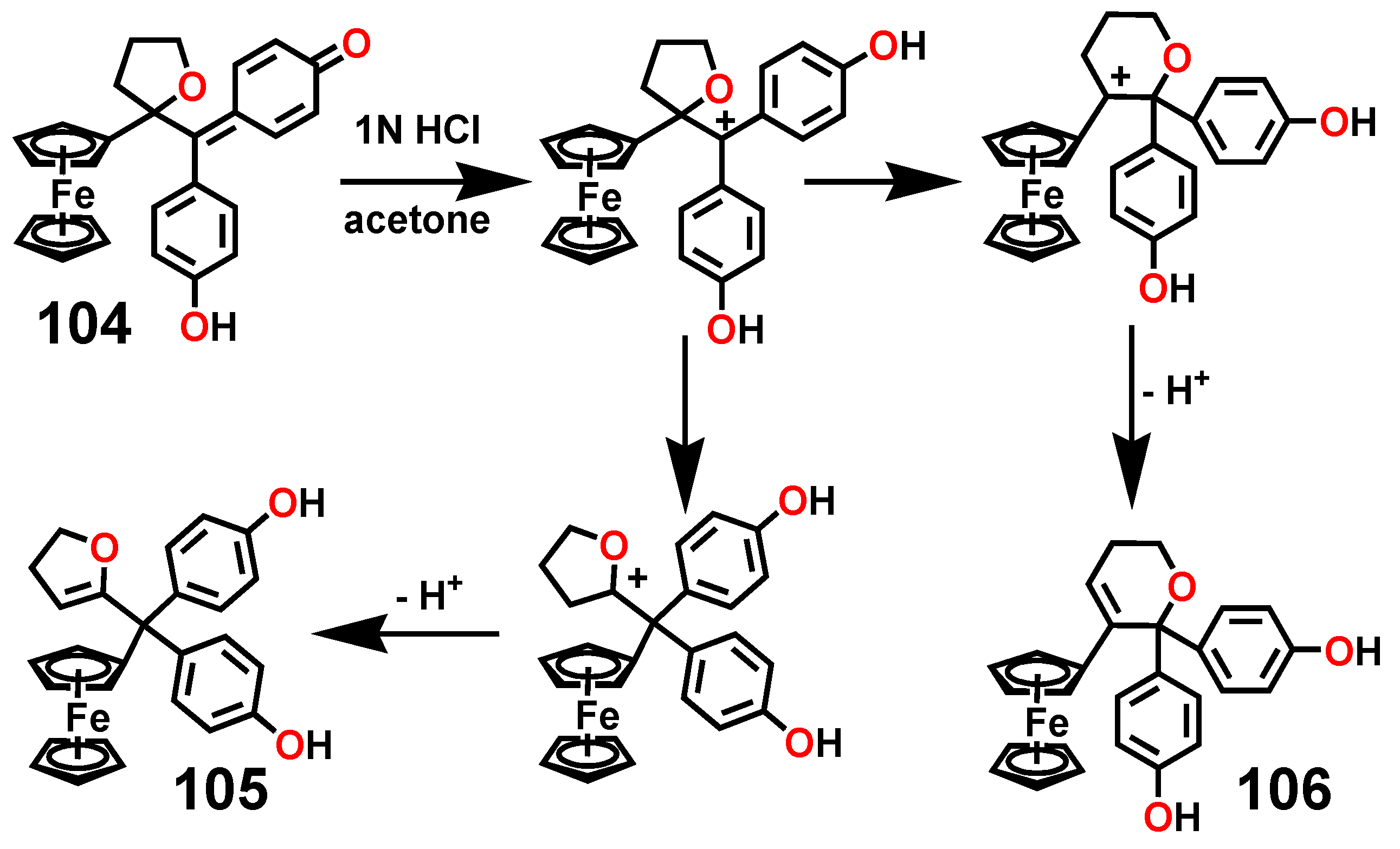
7.3. Formation and Rearrangement of a Ferrocenyl-Tetrahydrofuranyl Quinone Methide
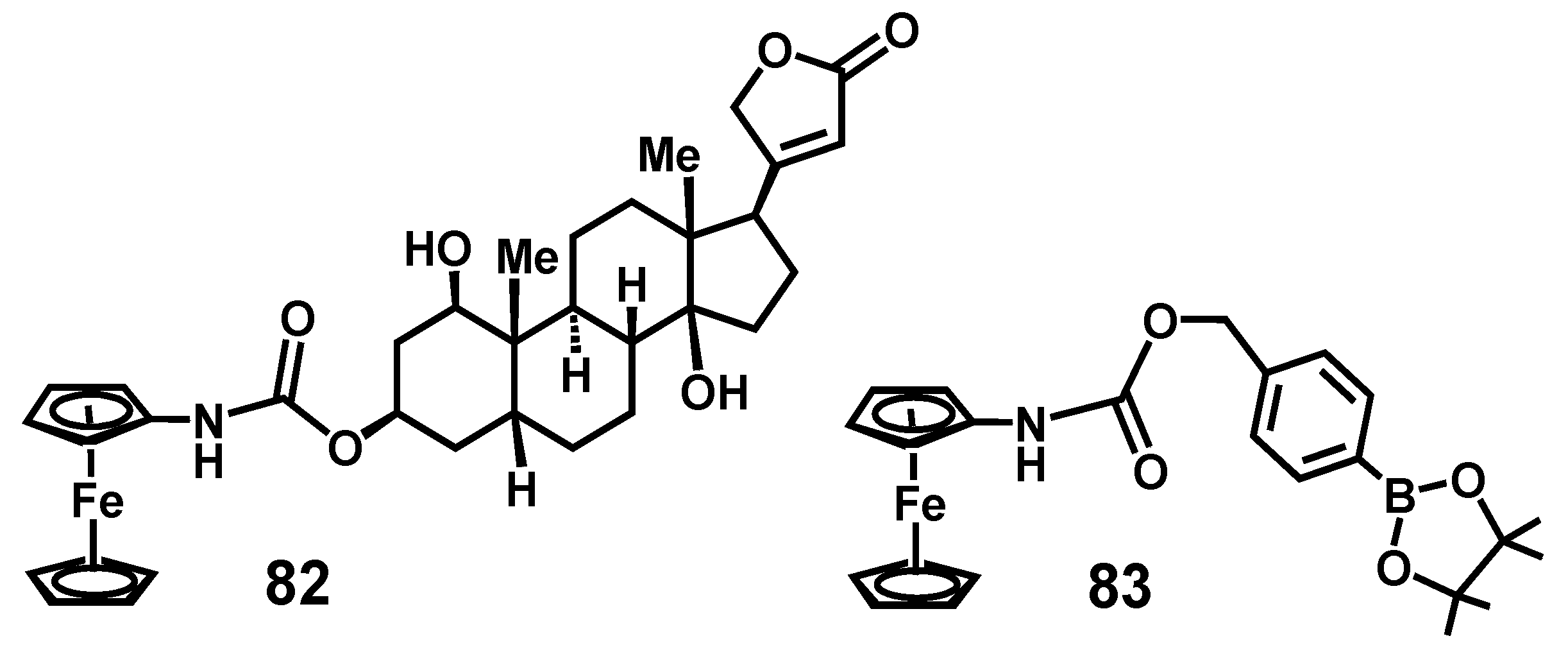
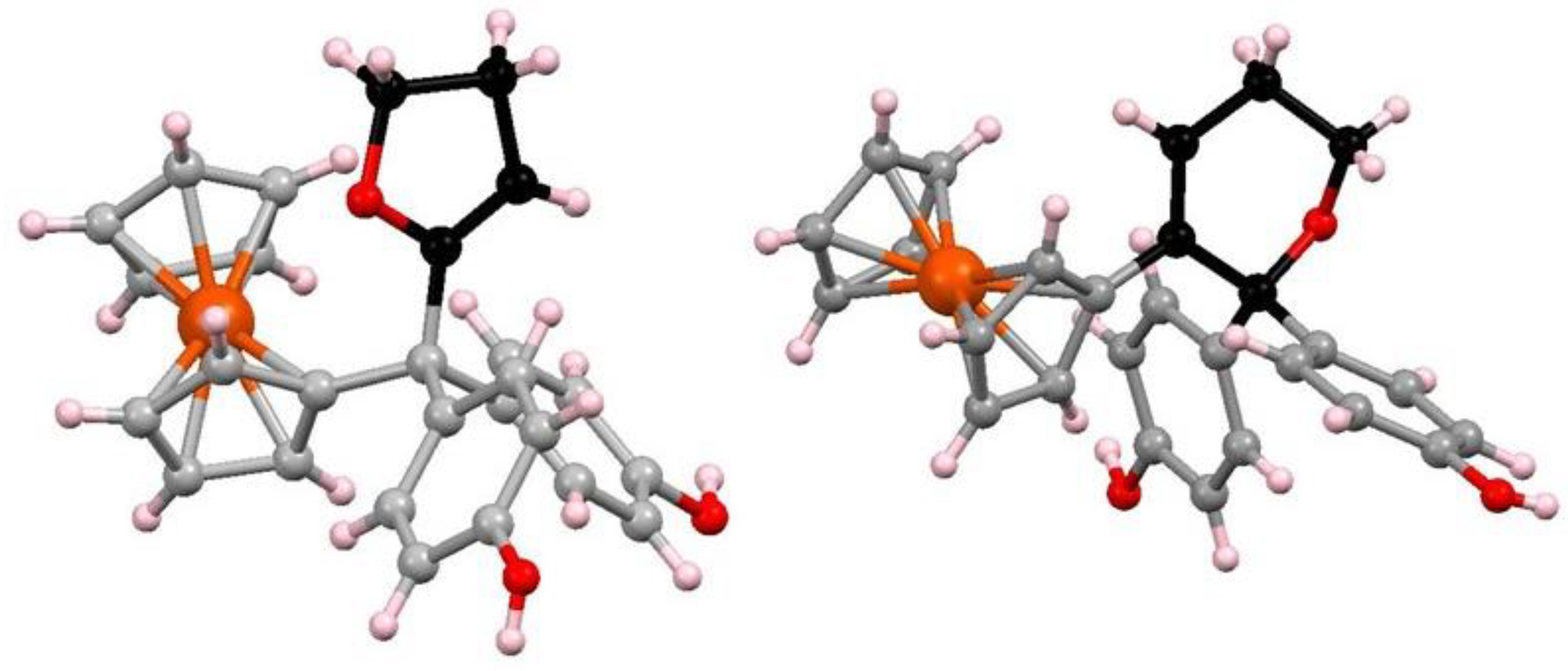
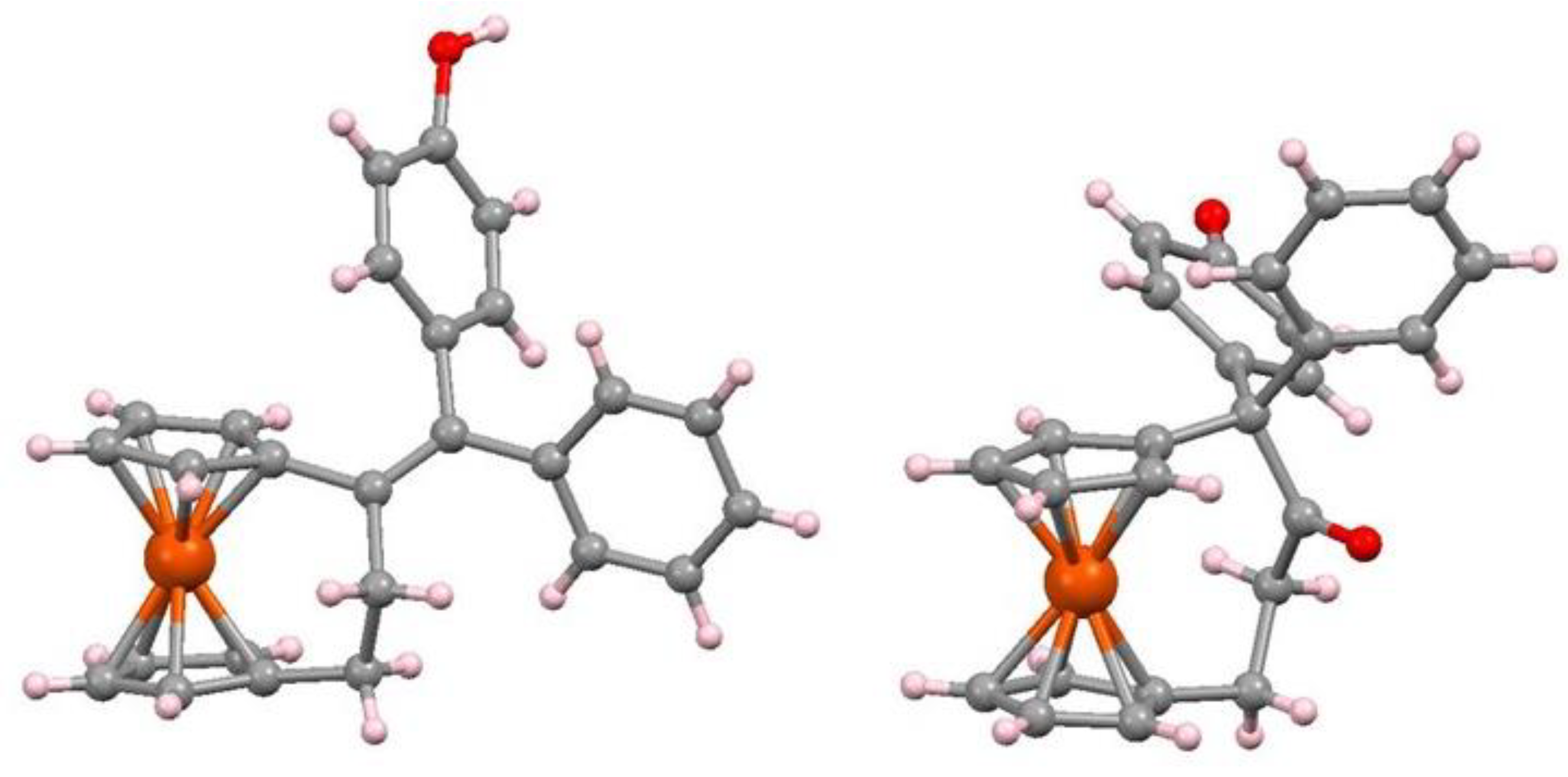
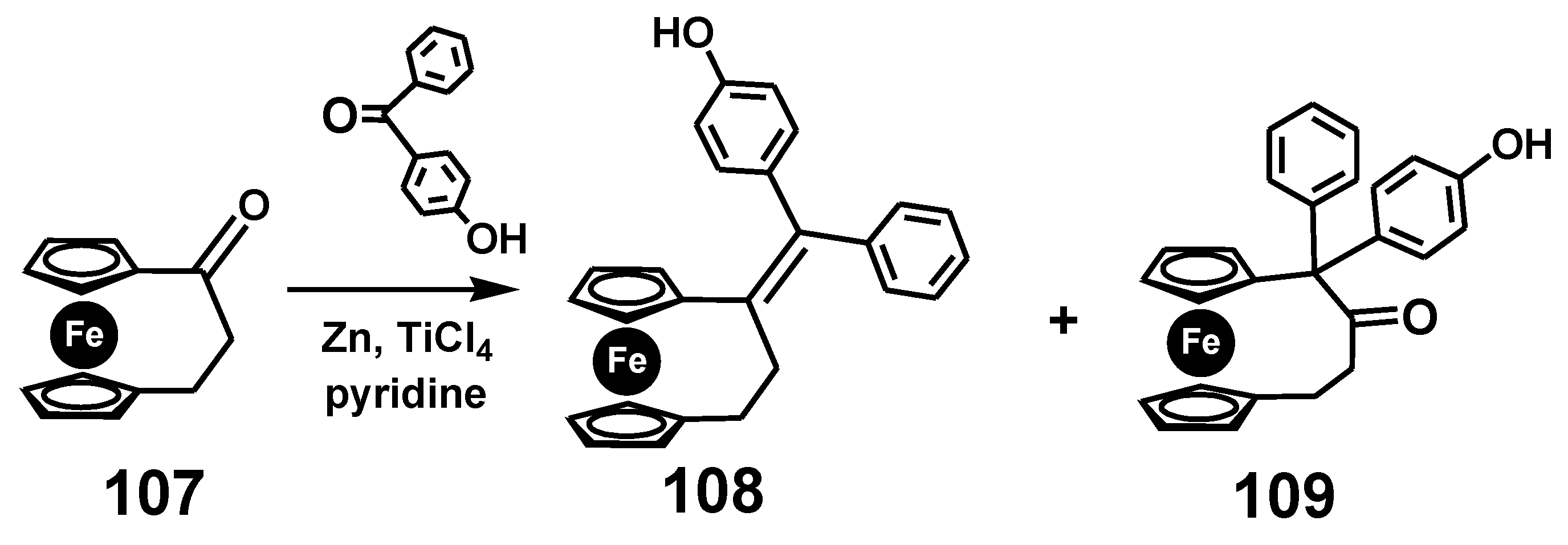
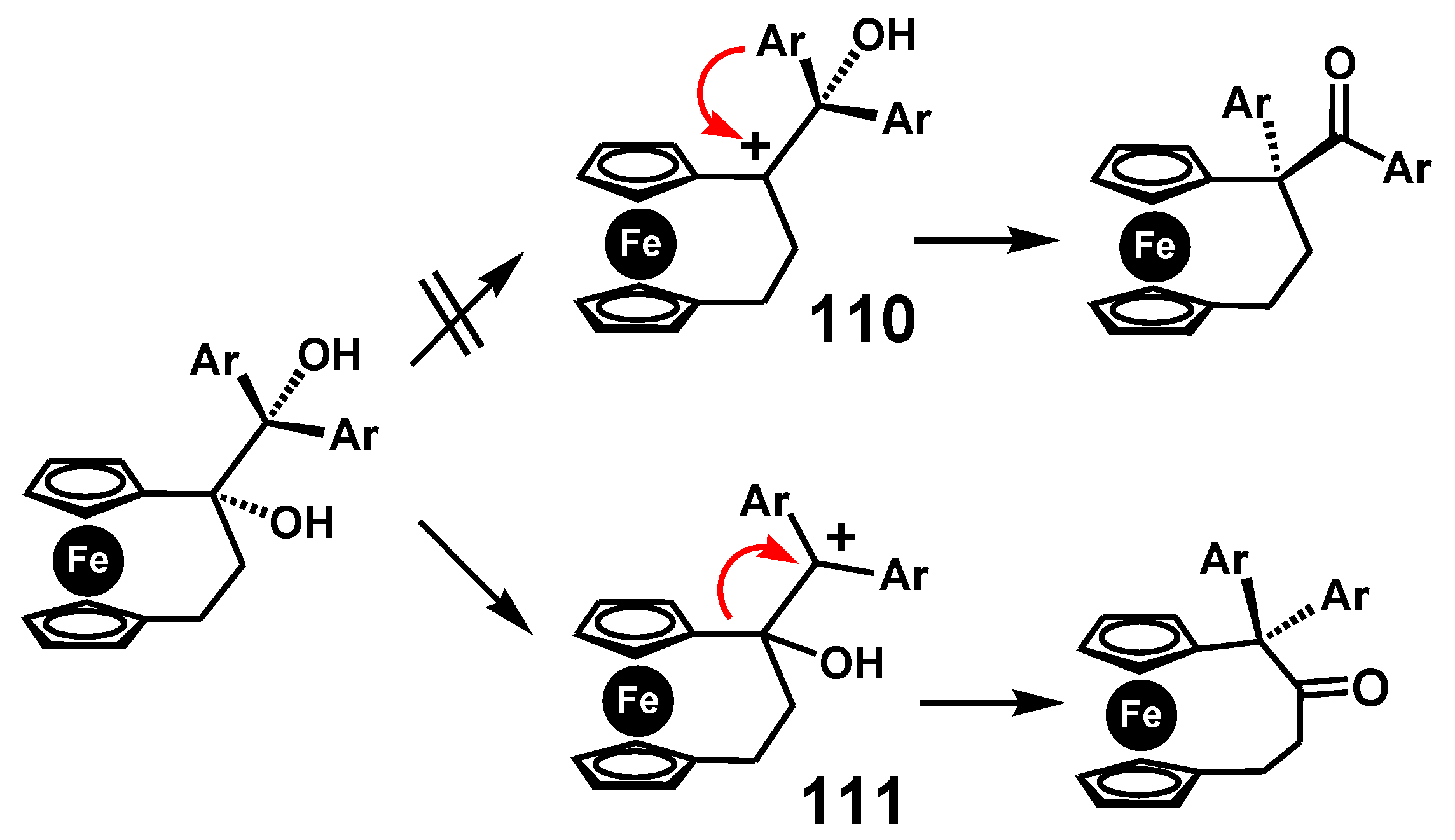
7.4. Pinacol-to-Pinacolone Rearrangement of Ansa-Ferrociphenols
8. Concluding Comments
Funding
Acknowledgments
Conflicts of Interest
References
- Richards, J.H.; Hill, E.A. Carbonium ion stabilization by metallocene nuclei. II. α-Metallocenylcarbonium ions. J. Am. Chem. Soc. 1961, 83, 3840–3846. [Google Scholar] [CrossRef]
- Lupan, S.; Kapon, M.; Cais, M.; Herbstein, F.H. Structure and properties of α,α-diferrocenylmethylium ion. Angew. Chem. Int. Ed. Engl. 1972, 11, 1025–1027. [Google Scholar] [CrossRef]
- Behrens, U. Transition metal-fulvene complexes. 14. Crystal and molecular structure of ferrocenyldiphenylcarbenium tetrafluoroborate—Fulvene-iron complex. J. Organometal. Chem. 1979, 182, 89–98. [Google Scholar] [CrossRef]
- Gleiter, R.; Seeger, R. Structure of ferrocenyl-methyl cation. Helv. Chim. Acta 1971, 54, 1217–1220. [Google Scholar] [CrossRef]
- Casper, L.A.; Osswald, S.; Anders, P.; Rosenbaum, L.-C.; Winter, R.F. Extremely electron-poor bis(diarylmethylium)-substituted ferrocenes and the first peroxoferrocenophane. Z. Anorg. Allg. Chem. 2020, 646, 1–15. [Google Scholar] [CrossRef]
- Turbitt, T.D.; Watts, W.E. Stable carbonium ions. Part IV. Torsional barriers in secondary and tertiary ferrocenylalkylium ions. J. Chem. Soc. 1974, 2, 177–184. [Google Scholar] [CrossRef]
- Gleiter, R.; Bleiholder, C.; Rominger, F. α-Metallocenylmethylium ions and isoelectronic fulvene complexes of d6 to d9 metals. Structural considerations. Organometallics 2007, 26, 4850–4859. [Google Scholar] [CrossRef]
- Cais, M.; Dannenberg, J.J.; Eisenstadt, A.; Levenberg, M.L.; Richards, J.H. Nuclear magnetic resonance spectra of ferrocenyl carbonium ions. Tetrahedron Lett. 1966, 7, 1695–1701. [Google Scholar] [CrossRef]
- Traylor, T.G.; Ware, J.C. The chemistry of metallocenes. Carbonium stabilization by the ferrocenyl group. J. Am. Chem. Soc. 1967, 89, 2304–2316. [Google Scholar] [CrossRef]
- Feinberg, J.; Rosenblum, M. The structure of α-ferrocenylcarbonium ions. J. Am. Chem. Soc. 1969, 91, 4324–4325. [Google Scholar] [CrossRef]
- Hisatome, M.; Yamakawa, K. Organometallic compounds IX. The structure of α-ferrocenylcarbonium ions. Tetrahedron 1971, 27, 2101–2110. [Google Scholar] [CrossRef]
- Watts, W.E. The organic chemistry of metal-coordinated cyclopentadienyl and arene ligands. In Comprehensive Organometallic Chemistry; Wilkinson, G., Stone, F.G.A., Abel, E.W., Eds.; Pergamon: Oxford, UK, 1982; Volume 8, Chapter 59; pp. 1051–1071. [Google Scholar]
- Turbitt, T.D.; Watts, W.E. Double-shift rearrangements of secondary ferrocenylcarbonium ions. J. Organometal. Chem. 1973, 57, C78–C79. [Google Scholar] [CrossRef]
- Abram, T.S.; Turbitt, T.D.; Watts, W.E. Stable carbocations. Part 15. Double-shift rearrangements of ferrocenylcarbenium ions. J. Chem. Soc. 1977, 13, 1536–1541. [Google Scholar] [CrossRef]
- Abram, T.S.; Watts, W.E. Stable carbocations. Part 13. Intra-ionic cyclisation reactions of ferrocenyl- and phenyl-stabilised vinyl cations. J. Chem. Soc. 1977, 13, 1527–1531. [Google Scholar] [CrossRef]
- Schottenberger, H.; Buchmeiser, M.R.; Angleitner, H.; Wurst, K.; Herber, R.H. Rearrangements and dimerizations of congested ferrocenyl allyl alcohols. J. Organometal. Chem. 2000, 605, 174–183. [Google Scholar] [CrossRef]
- Gupta, H.K.; Reginato, N.; Ogini, F.O.; Brydges, S.; McGlinchey, M.J. SiCl4-ethanol as a trimerization agent for organometallics: Convenient syntheses of the symmetrically substituted arenes 1,3,5-C6H3R3 where R is (C5H4)Mn(CO)3 and (C5H4)Fe(C5H5). Can. J. Chem. 2002, 80, 1546–1554. [Google Scholar] [CrossRef]
- Štěpnička, P.; Císařová, I.; Sedláček, J.; Vohlídal, J.; Polásek, M. Synthesis of triferrocenylbenzenes by tantalum(V)-catalyzed cyclotrimerization of ethynylferrocene. The crystal structure of 1,3,5-triferrocenylbenzene. Collect. Czech. Chem. Commum. 1997, 62, 1577–1584. [Google Scholar] [CrossRef]
- Fiorentini, S.; Floris, B.; Galloni, P.; Grepioni, F.; Polito, M.; Tagliatesta, P. The metal-catalyzed cyclotrimerization of ferocenylethyne—Preparation and characterization of 1,2,4-triferrocenylbenzene. Eur. J. Org. Chem. 2006, 7, 1726–1732. [Google Scholar] [CrossRef]
- Ogini, F.O.; Ortin, Y.; Mahmoudkhani, A.H.; Cozzolino, A.F.; McGlinchey, M.J.; Vargas-Baca, I. An investigation of the formation of 1,3,5-heterosubstituted benzene rings by cyclocondensation of acetyl-substituted organometallic complexes. J. Organometal. Chem. 2008, 693, 1957–1967. [Google Scholar] [CrossRef]
- Top, S.; Jaouen, G.; Sayer, B.G.; McGlinchey, M.J. Asymmetric induction via co-cordination to Cr(CO)3: Nucleophilic attack on acyclic carbenium ions. J. Chem. Soc. Chem. Comm. 1980, 23, 1110–1112. [Google Scholar] [CrossRef]
- Top, S.; Jaouen, G.; Sayer, B.G.; McGlinchey, M.J. Rotational barriers in diphenylmethyl anions stabilized by trimethylsilyl and tricarbonylchromium(0) moieties. J. Am. Chem. Soc. 1983, 105, 6426–6429. [Google Scholar] [CrossRef]
- Klimova, E.I.; Garcia, M.M.; Klimova, T.; Ramirez, L.R.; Meleshonkova, N.N. 3-Cyclo-butyl- 3-ferrocenylcyclopropene and 3-cyclobutylidene-3-ferrocenylpropyne. Synthesis and chemical properties. Russ. Chem. Bull. 1999, 48, 2153–2157. [Google Scholar] [CrossRef]
- Méndez, D.; Klimova, E.; Klimova, T.; Hernández, O.S.; Martínez, G.M. Synthesis of ferrocenylvinylcyclopropene and its transformation into cyclopentadiene. J. Organometal. Chem. 2003, 681, 115–119. [Google Scholar] [CrossRef]
- Harwood, L.M. Polar Rearrangements; Oxford University Press: Oxford, UK, 1992; pp. 9–10, 49. [Google Scholar]
- Bachmann, W.E.; Sternberger, H.R. The pinacol-pinacolone rearrangement. V. The rearrangement of unsymmetrical aromatic pinacols. J. Am. Chem. Soc. 1934, 56, 170–173. [Google Scholar] [CrossRef]
- Weliky, N.; Gould, E.S. Studies in the ferrocene series. I. Some reactions of compounds related to monobenzoylferrocene. J. Am. Chem. Soc. 1957, 79, 2742–2746. [Google Scholar] [CrossRef]
- Pauson, P.L.; Watts, W.E. Ferrocene derivatives. Part XII. Di-and tri-ferrocenylmethane derivatives. J. Chem. Soc. 1962, 3880–3886. [Google Scholar] [CrossRef]
- Moffett, L.R., Jr. Pinacol and pinacolone derivatives of some acylferrocenes. J. Org. Chem. 1964, 29, 3726–3727. [Google Scholar] [CrossRef]
- Rausch, M.D.; Adams, D.L. The Clemmensen reduction of benzoylferrocene. J. Org. Chem. 1967, 32, 4144–4145. [Google Scholar] [CrossRef]
- Goldberg, S.I.; Bailey, W.D. Evidence for appreciable phenyl migration in the rearrangements of threo and erythro-1,2-diferroceny-1,2-diphenylethane-1,2-diols. Chem. Commun. 1969, 18, 1059–1060. [Google Scholar] [CrossRef]
- Denifl, P.; Hradsky, A.; Bildstein, B.; Wurst, K. Trimethylchlorosilane-modified Clemmensen reduction of metallocenyl ketones: Trapping and X-ray structures of aliphatic, olefinic, silylated pinacolic, and rearranged pinacolonic products. J. Organometal. Chem. 1996, 523, 79–91. [Google Scholar] [CrossRef]
- Bildstein, B. Cationic and neutral cumulene sp-carbon chains with ferrocenyl termini. Coord. Chem. Rev. 2000, 206, 369–394. [Google Scholar] [CrossRef]
- Ortin, Y.; Grealis, J.; Scully, C.; Müller-Bunz, H.; Manning, A.R.; McGlinchey, M.J. McMurry reactions of (η5-acetylcyclopentadienyl)cobalt(η4-tetraphenylcyclobutadiene) with benzophenone: Ketone couplings and a pinacol/pinacolone rearrangement. J. Organometal. Chem. 2004, 689, 4683–4690. [Google Scholar] [CrossRef]
- Nikitin, K.; Ortin, Y.; Müller-Bunz, H.; Plamont, M.-A.; Jaouen, G.; Vessières, A.; McGlinchey, M.J. Organometallic SERMS [selective estrogen receptor modulators]: Cobaltifens, the (cyclobutadiene)cobalt analogues of hydroxytamoxifen. J. Organometal. Chem. 2010, 695, 595–608. [Google Scholar] [CrossRef]
- Jaouen, G.; Vessières, A.; Top, S. Ferrocifen Type Anti Cancer Drugs. Chem. Soc. Rev. 2015, 44, 8802–8817. [Google Scholar] [CrossRef] [PubMed]
- Bogdanovic, B.; Bolte, A. A comparative study of the McMurry reaction utilizing [HTiCl(THF)~0.5]x, TiCl3(DME)1.5-Zn(Cu) and TiCl2·LiCl as coupling reagents. J. Organometal. Chem. 1995, 502, 109–121. [Google Scholar] [CrossRef]
- Dams, R.; Malinowski, M.; Westdorp, I.; Geise, H.Y. On the mechanism of the titanium-induced reductive couplings to olefins. J. Org. Chem. 1982, 47, 248–259. [Google Scholar] [CrossRef]
- Härter, P.; Latzel, K.; Spiegler, M.; Herdtweck, E. Migration of the ferrocenyl moiety during the McMurry coupling of ferrocenyl ketones. Polyhedron 1998, 17, 1141–1148. [Google Scholar] [CrossRef]
- Ye, T.; McKervey, M.A. Organic synthesis with α-diazo carbonyl compounds. Chem. Rev. 1994, 94, 1091–1160. [Google Scholar] [CrossRef]
- Aguilar-Aguilar, A.; Allen, A.D.; Cabrera, E.P.; Federov, A.; Fu, N.; Henry-Riyad, H.; Leuninger, J.; Schmid, U.; Tidwell, T.T.; Verma, R. Ferrocenylketene and ferrocenyl-1,2-bisketenes: Direct observation and reactivity measurements. J. Org. Chem. 2005, 70, 9556–9561. [Google Scholar] [CrossRef]
- Inayama, S.; Mamoto, K.; Shibata, T.; Hirose, T. Structure and antitumor activity relationship of 2-arylidene-4-cyclopentene-1,3-diones and 2-arylideneindan-1,3-diones. J. Med. Chem. 1976, 19, 433–436. [Google Scholar] [CrossRef]
- Zora, M.; Kokturk, M.; Eralp, T. Synthesis of 2-ferrocenylidene-4-cyclopentene-1,3-diones. Tetrahedron 2006, 62, 10344–10351. [Google Scholar] [CrossRef]
- Banide, E.V.; Ortin, Y.; Chamiot, B.; Cassidy, A.; Niehaus, J.; Moore, A.; Seward, C.M.; Müller-Bunz, H.; McGlinchey, M.J. Syntheses, structures and dimerizations of ferrocenyl-and fluorenylideneallenes: Push-pull multiple bonds? Organometallics 2008, 27, 4173–4182. [Google Scholar] [CrossRef]
- Yamakawa, K.; Hisatome, M. The stereochemistry of acylferrocene oxime derivatives. Tetrahedron 1970, 26, 4483–4489. [Google Scholar] [CrossRef]
- Schlögl, K.; Mechtler, H. Über die Umlagerung von Acylferrocen-oximen bei der Reduktion mit Lithiumalanat-Aluminiumchlorid. Monatsh. 1966, 97, 150–167. [Google Scholar] [CrossRef]
- Hetnarski, B.; Lajtha, A.; Wisniewski, H.M. On some derivatives of ferrocene, novel acetylcholinesterase inhibitors. J. Neurosci. Res. 1980, 5, 1–5. [Google Scholar] [CrossRef]
- Schlögl, K.; Seiler, H. Ferrocenyl-isocyanat. Naturwiss 1958, 45, 337. [Google Scholar] [CrossRef]
- Shimada, K.; Orii, S.; Tanaka, M.; Nambara, T. New ferrocene reagents for derivatization of alcohols in high-performance liquid chromatography with electrochemical detection. J. Chromatogr. 1986, 352, 329–335. [Google Scholar] [CrossRef]
- Goggins, S.; Apsey, E.A.; Mahon, M.F.; Frost, C.G. Ratiometric electrochemical detection of hydrogen peroxide and glucose. Org. Biomol. Chem. 2017, 15, 2458–2466. [Google Scholar] [CrossRef]
- Nesmeyanov, A.N.; Perevalova, E.G.; Nikitina, T.V.; Kuznetsova, N.I. Behavior of 3- and 4-ferrocenyl-hydrazobenzenes under the conditions of the benzidine rearrangement. Izv. Akad. Nauk Ser. Khim. 1965, 14, 2120–2124. [Google Scholar]
- Nesmeyanov, A.N.; Perevalova, E.G.; Nikitina, T.V.; Kuznetsova, N.I. Action of hydrochloric acid on azo derivatives of ferrocene. Izv. Akad. Nauk Ser. Khim. 1965, 14, 2124–2128. [Google Scholar] [CrossRef]
- Kuebrich, J.P.; Schowen, R.L.; Wang, M.-S.; Lupes, M.E. Mechanism of the benzoin condensation. J. Am. Chem. Soc. 1971, 93, 1214–1220. [Google Scholar] [CrossRef]
- Gupta, H.K.; Brydges, S.; McGlinchey, M.J. Diels-Alder reactions of 3-ferrocenyl-2,4,5-triphenyl-cyclopentadienone: Syntheses of C6Ph5Fc, C7Ph6FcH and [C7Ph6FcH][SbCl6]. Organometallics 1999, 18, 115–122. [Google Scholar] [CrossRef]
- Rinehart, K.L., Jr.; Michejda, C.J.; Kittle, P.A. 1,2-Diferrocenylethane from an unusual reaction. J. Am. Chem. Soc. 1959, 81, 3162–3163. [Google Scholar] [CrossRef]
- Rinehart, K.L., Jr.; Ellis, A.F.; Michejda, C.J.; Kittle, P.A. Acyl- from alkyl-ferrocenes by manganese dioxide oxidation. Ferrocobenzoquinone. J. Am. Chem. Soc. 1960, 82, 4112–4113. [Google Scholar] [CrossRef]
- Nesmeyanov, A.N.; Perevalova, E.G.; Tsiskaridze, T.T. Diferrocenoyl and diferrocenylethylene. Izv. Akad. Nauk Ser. Khim. 1966, 15, 2209–2211. [Google Scholar]
- Harrington, L.E.; Britten, J.F.; McGlinchey, M.J. Ferrocenyl-penta(β-naphthyl)benzene: Synthesis, structure and molecular dynamics. Can. J. Chem. 2003, 81, 1180–1186. [Google Scholar] [CrossRef]
- McGlinchey, M.J. Diels-Alder additions as mechanistic probes–interception of silyl-isoindenes: Organometallic derivatives of polyphenylated cycloheptatrienes and related seven-membered rings. Molecules 2020, 25, 4730. [Google Scholar] [CrossRef]
- Hauser, C.R.; Lindsay, J.K.; Lednicer, D.; Cain, C.E. Rearrangement of the methiodide of N,N-dimethyl-amino-methylferrocene by potassium amide in liquid ammonia. J. Org. Chem. 1958, 23, 358–360. [Google Scholar] [CrossRef]
- March, J. Chapter 18. Rearrangements. In Advanced Organic Chemistry, Reactions, Mechanisms and Structure, 4th ed.; John Wiley & Sons: New York, NY, USA, 1992; pp. 1100–1102. [Google Scholar]
- Top, S.; Jaouen, G.; Vessières, A.; Abjean, J.P.; Davoust, D.; Rodger, C.A.; Sayer, B.G.; McGlinchey, M.J. Chromium tricarbonyl complexes of estradiol derivatives: Differentiation of α- and β-diastereomers using one- and two-dimensional NMR spectroscopy at 500 MHz. Organometallics 1985, 4, 2143–2150. [Google Scholar] [CrossRef]
- Jaouen, G.; Beck, W.; McGlinchey, M.J. A novel field of research: Bioorganometallic chemistry, origins and founding principles. In Bioorganometallics, Biomolecules, Labeling, Medicine; Jaouen, G., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 1–37. [Google Scholar]
- Jaouen, G.; Top, S.; Vessières, A.; Leclercq, G.; McGlinchey, M.J. The first organometallic Selective Estrogen Receptor Modulators (SERMs) and their relevance to breast cancer. Curr. Med. Chem. 2004, 11, 2505–2517. [Google Scholar] [CrossRef]
- Wang, Y.; Pigeon, P.; Top, S.; McGlinchey, M.J.; Jaouen, G. Organometallic antitumour compounds: Ferrocifens as precursors to quinone methides. Angew. Chem. Int. Ed. 2015, 54, 10230–10233. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dansette, P.M.; Pigeon, P.; Top, S.; McGlinchey, M.J.; Mansuy, D.; Jaouen, G. A new generation of ferrociphenols leads to a great diversity of reactive intermediates, and exhibits remarkable antiproliferative properties. Chem. Sci. 2018, 9, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Vessières, A.; Wang, Y.; McGlinchey, M.J.; Jaouen, G. Multifaceted chemical behaviour of metallocene (M = Fe, Os) quinone methides. Their contribution to biology. Coord. Chem. Rev. 2020. In Press. [Google Scholar] [CrossRef]
- Richard, M.-A.; Hamels, D.; Pigeon, P.; Top, S.; Dansette, P.M.; Lee, H.Z.S.; Vessières, A.; Mansuy, D.; Jaouen, G. Oxidative metabolism of ferrocene analogues of tamoxifen: Characterization and anti-proliferative activities of the metabolites. ChemMedChem 2015, 10, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Görmen, M.; Pigeon, P.; Hillard, E.A.; Vessières, A.; Huché, M.; Richard, M.-A.; McGlinchey, M.J.; Top, S.; Jaouen, G. Synthesis and antiproliferative effects of [3]ferrocenophane transposition products and pinacols obtained from McMurry cross-coupling reactions. Organometallics 2012, 31, 5856. [Google Scholar] [CrossRef]
- Dubar, F.; Biot, C. On the molecular mechanisms of the antimalarial action of ferroquine. In Bioorganometallic Chemistry, Applications in Drug Discovery, Biocatalysis, and Imaging; Jaouen, G., Salmain, M., Eds.; Wiley-VCH: Weinheim, Germany, 2015; pp. 141–164. [Google Scholar]










































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGlinchey, M.J. Ferrocenyl Migrations and Molecular Rearrangements: The Significance of Electronic Charge Delocalization. Inorganics 2020, 8, 68. https://doi.org/10.3390/inorganics8120068
McGlinchey MJ. Ferrocenyl Migrations and Molecular Rearrangements: The Significance of Electronic Charge Delocalization. Inorganics. 2020; 8(12):68. https://doi.org/10.3390/inorganics8120068
Chicago/Turabian StyleMcGlinchey, Michael J. 2020. "Ferrocenyl Migrations and Molecular Rearrangements: The Significance of Electronic Charge Delocalization" Inorganics 8, no. 12: 68. https://doi.org/10.3390/inorganics8120068
APA StyleMcGlinchey, M. J. (2020). Ferrocenyl Migrations and Molecular Rearrangements: The Significance of Electronic Charge Delocalization. Inorganics, 8(12), 68. https://doi.org/10.3390/inorganics8120068