Theoretical Studies of Nickel-Dependent Enzymes


Abstract
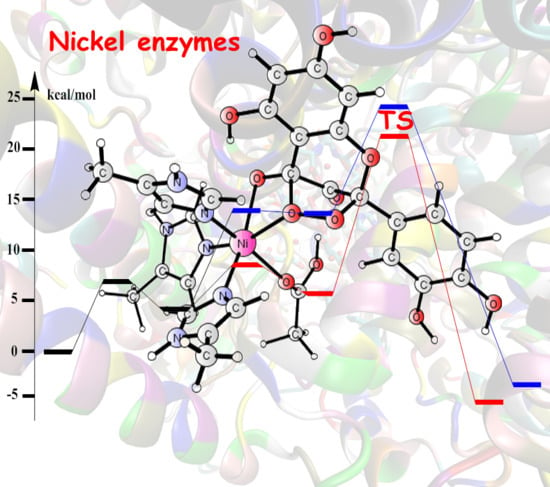
1. Introduction
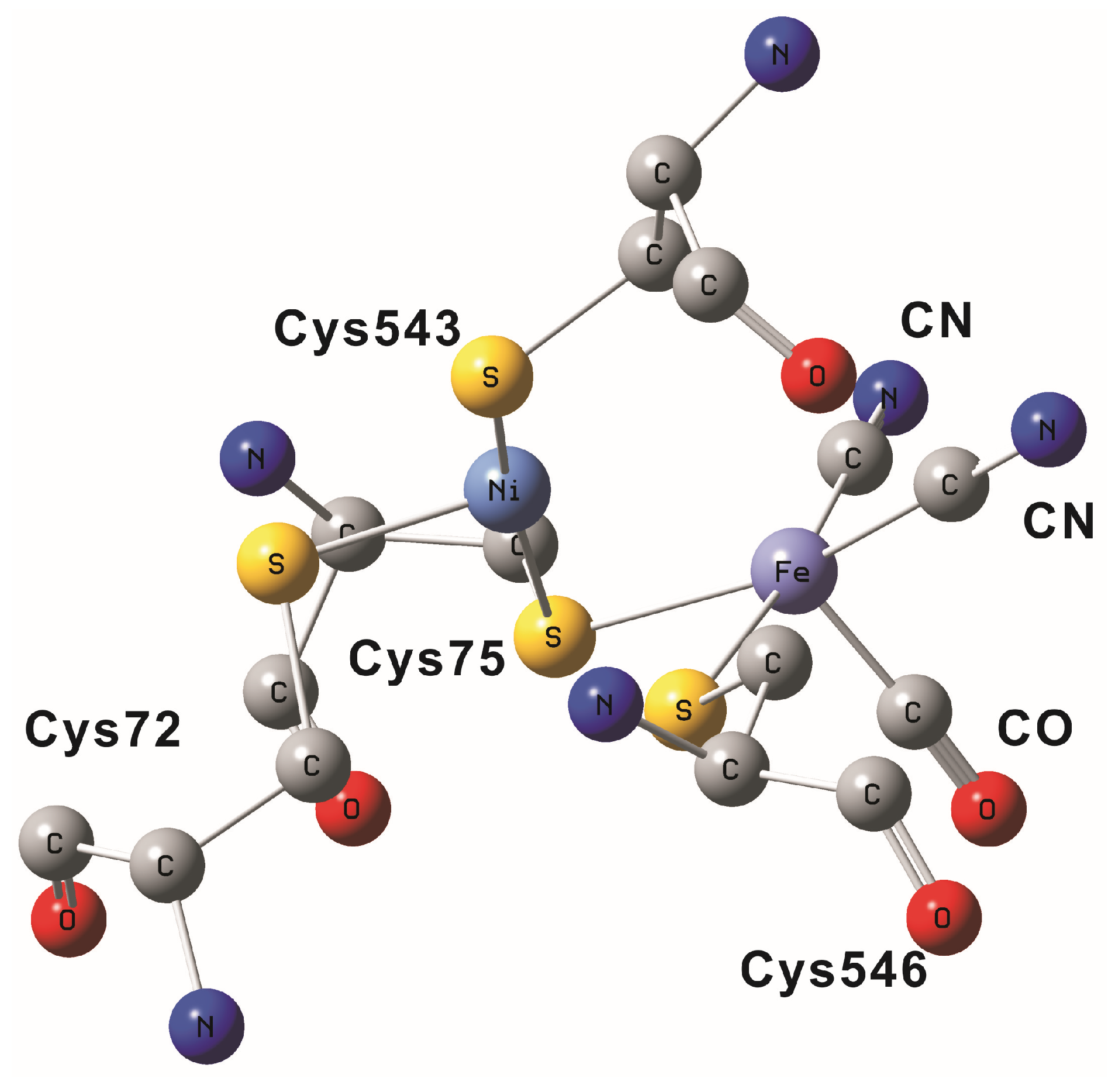
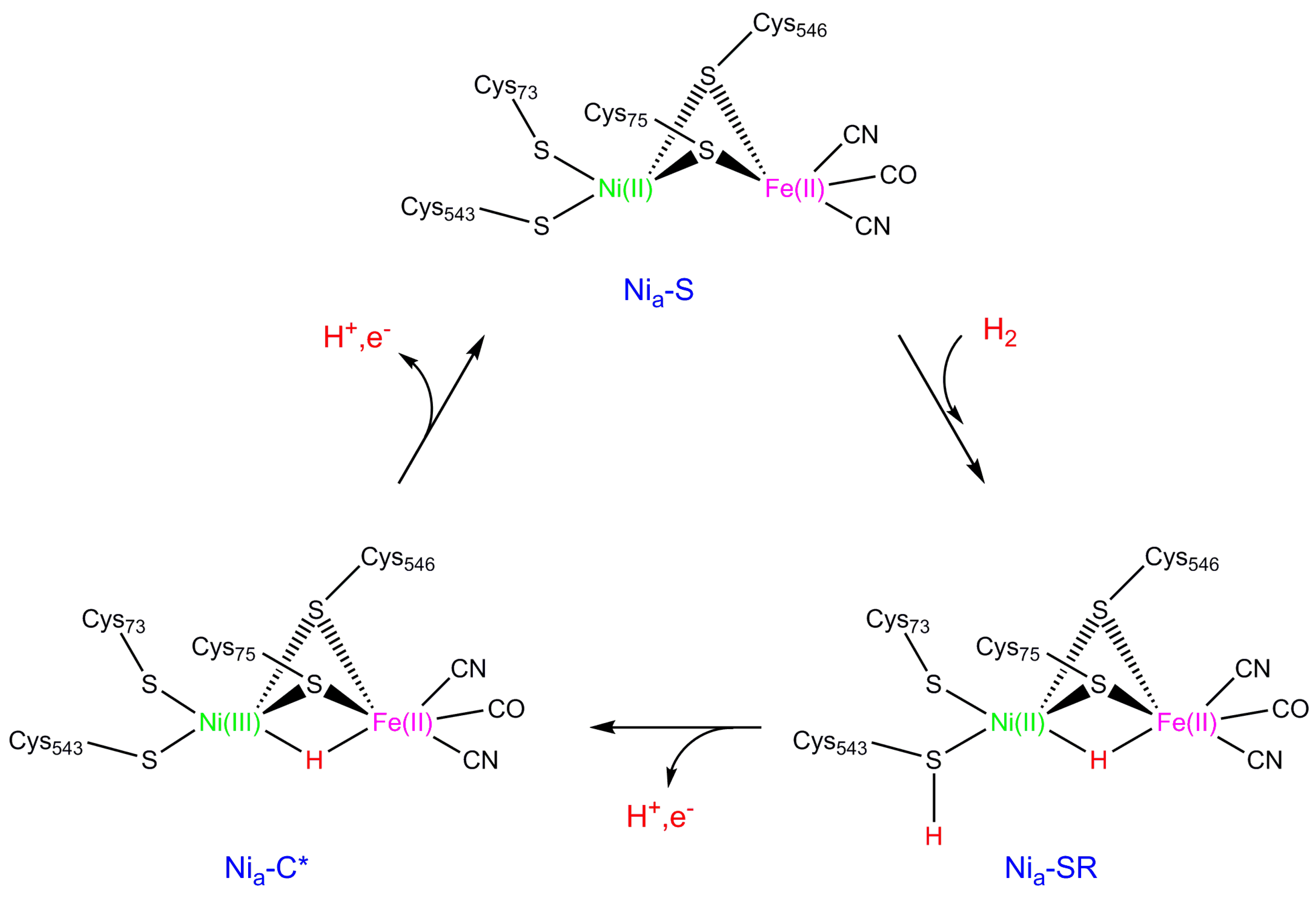
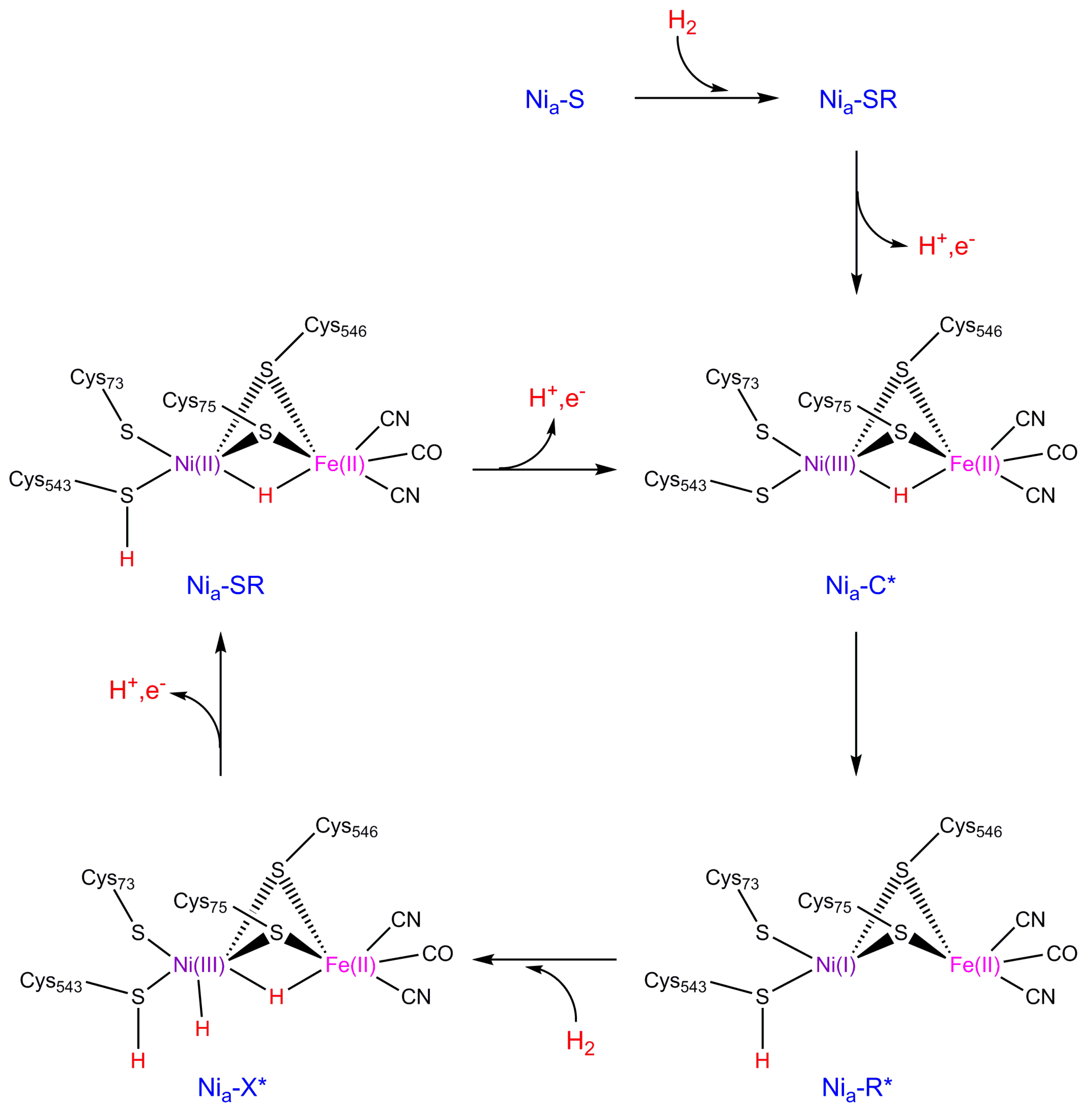
2. NiFe Hydrogenase
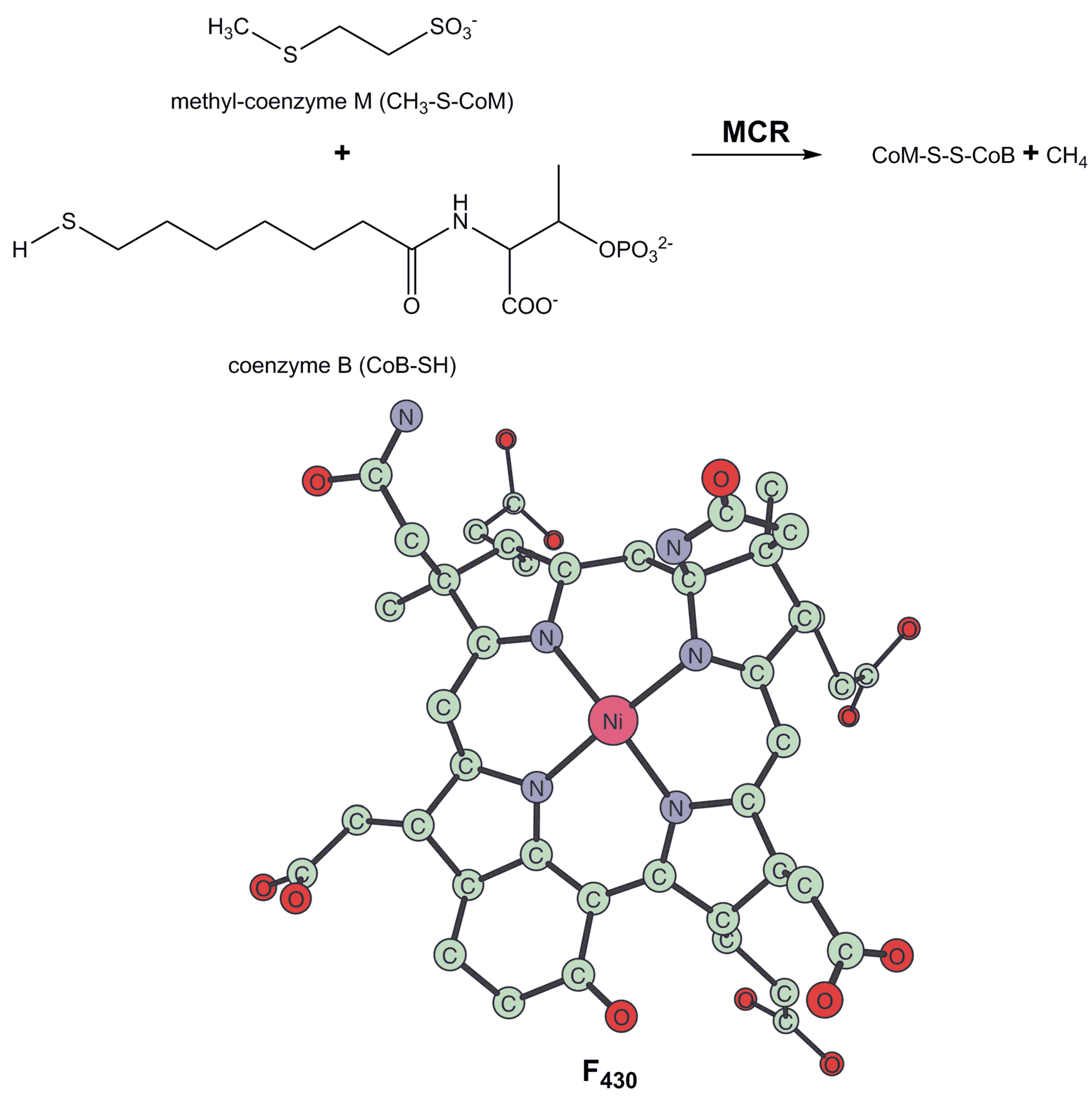
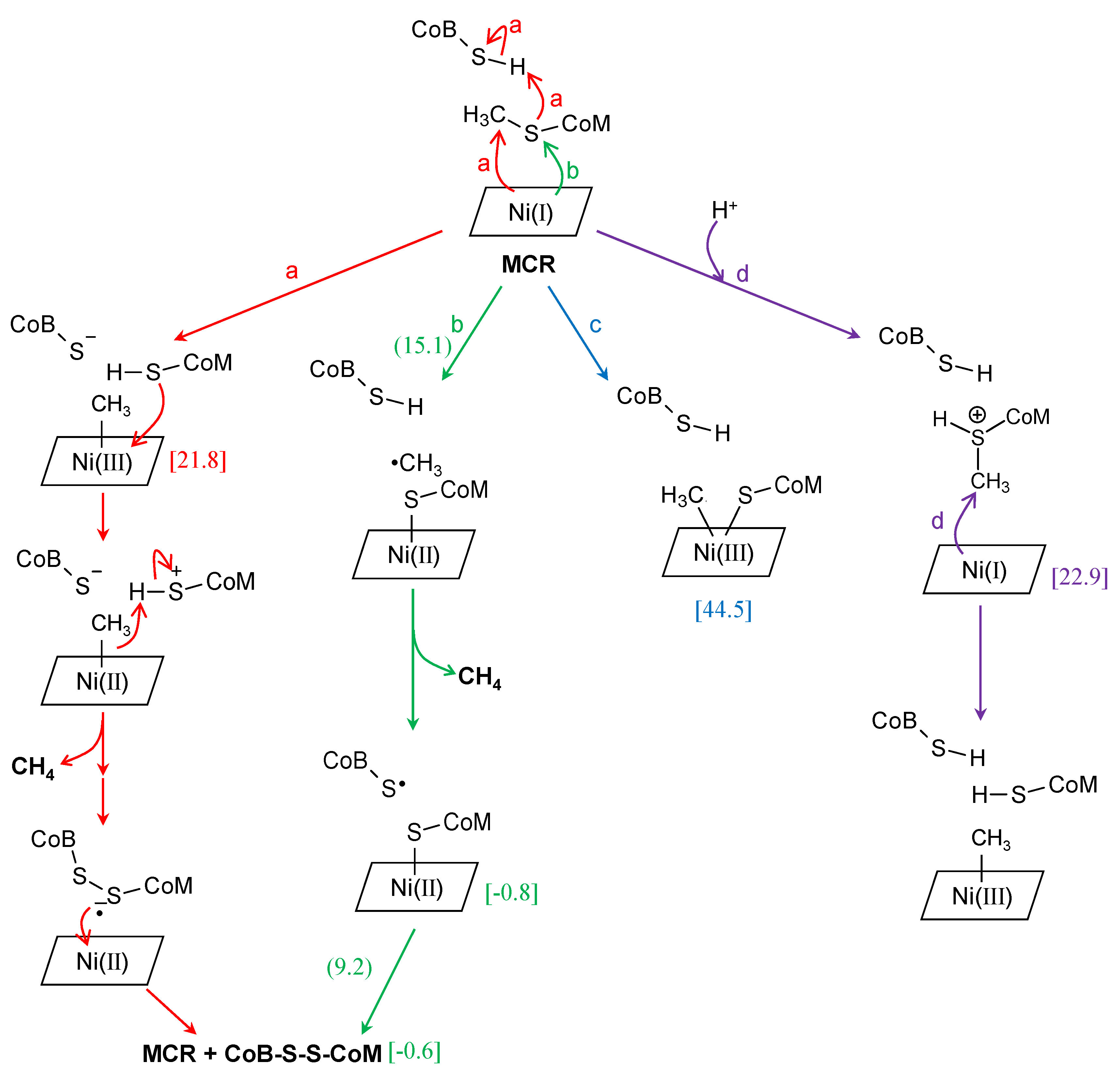
3. Methyl-Coenzyme M Reductase
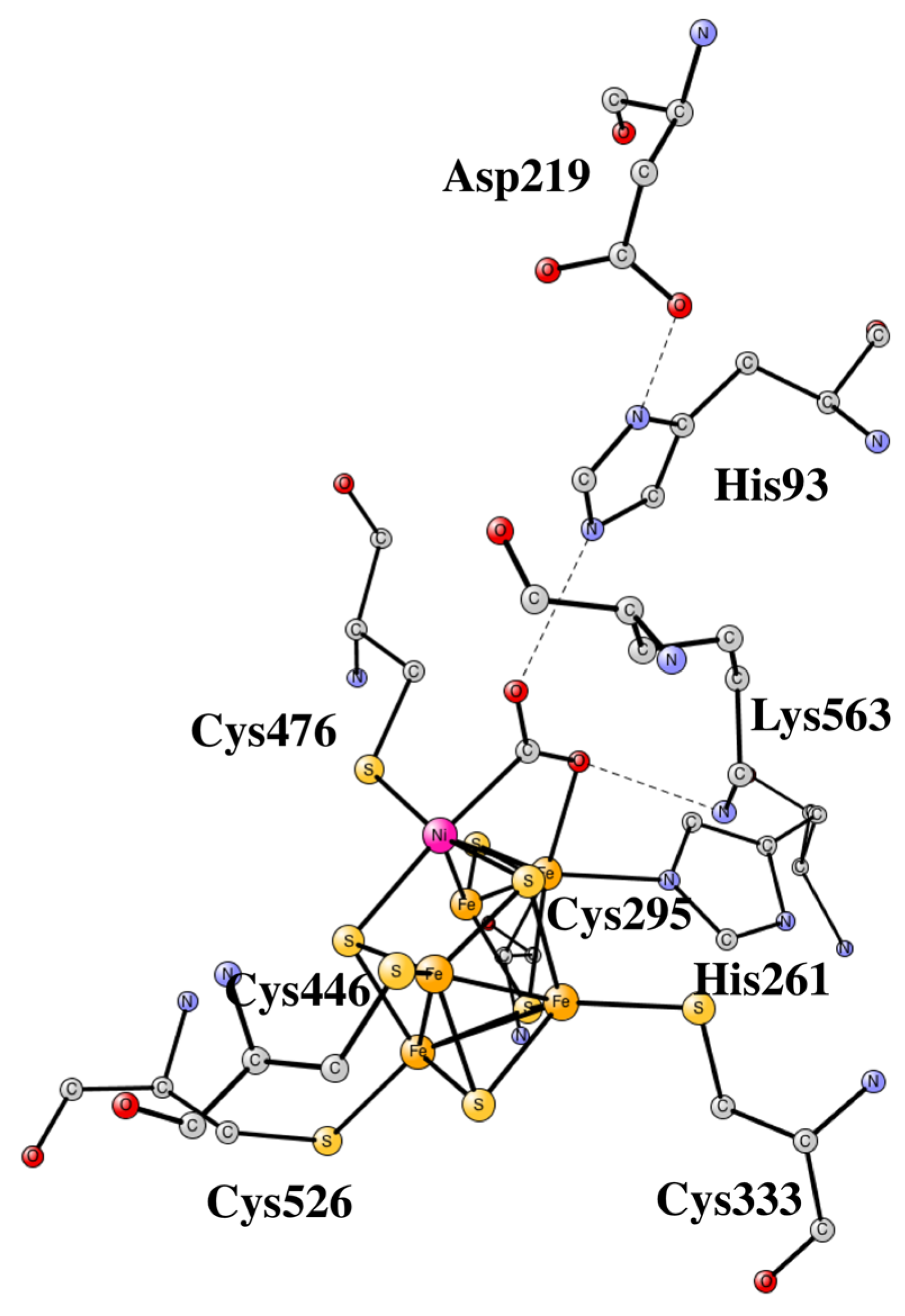
4. Nickel CO Dehydrogenase and Acetyl CoA Synthase
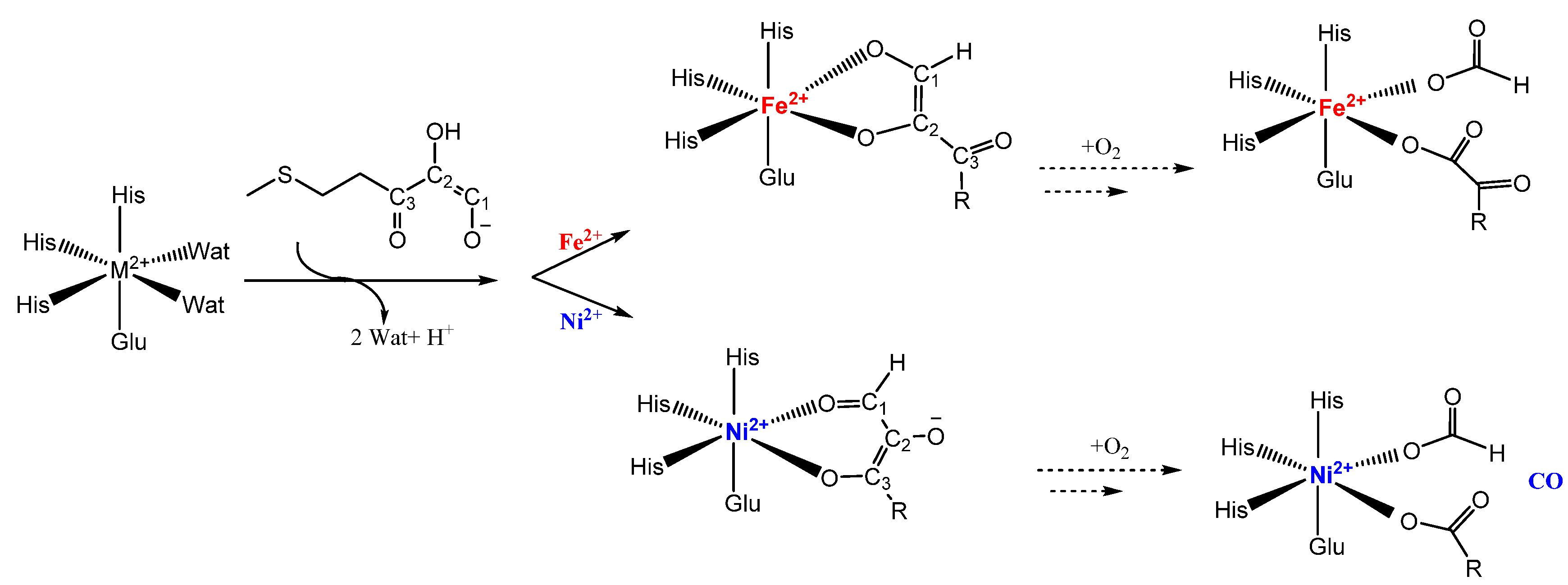
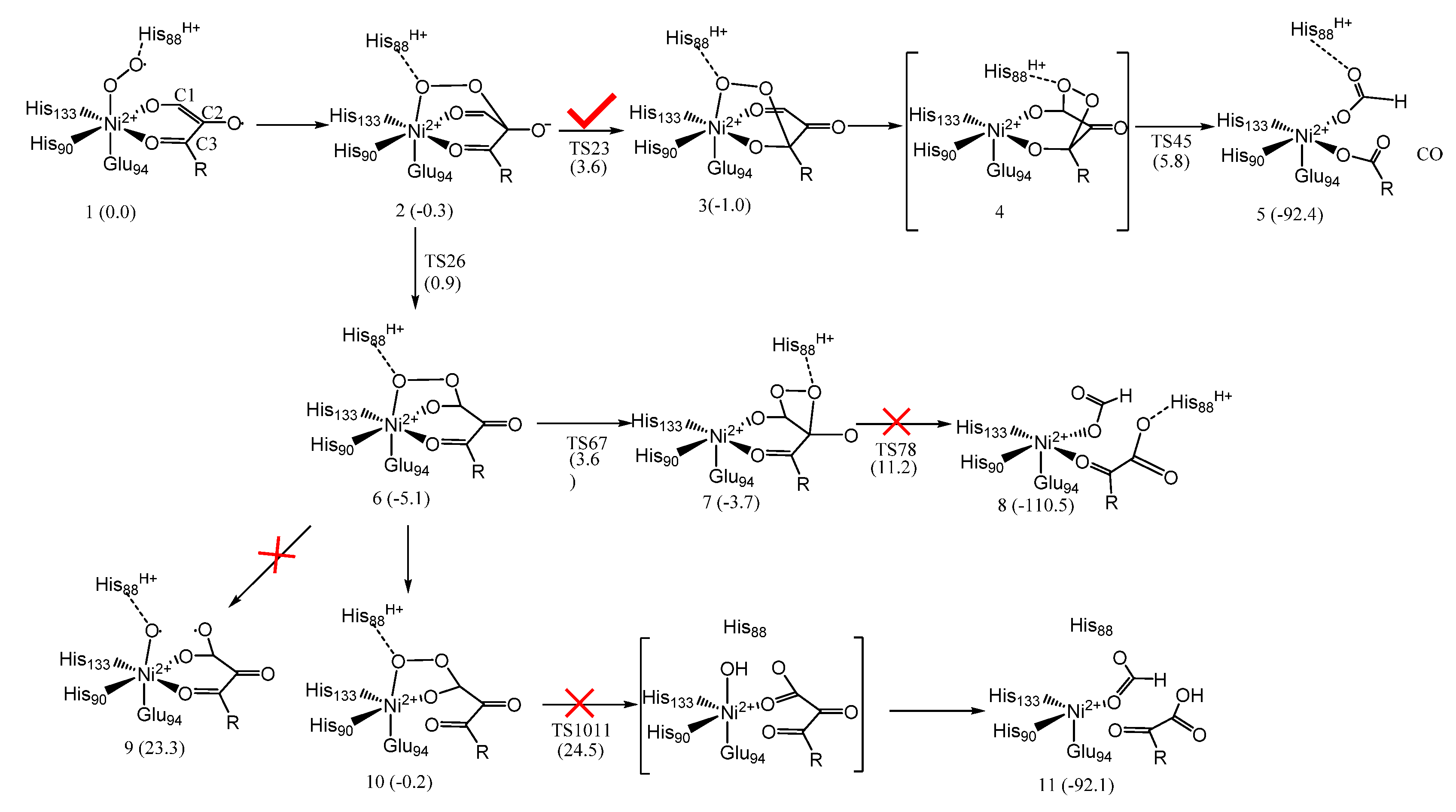
5. Acireductone Dioxygenase
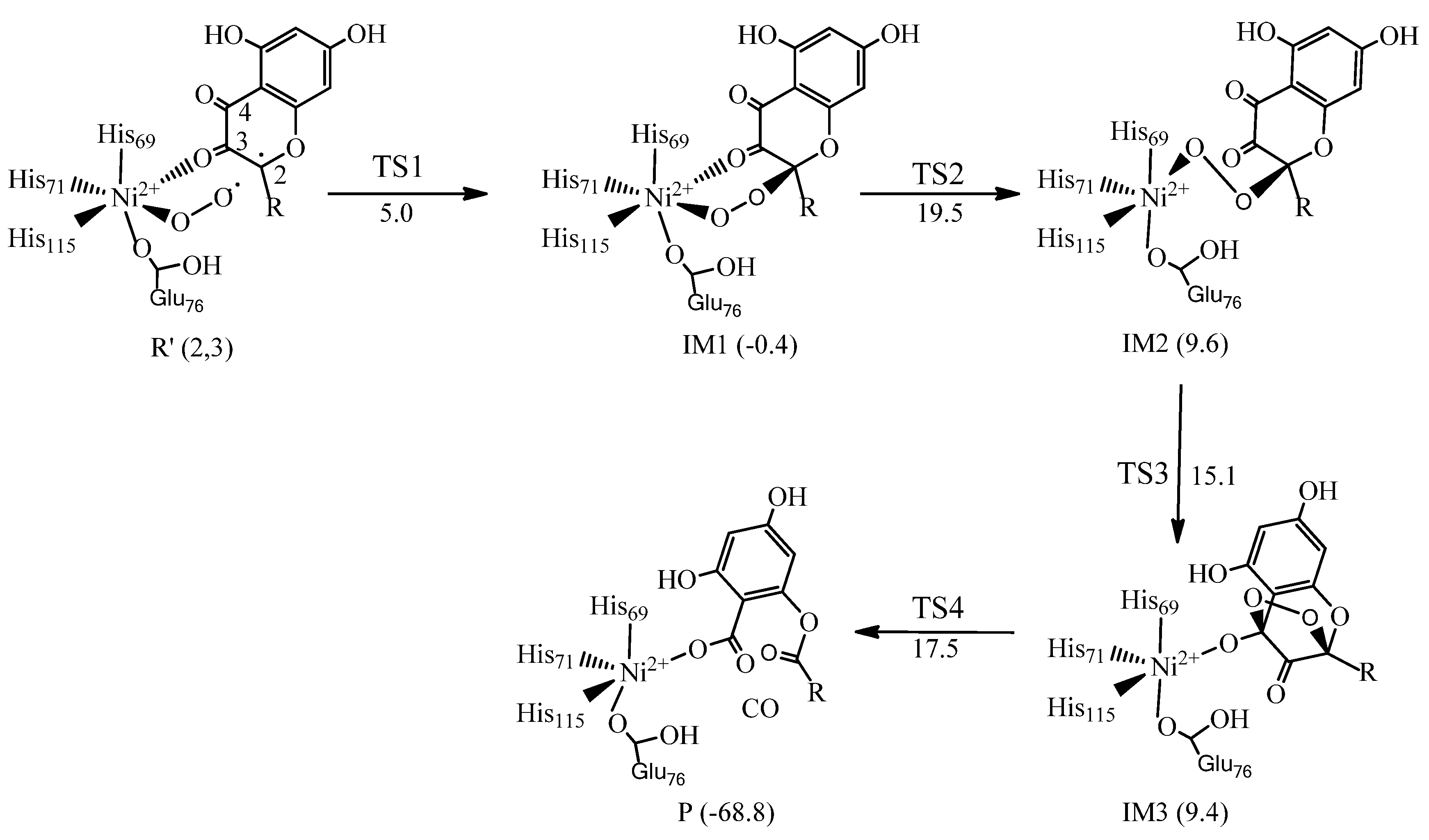
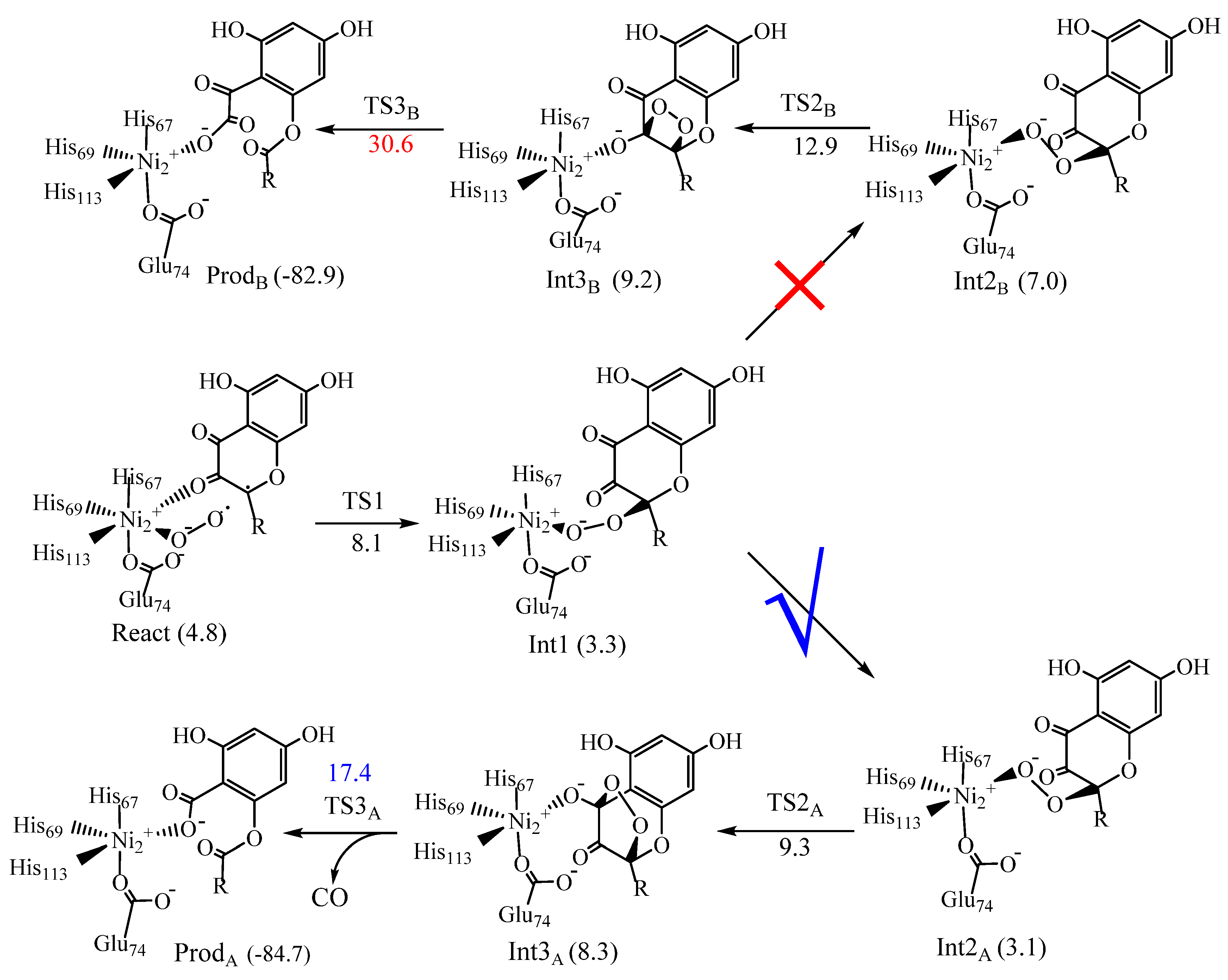
6. Quercetin 2,4-Dioxygenase
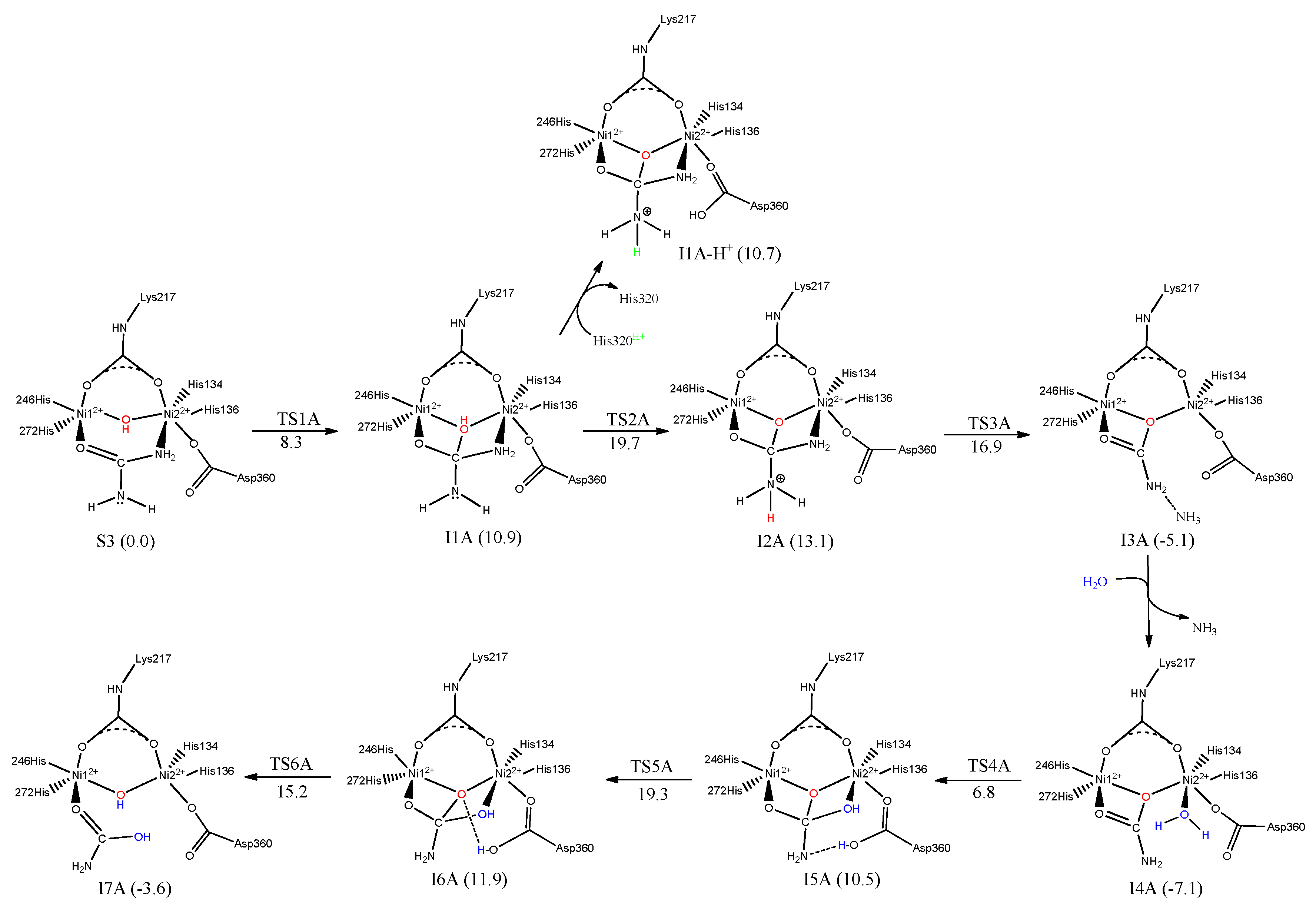
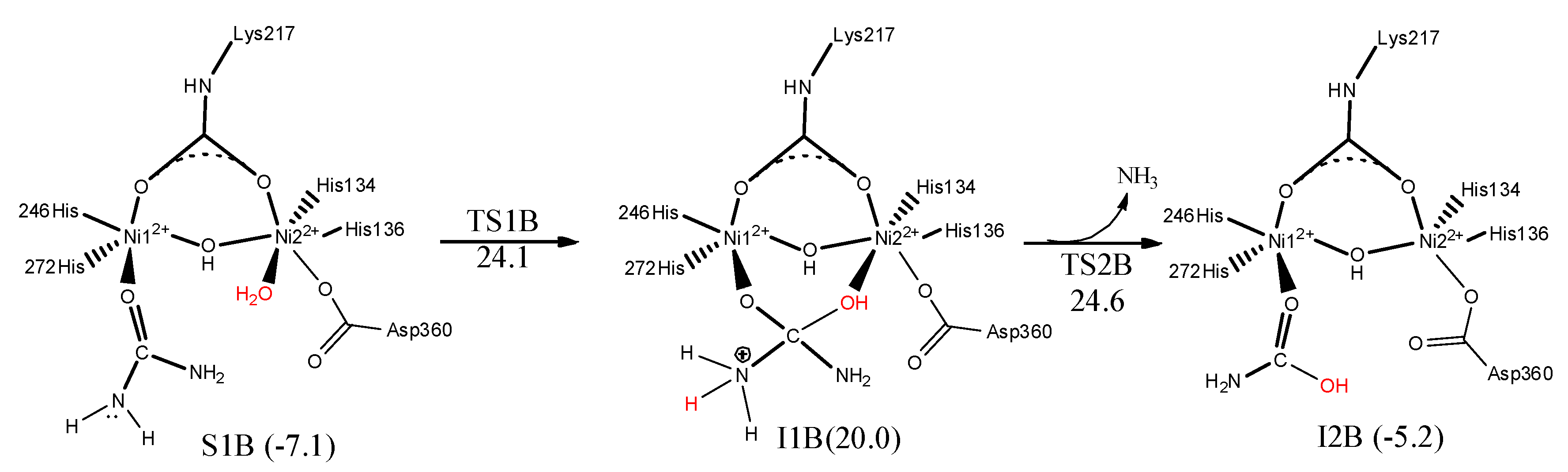
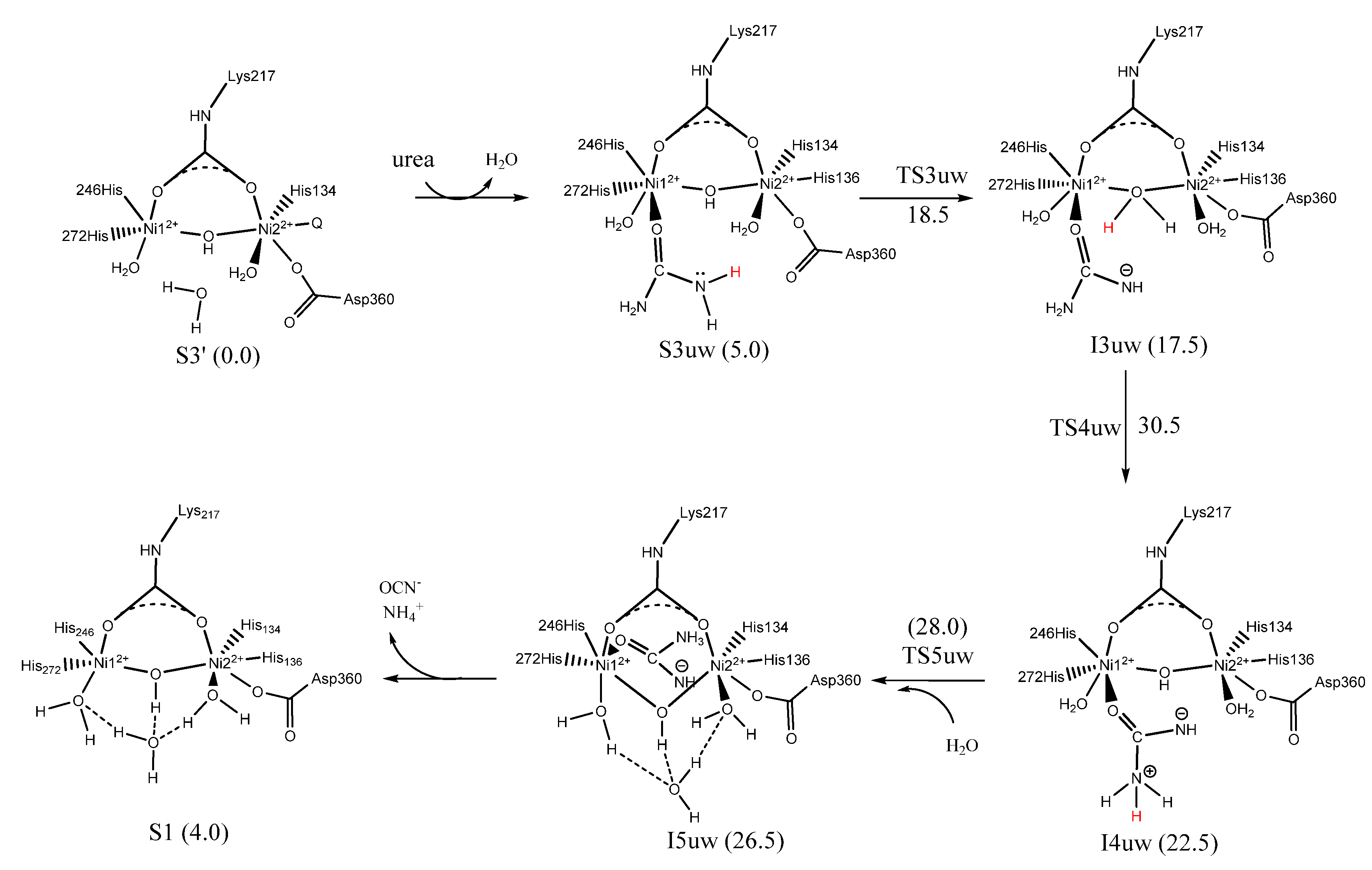
7. Urease
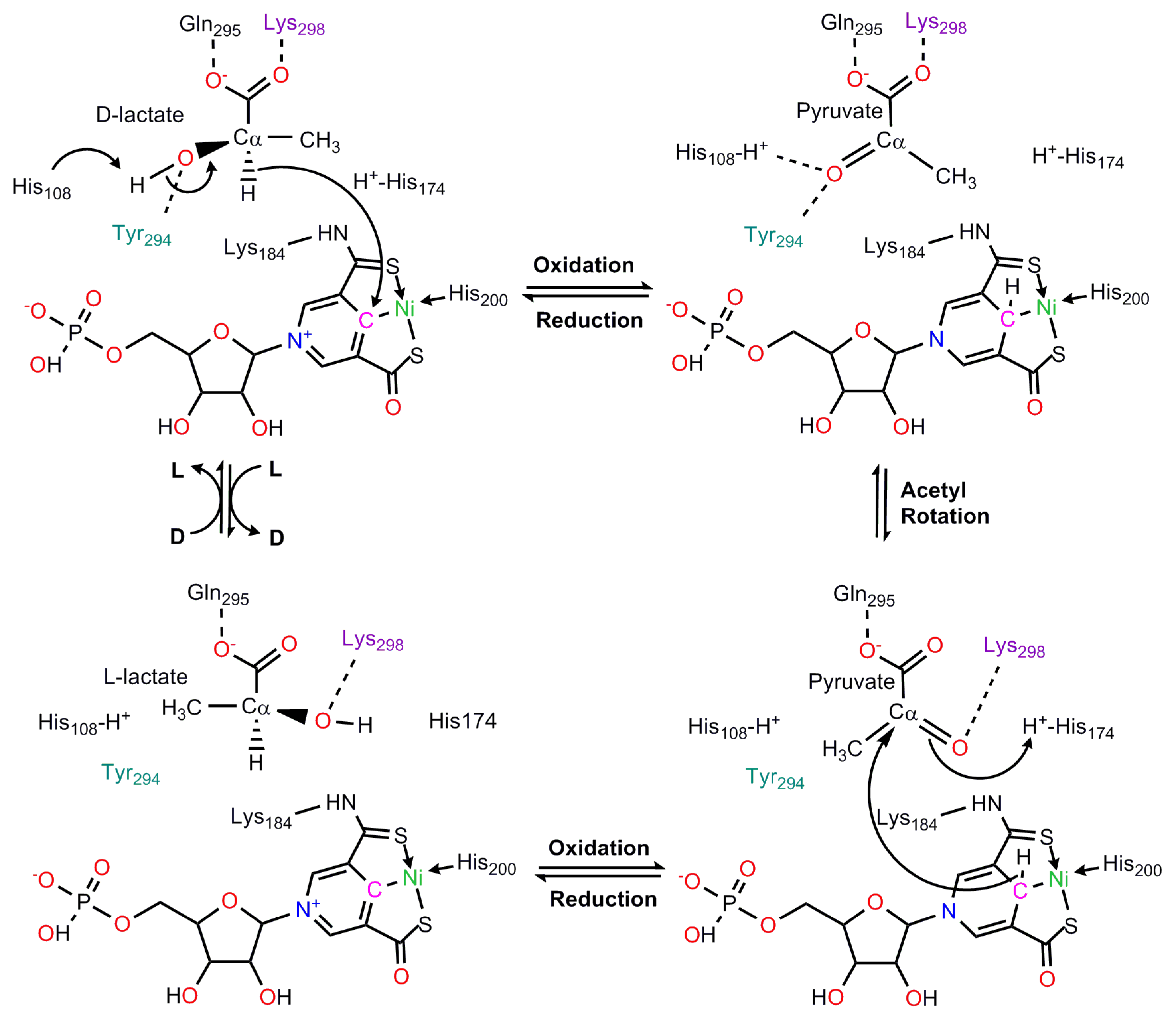
8. Lactate Racemase
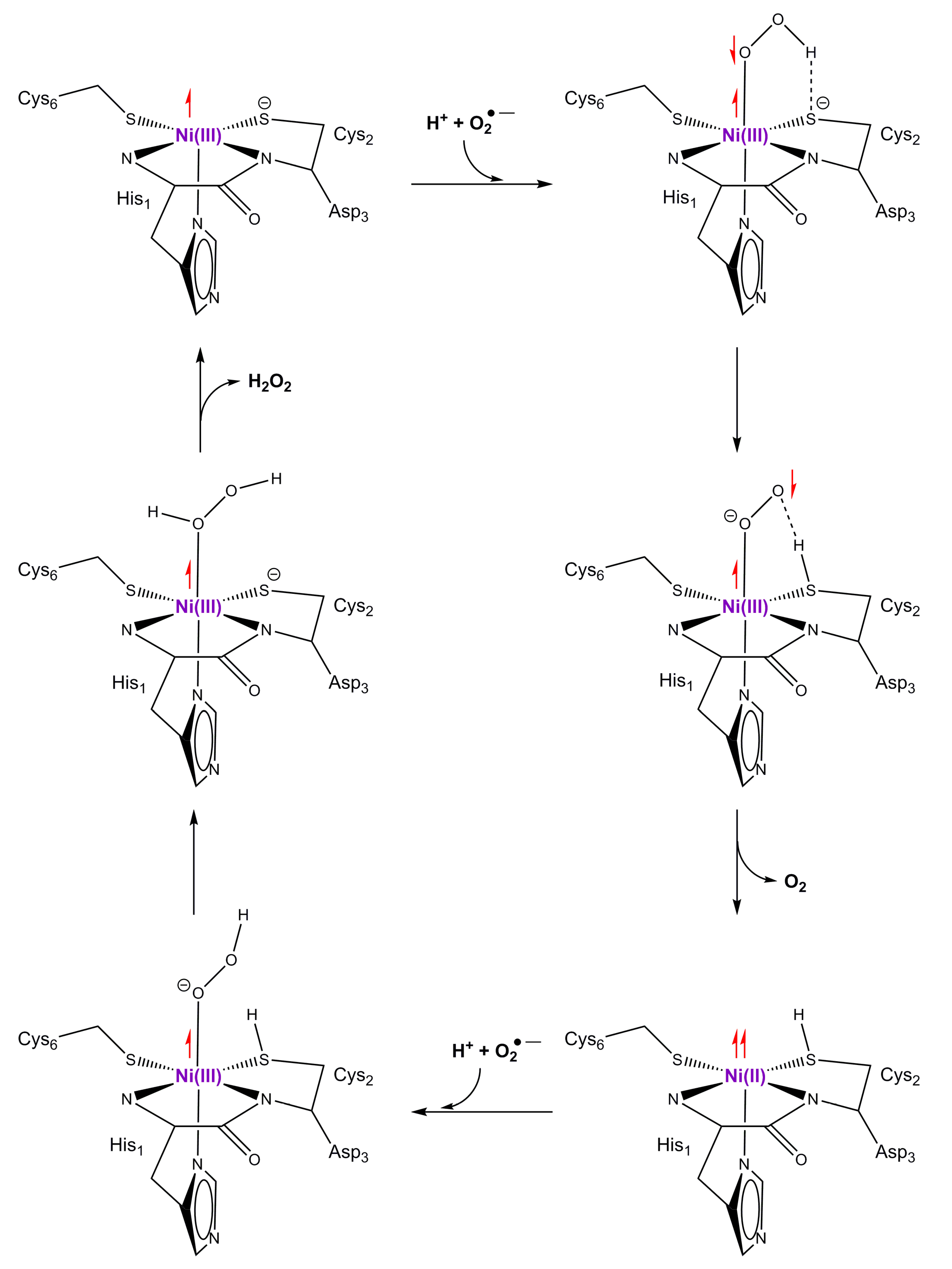
9. Superoxide Dismutase
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Walsh, C.T.; Orme-Johnson, W.H. Nickel Enzymes. Biochemistry 1987, 26, 4901–4906. [Google Scholar] [CrossRef] [PubMed]
- Ermler, U.; Grabarse, W.; Shima, S.; Goubeaud, M.; Thauer, R.K. Active sites of transition-metal enzymes with a focus on nickel. Curr. Opin. Struct. Biol. 1998, 8, 749–758. [Google Scholar] [CrossRef]
- Maroney, M.J. Structure/function relationships in nickel metallobiochemistry. Curr. Opin. Chem. Biol. 1999, 3, 188–199. [Google Scholar] [CrossRef]
- Mulrooney, S.B.; Hausinger, R.P. Nickel uptake and utilization by microorganisms. FEMS Microbiol. Rev. 2003, 27, 239–261. [Google Scholar] [CrossRef]
- Li, Y.; Zamble, D.B. Nickel Homeostasis and Nickel Regulation: An Overview. Chem. Rev. 2009, 109, 4617–4643. [Google Scholar] [CrossRef]
- Ragsdale, S.W. Nickel-based Enzyme Systems. J. Biol. Chem. 2009, 284, 18571–18575. [Google Scholar] [CrossRef]
- Fontecilla-Camps, J.C.; Amara, P.; Cavazza, C.; Nicolet, Y.; Volbeda, A. Structure–function relationships of anaerobic gas-processing metalloenzymes. Nature 2009, 460, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Maroney, M.J.; Ciurli, S. Nonredox Nickel Enzymes. Chem. Rev. 2014, 114, 4206–4228. [Google Scholar] [CrossRef] [PubMed]
- Boer, J.L.; Mulrooney, S.B.; Hausinger, R.P. Nickel-dependent metalloenzymes. Arch. Biochem. Biophys. 2014, 544, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Dixon, N.E.; Gazzola, C.; Blakeley, R.L.; Zerner, B. Jack Bean Urease (EC 3.5.1.5). A Metalloenzyme. A Simple Biological Role for Nickel? J. Am. Chem. Soc. 1975, 97, 4131–4133. [Google Scholar] [CrossRef]
- Volbeda, A.; Charon, M.H.; Piras, C.; Hatchikian, E.C.; Frey, M.; Fontecilla-Camps, J.C. Crystal structure of the nickel–iron hydrogenase from Desulfovibrio gigas. Nature 1995, 373, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Ermler, U.; Grabarse, W.; Shima, S.; Goubeaud, M.; Thauer, R.K. Crystal Structure of Methyl-Coenzyme M Reductase: The Key Enzyme of Biological Methane Formation. Science 1997, 278, 1457–1462. [Google Scholar] [CrossRef] [PubMed]
- Jeoung, J.H.; Dobbek, H. Carbon dioxide activation at the Ni, Fe-cluster of anaerobic carbon monoxide dehydrogenase. Science 2007, 318, 1461–1464. [Google Scholar] [CrossRef] [PubMed]
- Doukov, T.; Iverson, T.M.; Seravalli, J.; Ragsdale, S.W.; Drennan, C.L. A Ni–Fe–Cu Center in a Bifunctional Carbon Monoxide Dehydrogenase/Acetyl-CoA Synthase. Science 2002, 298, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Pochapsky, T.C.; Pochapsky, S.S.; Ju, T.; Hoefler, C.; Liang, J. A refined model for the structure of acireductone dioxygenase from Klebsiella ATCC 8724 incorporating residual dipolar couplings. J. Biomol. NMR. 2006, 34, 117–127. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jeoung, J.H.; Nianios, D.; Fetzner, S.; Dobbek, H. Quercetin 2,4-Dioxygenase Activates Dioxygen in a Side-On O2–Ni Complex. Angew. Chem. Int. Ed. 2016, 55, 3281–3284. [Google Scholar] [CrossRef]
- Wuerges, J.; Lee, J.W.; Yim, Y.I.; Yim, H.S.; Kang, S.O.; Carugo, K.D. Crystal structure of nickel-containing superoxide dismutase reveals another type of active site. Proc. Natl Acad. Sci. USA 2004, 101, 8569–8574. [Google Scholar] [CrossRef]
- He, M.M.; Clugston, S.L.; Honek, J.F.; Matthews, B.W. Determination of the Structure of Escherichia coli Glyoxalase I Suggests a Structural Basis for Differential Metal Activation. Biochemistry 2000, 39, 8719–8727. [Google Scholar] [CrossRef]
- Desguin, B.; Zhang, T.; Soumillion, P.; Hols, P.; Hu, J.; Hausinger, R.P. A tethered niacin-derived pincer complex with a nickel–carbon bond in lactate racemase. Science 2015, 349, 66–69. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M.; Blomberg, M.R. Transition-metal systems in biochemistry studied by high-accuracy quantumchemical methods. Chem. Rev. 2000, 100, 421–437. [Google Scholar] [CrossRef]
- Himo, F.; Siegbahn, P.E.M. Quantum chemical studies of radical containing enzymes. Chem. Rev. 2003, 103, 2421–2456. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M.; Borowski, T. Modeling enzymatic reactions involving transition metals. Acc. Chem. Res. 2006, 39, 729–738. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M.; Himo, F. Recent developments of the quantum chemical cluster approach for modeling enzyme reactions. J. Biol. Inorg. Chem. 2009, 14, 643–651. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M.; Himo, F. The quantum chemical cluster approach for modeling enzyme reactions. WIREs Comput. Mol. Sci. 2011, 1, 323–336. [Google Scholar] [CrossRef]
- Himo, F. Recent trends in quantum chemical modeling of enzymatic reactions. J. Am. Chem. Soc. 2017, 139, 6780–6786. [Google Scholar] [CrossRef]
- Blomberg, M.R.; Borowski, T.; Himo, F.; Liao, R.Z.; Siegbahn, P.E.M. Quantum chemical studies of mechanisms for metalloenzymes. Chem. Rev. 2014, 114, 3601–3658. [Google Scholar] [CrossRef]
- Warshel, A.; Levitt, M. Theoretical studies of enzymatic reactions dielectric, electrostatic, and steric stabilization of carbonium-ion in reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. [Google Scholar] [CrossRef]
- Liao, R.Z.; Thiel, W. Comparison of QM-only and QM/MM models for the mechanism of tungsten-dependent acetylene hydratase. J. Chem. Theory Comput. 2012, 8, 3793–3803. [Google Scholar] [CrossRef]
- Liao, R.Z.; Thiel, W. Convergence in the QM-only and QM/MM modeling of enzymatic reactions: A case study for acetylene hydratase. J. Comput. Chem. 2013, 34, 2389–2397. [Google Scholar] [CrossRef]
- Volbeda, A.; Martin, L.; Cavazza, C.; Matho, M.; Faber, B.; Roseboom, W.; Albracht, S.; Garcin, E.; Rousset, M.; Fontecilla-Camps, J.C. Structural differences between the ready and unready oxidized states of [NiFe] hydrogenases. J. Biol. Inorg. Chem. 2005, 10, 239–249. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M.; Tye, J.W.; Hall, M.B. Computational studies of [NiFe] and [FeFe] hydrogenases. Chem. Rev. 2007, 107, 4414–4435. [Google Scholar] [CrossRef]
- Bruschi, M.; Zampella, G.; Greco, Y.; Bertini, L.; Fantucci, P.; Gioia, D.L. Computational Inorganic and Bioinorganic Chemistry; Solomon, E.I., King, B., Scott, R., Eds.; John and Wiley and Sons: Chichester, UK, 2009. [Google Scholar]
- Siegbahn, P.E.M.; Blomberg, M.R.A. Quantum Chemical Studies of Proton-Coupled Electron Transfer in Metalloenzymes. Chem. Rev. 2010, 110, 7040–7061. [Google Scholar] [CrossRef]
- Kurkin, S.; George, S.J.; Thorneley, R.N.F.; Albracht, S.P.J. Hydrogen-induced activation of the [NiFe]-hydrogenase from Allochromatium vinosum as studied by stopped-flow infrared spectroscopy. Biochemistry 2004, 43, 6820–6831. [Google Scholar] [CrossRef]
- Nilsson-Lill, S.O.; Siegbahn, P.E.M. An autocatalytic Mechanism for NiFe-Hydrogenase: Reduction to Ni(I) Followed by Oxidative Addition. Biochemistry 2009, 48, 1056–1066. [Google Scholar] [CrossRef]
- Ogata, H.; Hirota, S.; Nakahara, A.; Komori, H.; Shibata, N.; Kato, T.; Kano, K.; Higuchi, Y. Activation process of [NiFe] hydrogenase elucidated by high-resolution X-ray analyses: Conversion of the ready to the unready state. Structure 2005, 13, 1635–1642. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M. Hybrid density functional study of the oxidized states of NiFe-hydrogenase. C. R. Chim. 2007, 10, 766–774. [Google Scholar] [CrossRef]
- Pardo, A.; Lacey, A.L.D.; Fernandez, V.M.; Fan, Y.B.; Hall, M.B. Characterization of the active site of catalytically inactive forms of [NiFe] hydrogenases by density functional theory. J. Biol. Inorg. Chem. 2007, 12, 751–760. [Google Scholar] [CrossRef]
- Söderhjelm, P.; Ryde, U. Combined computational and crystallographic study of the oxidised states of [NiFe] hydrogenase. Theochem 2006, 770, 108–116. [Google Scholar] [CrossRef]
- Volbeda, A.; Martin, L.; Barbier, E.; Gutiérrez-Sanz, O.; De Lacey, A.L.; Liebgott, P.P.; Dementin, S.; Rousset, M.; Fontecilla-Camps, J.C. Crystallographic studies of [NiFe]-hydrogenase mutants: Towards consensus structures for the elusive unready oxidized states. J. Biol. Inorg. Chem. 2015, 20, 11–22. [Google Scholar] [CrossRef]
- Breglia, R.; Greco, C.; Fantucci, P.; De Gioia, L.; Bruschi, M. Theoretical investigation of aerobic and anaerobic oxidative inactivation of the [NiFe]-hydrogenase active site. Phys. Chem. Chem. Phys. 2018, 20, 1693–1706. [Google Scholar] [CrossRef]
- Breglia, R.; Greco, C.; Fantucci, P.; De Gioia, L. Reactivation of the Ready and Unready Oxidized States of [NiFe]-Hydrogenases: Mechanistic Insights from DFT Calculations. Inorg. Chem. 2019, 58, 279–293. [Google Scholar] [CrossRef]
- Goris, T.; Wait, A.F.; Saggu, M.; Fritsch, J.; Heidary, N.; Stein, M.; Zebger, I.; Lendzian, F.; Armstrong, F.A.; Friedrich, B.; et al. A unique iron–sulfur cluster is crucial for oxygen tolerance of a [NiFe]-hydrogenase. Nat. Chem. Biol. 2011, 7, 310–318. [Google Scholar] [CrossRef]
- Mouesca, J.M.; Fontecilla-Camps, J.C.; Amara, P. The Structural Plasticity of the Proximal [4Fe3S] Cluster is Responsible for the O2 Tolerance of Membrane-Bound [NiFe] Hydrogenases. Angew. Chem. Int. Ed. 2013, 52, 2002–2006. [Google Scholar] [CrossRef]
- Pandelia, M.E.; Bykov, D.; Izsak, R.; Infossi, P.; Giudici-Orticoni, M.T.; Bill, E.; Neese, F.; Lubitz, W. Electronic structure of the unique [4Fe–3S] clusterin O2-tolerant hydrogenases characterized by 57Fe Mössbauer and EPR spectroscopy. Proc. Natl Acad. Sci. USA 2013, 110, 483–488. [Google Scholar] [CrossRef]
- Pelmenschikov, V.; Kaupp, M. Redox-Dependent Structural Transformations of the [4Fe–3S] Proximal Cluster in O2-Tolerant Membrane-Bound [NiFe]-Hydrogenase: A DFT Study. J. Am. Chem. Soc. 2013, 135, 11809–11823. [Google Scholar] [CrossRef]
- Delcey, M.G.; Pierloot, K.; Quan, M.P.; Vancoille, S.; Lind, R.; Ryde, U. Accurate calculations of geometries and singlet–triplet energy differences for active-site models of [NiFe] hydrogenase. Phys. Chem. Chem. Phys. 2014, 16, 7927–7938. [Google Scholar] [CrossRef]
- Dong, G.; Quan, M.P.; Hallaert, S.D.; Pierloot, K.; Ryde, U. H2 binding to the active site of [NiFe] hydrogenase studied by multiconfigurational and coupled-cluster methods. Phys. Chem. Chem. Phys. 2017, 19, 10590–10601. [Google Scholar] [CrossRef]
- Dong, G.; Ryde, U. Protonation states of intermediates in the reaction mechanism of [NiFe] hydrogenase studied by computational methods. J. Biol. Inorg. Chem. 2016, 21, 383–394. [Google Scholar] [CrossRef]
- Dong, G.; Ryde, U.; Jensen, H.J.; Hedegård, E.D. Exploration of H2 binding to the [NiFe]-hydrogenase active site with multiconfigurational density functional methods. Phys. Chem. Chem. Phys. 2018, 20, 794–801. [Google Scholar] [CrossRef]
- Dong, G.; Quan, M.P.; Pierloot, K.; Ryde, U. Reaction Mechanism of [NiFe] Hydrogenase Studied by Computational Methods. Inorg. Chem. 2018, 57, 15289–15298. [Google Scholar] [CrossRef]
- Murphy, B.J.; Hidalgo, R.; Roessler, M.M.; Evans, R.M.; Ash, P.A.; Myers, W.K.; Vincent, K.A.; Armstrong, F.A. Discovery of Dark pH-Dependent H+ Migration in a [NiFe]-Hydrogenase and Its Mechanistic Relevance: Mobilizing the Hydrido Ligand of the Ni–C Intermediate. J. Am. Chem. Soc. 2015, 137, 8484–8489. [Google Scholar] [CrossRef]
- Murrell, J.C.; Jetten, M.S.M. The microbial methane cycle. Environ. Microbiol. Rep. 2009, 1, 279–284. [Google Scholar] [CrossRef]
- Hogan, K.B.; Hoffman, J.S.; Thompson, A.M. Methane on the greenhouse agenda. Nature 1991, 354, 181–182. [Google Scholar] [CrossRef]
- Conrad, R. The global methane cycle: Recent advances in understanding the microbial processes involved. Environ. Microbiol. Rep. 2009, 1, 285–292. [Google Scholar] [CrossRef]
- Evans, P.N.; Boyd, J.A.; Leu, A.O.; Woodcroft, B.J.; Parks, D.H.; Hugenholtz, P.; Tyson, G.W. Methane on the greenhouse agenda. Nat. Rev. Microbiol. 2019, 17, 219–232. [Google Scholar] [CrossRef]
- Thauer, R.K.; Kaster, A.K.; Seedorf, H.; Buckel, W.; Hedderich, R. Methanogenic archaea: Ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 2008, 6, 579–591. [Google Scholar] [CrossRef]
- Nauhaus, K.; Albrecht, M.; Elvert, M.; Boetius, A.; Widdel, F. In vitro cell growth of marine archaeal-bacterial consortia during anaerobic oxidation of methane with sulfate. Environ. Microbiol. 2007, 9, 187–196. [Google Scholar] [CrossRef]
- Nauhaus, K.; Boetius, A.; Krüger, M.; Widdel, F. In vitro demonstration of anaerobic oxidation of methane coupled to sulphate reduction in sediment from a marine gas hydrate area. Environ. Microbiol. 2002, 4, 296–305. [Google Scholar] [CrossRef]
- Ettwig, K.F.; Zhu, B.; Speth, D.; Keltjens, J.T.; Jetten, M.S.M.; Kartal, B. Archaea catalyze iron-dependent anaerobic oxidation of methane. Proc. Natl Acad. Sci. USA 2016, 113, 12792–12796. [Google Scholar] [CrossRef]
- Beal, E.J.; House, C.H.; Orphan, V.J. Manganese-and iron-dependent marine methane oxidation. Science 2009, 325, 184–187. [Google Scholar] [CrossRef]
- Haroon, M.F.; Hu, S.; Shi, Y.; Imelfort, M.; Keller, J.; Hugenholtz, P.; Yuan, Z.; Tyson, G.W. Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage. Nature 2013, 500, 567–570. [Google Scholar] [CrossRef]
- Scheller, S.; Yu, H.; Chadwick, G.L.; McGlynn, S.E.; Orphan, V.J. Artificial electron acceptors decouple archaeal methane oxidation from sulfate reduction. Science 2016, 351, 703–707. [Google Scholar] [CrossRef]
- Rotaru, A.E.; Thamdrup, B. A new diet for methane oxidizers. Science 2016, 351, 658–659. [Google Scholar] [CrossRef]
- Shen, B.; Dong, L.; Xiao, S.H.; Lang, X.G.; Huang, K.J.; Peng, Y.B.; Zhou, C.M.; Ke, S.; Liu, P.J. Molar tooth carbonates and benthic methane fluxes in Proterozoic oceans. Nat. Commun. 2016, 7, 10317. [Google Scholar] [CrossRef]
- Planavsky, N.J.; Reinhard, C.T.; Wang, X.; Thomson, D.; McGoldrick, P.; Rainbird, R.H.; Johnson, T.; Fischer, W.W.; Lyons, T.W. Low Mid-Proterozoic atmospheric oxygen levels and the delayed rise of animals. Science 2014, 346, 635–638. [Google Scholar] [CrossRef]
- Thauer, R.K. The Wolfe cycle comes full circle. Proc. Natl Acad. Sci. USA 2012, 109, 15084–15085. [Google Scholar] [CrossRef]
- Chen, S.C.; Musat, N.; Lechtenfeld, O.J.; Paschke, H.; Schmidt, M.; Said, N.; Popp, D.; Calabrese, F.; Stryhanyuk, H.; Jaekel, U.; et al. Anaerobic oxidation of ethane by archaea from a marine hydrocarbon seep. Nature 2019, 568, 108–111. [Google Scholar] [CrossRef]
- Pérez, R.L.; Wegener, G.; Knittel, K.; Widdel, F.; Harding, K.J.; Krukenberg, V.; Meier, D.V.; Richter, M.; Tegetmeyer, H.E.; Riedel, D.; et al. Thermophilic archaea activate butane via alkyl-coenzyme M formation. Nature 2016, 539, 396–401. [Google Scholar] [CrossRef]
- Yang, N.; Reiher, M.; Wang, M.; Harmer, J.; Duin, E.C. Formation of a nickel-methyl species in methyl-coenzyme M reductase, an enzyme catalyzing methane formation. J. Am. Chem. Soc. 2007, 129, 11028–11029. [Google Scholar] [CrossRef]
- Dey, M.; Telser, J.; Kunz, R.C.; Lees, N.S.; Ragsdale, S.W.; Hoffman, B.M. Biochemical and spectroscopic studies of the electronic structure and reactivity of a methyl-Ni species formed on methyl-coenzyme m reductase. J. Am. Chem. Soc. 2007, 129, 11030–11032. [Google Scholar] [CrossRef]
- Pelmenschikov, V.; Blomberg, M.R.A.; Siegbahn, P.E.M.; Crabtree, R.H. A mechanism from quantum chemical studies for methane formation in methanogenesis. J. Am. Chem. Soc. 2002, 124, 4039–4049. [Google Scholar] [CrossRef]
- Chen, S.L.; Pelmenschikov, V.; Blomberg, M.R.A.; Siegbahn, P.E.M. Is There a Ni-Methyl Intermediate in the Mechanism of Methyl-Coenzyme M Reductase? J. Am. Chem. Soc. 2009, 131, 9912–9913. [Google Scholar] [CrossRef]
- Chen, S.L.; Blomberg, M.R.A.; Siegbahn, P.E.M. How is methane formed and oxidized reversibly when catalyzed by Ni-containing methyl-coenzyme M reductase? Chem. Eur. J. 2012, 18, 6309–6315. [Google Scholar] [CrossRef]
- Chen, S.L.; Blomberg, M.R.A.; Siegbahn, P.E.M. An investigation of possible competing mechanisms for Ni-containing methyl-coenzyme M reductase. Phys. Chem. Chem. Phys. 2014, 16, 14029–14035. [Google Scholar] [CrossRef]
- Pelmenschikov, V.; Siegbahn, P.E.M. Catalysis by methyl-coenzyme M reductase: A theoretical study for heterodisulfide product formation. J. Biol. Inorg. Chem. 2003, 8, 653. [Google Scholar] [CrossRef]
- Scheller, S.; Goenrich, M.; Thauer, R.K.; Jaun, B. Methyl-coenzyme M reductase from methanogenic archaea: Isotope effects on label exchange and ethane formation with the homologous substrate ethyl-coenzyme M. J. Am. Chem. Soc. 2013, 135, 14975–14984. [Google Scholar] [CrossRef]
- Ahn, Y.; Krzycki, J.A.; Floss, H.G. Steric course of the reduction of ethyl coenzyme M to ethane catalyzed by methyl coenzyme m reductase from methanosarcina barkeri. J. Am. Chem. Soc. 1991, 113, 4700–4701. [Google Scholar] [CrossRef]
- Scheller, S.; Goenrich, M.; Boecher, R.; Thauer, R.K.; Jaun, B. The key nickel enzyme of methanogenesis catalyses the anaerobic oxidation of methane. Nature 2010, 465, 606–609. [Google Scholar] [CrossRef]
- Scheller, S.; Goenrich, M.; Mayr, S.; Thauer, R.K.; Jaun, B. Intermediates in the Catalytic Cycle of Methyl Coenzyme? M Reductase: Isotope Exchange is Consistent with Formation of a σ-Alkane-Nickel Complex. Angew. Chem. Int. Ed. 2010, 49, 8112–8115. [Google Scholar] [CrossRef]
- Wongnate, T.; Sliwa, D.; Ginovska, B.; Smith, D.; Wolf, M.W.; Lehnert, N.; Raugei, S.; Ragsdale, S.W. The radical mechanism of biological methane synthesis by methyl-coenzyme M reductase. Science 2016, 352, 953–958. [Google Scholar] [CrossRef]
- Lawton, T.J.; Rosenzweig, A.C. Methane-make it or break it. Science 2016, 352, 892–893. [Google Scholar] [CrossRef]
- Duin, E.C.; Mckee, M.L. A new mechanism for methane production from methyl-coenzyme M reductase as derived from density functional calculations. J. Phys. Chem. B 2008, 112, 2466–2482. [Google Scholar] [CrossRef]
- Harmer, J.; Finazzo, C.; Piskorski, R.; Ebner, S.; Duin, E.C.; Goenrich, M.; Thauer, R.K.; Reiher, M.; Schweiger, A.; Hinderberger, D.; et al. A nickel hydride complex in the active site of methyl-coenzyme m reductase: Implications for the catalytic cycle. J. Am. Chem. Soc. 2008, 130, 10907–10920. [Google Scholar] [CrossRef]
- Grabarse, W.; Mahlert, F.; Duin, E.C.; Goubeaud, M.; Shima, S.; Thauer, R.K.; Lamzin, V.; Ermler, U. On the mechanism of biological methane formation: Structural evidence for conformational changes in methyl-coenzyme M reductase upon substrate binding. J. Mol. Biol. 2001, 309, 315–330. [Google Scholar] [CrossRef]
- Horng, Y.C.; Becker, D.F.; Ragsdale, S.W. Mechanistic studies of methane biogenesis by methyl-coenzyme M reductase: Evidence that coenzyme B participates in cleaving the C–S bond of methyl-coenzyme M. Biochemistry 2001, 40, 12875–12885. [Google Scholar] [CrossRef]
- Borman, S. Carbon dioxide hydrogenated to methanol on large scale. Chem. Eng. News 2016, 94, 7. [Google Scholar]
- Shima, S. The Biological Methane-Forming Reaction: Mechanism Confirmed Through Spectroscopic Characterization of a Key Intermediate. Angew. Chem. Int. Ed. 2016, 55, 13648–13649. [Google Scholar] [CrossRef]
- Dobbek, H.; Svetlitchnyi, V.; Gremer, L.; Huber, R.; Meyer, O. Crystal Structure of a Carbon Monoxide Dehydrogenase Reveals a [Ni–4Fe–5S] Cluster. Science 2001, 293, 1281–1285. [Google Scholar] [CrossRef]
- Darnault, C.; Volbeda, A.; Kim, E.J.; Legrand, P.; Vernede, X.; Lindahl, P.A.; Fontecilla-Camps, J.C. Ni–Zn–[Fe4–S4] and Ni–Ni–[Fe4–S4] clusters in closed and open α subunits of acetyl-CoA synthase/carbon monoxide dehydrogenase. Nat. Struct. Biol. 2003, 10, 271–279. [Google Scholar] [CrossRef]
- Appel, A.M.; Bercaw, J.E.; Bocarsly, A.B.; Dobbek, H.; DuBois, D.L.; Dupuis, M.; Ferry, J.G.; Fujita, E.; Hille, R.; Kenis, P.J.A.; et al. Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem. Rev. 2013, 113, 6621–6658. [Google Scholar] [CrossRef]
- Can, M.; Armstrong, F.A.; Ragsdale, S.W. Structure, Function, and Mechanism of the Nickel Metalloenzymes, CO Dehydrogenase, and Acetyl-CoA Synthase. Chem. Rev. 2014, 114, 4149–4174. [Google Scholar] [CrossRef]
- Lindahl, P.A.; Munck, E.; Ragsdale, S.W. CO Dehydrogenase from Clostridium thermoaceticum: EPR and electrochemical studies in CO2 and argon atmospheres. J. Biol. Chem. 1990, 265, 3873–3879. [Google Scholar]
- Cao, Z.-X.; Mo, Y.-R. Computational Characterization of the Elusive C-Cluster of Carbon Monoxide Dehydrogenase. J. Theor. Comput. Chem. 2008, 7, 473–484. [Google Scholar] [CrossRef]
- Xie, H.-J.; Cao, Z.-X. Computational Characterization of Reactive Intermediates of Carbon Monoxide Dehydrogenase. Chin. J. Struct. Chem. 2009, 28, 1525–1532. [Google Scholar]
- Amara, P.; Mouesca, J.-M.; Volbeda, A.; Fontecilla-Camps, J.C. Carbon Monoxide Dehydrogenase Reaction Mechanism: A Likely Case of Abnormal CO2 Insertion to a Ni–H− Bond. Inorg. Chem. 2011, 50, 1868–1878. [Google Scholar] [CrossRef]
- Liao, R.-Z.; Siegbahn, P.E.M. Energetics for the mechanism of nickel-containing CO-dehydrogenase. Inorg. Chem. 2019. [Google Scholar] [CrossRef]
- Amara, P.; Volbeda, A.; Fontecilla-Camps, J.C.; Field, M.J. A quntum chemical study of the reaction mechanism of Acetyl-Coenzyme A synthase. J. Am. Chem. Soc. 2005, 127, 2776–2784. [Google Scholar] [CrossRef]
- Chmielowska, A.; Lodowski, P.; Jaworska, M. Redox Potentials and Protonation of the A-Cluster from Acetyl-CoA Synthase. A Density Functional Theory Study. J. Phys. Chem. 2013, 117, 12484–12496. [Google Scholar] [CrossRef]
- Schenker, R.P.; Brunold, T.C. Computational studies on the A cluster of acetyl-coenzyme A synthase: Geometric and electronic properties of the NiFeC species and mechanistic implications. J. Am. Chem. Soc. 2003, 125, 13962–13963. [Google Scholar] [CrossRef]
- Dai, Y.; Wensink, P.C.; Abeles, R.H.J. One protein, two enzymes. Biol. Chem. 1999, 274, 1193–1195. [Google Scholar] [CrossRef]
- Wray, J.W.; Abeles, R.H. The Methionine Salvage Pathway in Klebsiella pneumoniae and Rat Liver identification and characterization of two novel dioxygenases. J. Biol. Chem. 1995, 270, 3147–3153. [Google Scholar] [CrossRef]
- Dai, Y.; Pochapsky, T.C.; Abeles, R.H. Mechanistic studies of two dioxygenases in the methionine salvage pathway of Klebsiella pneumonia. Biochemistry 2001, 40, 6379–6387. [Google Scholar] [CrossRef]
- Sparta, M.; Valdez, C.E.; Alexandrova, A.N. Metal-dependent activity of Fe and Ni acireductone dioxygenases: How two electrons reroute the catalytic pathway. J. Mol. Biol. 2013, 425, 3007–3018. [Google Scholar] [CrossRef]
- Miłaczewska, A.; Kot, E.; Amaya, J.A.; Makris, T.M.; Zając, M.; Korecki, J.; Chumakov, A.; Trzewik, B.; Kędracka-Krok, S.; Minor, W.; et al. On the Structure and Reaction Mechanism of Human Acireductone Dioxygenase. Chem. Eur. J. 2018, 24, 5225–5237. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M. Hybrid DFT study of the mechanism of quercetin 2,3-dioxygenase. Inorg. Chem. 2004, 43, 5944–5953. [Google Scholar] [CrossRef]
- Wang, W.J.; Wei, W.J.; Liao, R.Z. Deciphering the chemoselectivity of nickel-dependent quercetin 2, 4-dioxygenase. Phys. Chem. Chem. Phys. 2018, 20, 15784–15794. [Google Scholar] [CrossRef]
- Merkens, H.; Kappl, R.; Jakob, R.P.; Schmid, F.X.; Fetzner, S. Quercetinase QueD of Streptomyces sp. FLA, a Monocupin Dioxygenase with a Preference for Nickel and Cobalt. Biochemistry 2008, 47, 12185–12196. [Google Scholar] [CrossRef]
- Li, H.; Wang, X.; Tian, G.; Liu, Y. Insights into the dioxygen activation and catalytic mechanism of the nickel-containing quercetinase. Catal. Sci. Technol. 2018, 8, 2340–2351. [Google Scholar] [CrossRef]
- Karplus, P.A.; Pearson, M.A.; Hausinger, R.P. 70 years of crystalline urease: What have we learned? Acc. Chem. Res. 1997, 30, 330–337. [Google Scholar] [CrossRef]
- Estiu, G.; Merz, K.M. The hydrolysis of urea and the proficiency of urease. J. Am. Chem. Soc. 2004, 126, 6932–6944. [Google Scholar] [CrossRef]
- Pearson, M.A.; Park, I.S.; Schaller, R.A.; Michel, L.O.; Karplus, P.A.; Hausinger, R.P. Kinetic and Structural Characterization of Urease Active Site Variants. Biochemistry 2000, 39, 8575–8584. [Google Scholar] [CrossRef]
- Benini, S.; Rypniewski, W.R.; Wilson, K.S.; Miletti, S.; Ciurli, S.; Mangani, S. A new proposal for urease mechanism based on the crystal structures of the native and inhibited enzyme from Bacillus pasteurii: Why urea hydrolysis costs two nickels. Structure 1999, 7, 205–216. [Google Scholar] [CrossRef]
- Barrios, A.M.; Lippard, S.J. Interaction of urea with a hydroxide-bridged dinuclear nickel center: An alternative model for the mechanism of urease. J. Am. Chem. Soc. 2000, 122, 9172–9177. [Google Scholar] [CrossRef]
- Blakeley, R.L.; Hinds, J.A.; Kunze, H.E.; Webb, E.C.; Zerner, B. Jack bean urease (EC 3.5.1.5) demonstration of a carbamoyl-transfer reaction and inhibition by hydroxamic acids. Biochemistry 1969, 8, 1991–2000. [Google Scholar] [CrossRef]
- Smyj, R.P. A conformational analysis study of a nickel(II) enzyme: Urease. J. Mol. Struct. THEOCHEM 1997, 391, 207–223. [Google Scholar] [CrossRef]
- Zimmer, M. Molecular mechanics evaluation of the proposed mechanisms for the degradation of urea by urease. J. Biomol. Struct. Dyn. 2000, 17, 787–797. [Google Scholar] [CrossRef]
- Zimmer, M. Are classical molecular mechanics calculations still useful in bioinorganic simulations? Coord. Chem. Rev. 2009, 253, 817–826. [Google Scholar] [CrossRef]
- Musiani, F.; Arnofi, E.; Casadio, R.; Ciurli, S. Structure-based computational study of the catalytic and inhibition mechanisms of urease. J. Biol. Inorg. Chem. 2001, 6, 300–314. [Google Scholar] [CrossRef]
- Suárez, D.; Díaz, N.; Merz, K.M. Ureases: Quantum chemical calculations on cluster models. J. Am. Chem. Soc. 2003, 125, 15324–15337. [Google Scholar] [CrossRef]
- Estiu, G.; Merz, K.M. Catalyzed decomposition of urea. Molecular dynamics simulations of the binding of urea to urease. Biochemistry 2006, 45, 4429–4443. [Google Scholar] [CrossRef]
- Estiu, G.; Merz, K.M. Competitive hydrolytic and elimination mechanisms in the urease catalyzed decomposition of urea. J. Phys. Chem. B 2007, 111, 10263–10274. [Google Scholar] [CrossRef]
- Carlsson, H.; Nordlander, E. Computational Modeling of the Mechanism of Urease. Bioinorg. Chem. Appl. 2010, 2010, 8. [Google Scholar] [CrossRef]
- Mazzei, L.; Cianci, M.; Benini, S.; Ciurli, S. The impact of pH on catalytically critical protein conformational changes; the case of the urease, a nickel enzyme. Chem. Eur. J. 2019. [Google Scholar] [CrossRef]
- Mazzei, L.; Cianci, M.; Benini, S.; Ciurli, S. The structure of the elusive urease-urea complex unveils the mechanism of a paradigmatic nickel-dependent enzyme. Angew. Chem. Int. Ed. 2019, 131, 7493–7497. [Google Scholar] [CrossRef]
- Desguin, B.; Goffin, P.; Viaene, E.; Kleerebezem, M.; Diaconescu, V.M.; Maroney, M.J.; Declercq, J.P.; Soumillion, P.; Hols, P. Lactate racemase is a nickel-dependent enzyme activated by a widespread maturation system. Nat. Commun. 2014, 5, 3615. [Google Scholar] [CrossRef]
- Xu, T.; Bauer, G.; Hu, X. A novel nickel pincer complex in the active site of lactate racemase. ChemBioChem 2016, 17, 31–32. [Google Scholar] [CrossRef][Green Version]
- Zamble, D. It costs more than a nickel. Science 2015, 349, 35–36. [Google Scholar] [CrossRef]
- Yu, M.J.; Chen, S.L. From NAD+ to Nickel Pincer Complex: A Significant Cofactor Evolution Presented by Lactate Racemase. Chem. Eur. J. 2017, 23, 7545–7557. [Google Scholar] [CrossRef]
- Zhang, X.; Chung, L.W. Alternative Mechanistic Strategy for Enzyme Catalysis in a Ni-Dependent Lactate Racemase (LarA): Intermediate Destabilization by the Cofactor. Chem. Eur. J. 2017, 23, 3623–3630. [Google Scholar] [CrossRef]
- Qiu, B.; Yang, X.Z. A bio-inspired design and computational prediction of scorpion-like SCS nickel pincer complexes for lactate racemization. Chem. Commun. 2017, 53, 11410–11413. [Google Scholar] [CrossRef]
- Wang, B.; Shaik, S. The Nickel-Pincer Complex in Lactate Racemase Is an Electron Relay and Sink that acts through Proton-Coupled Electron Transfer. Angew. Chem. Int. Ed. 2017, 56, 10098–10102. [Google Scholar] [CrossRef]
- Zhang, T.H.; Zhang, X.Y.; Chung, L.W. Computational Insights into the Reaction Mechanisms of Nickel-Catalyzed Hydrofunctionalizations and Nickel-Dependent Enzymes. Asian J. Organ. Chem. 2018, 7, 522–536. [Google Scholar] [CrossRef]
- Pelmenschikov, V.; Siegbahn, P.E.M. Nickel superoxide dismutase reaction mechanism studied by hybrid density functional methods. J. Am. Chem. Soc. 2006, 128, 7466–7475. [Google Scholar] [CrossRef]
- Prabhakar, R.; Morokuma, K.; Musaev, D.G. A DFT study of the mechanism of Ni superoxide dismutase (NiSOD): Role of the active site cysteine-6 residue in the oxidative half-reaction. J. Comput. Chem. 2006, 27, 1438–1445. [Google Scholar] [CrossRef]
- Barondeau, D.P.; Kassmann, C.J.; Bruns, C.K.; Tainer, J.A.; Getzoff, E.D. Nickel superoxide dismutase structure and mechanism. Biochemistry 2004, 43, 8038–8047. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, D.; Liu, X.; Zhang, L.F. Theoretical mechanisms of the superoxide radical anion catalyzed by the nickel superoxide dismutase. Comput. Theor. Chem. 2011, 966, 357–363. [Google Scholar] [CrossRef]
- Shearer, J.; Schmitt, J.C.; Clewett, H.S. Adiabaticity of the Proton-Coupled Electron-Transfer Step in the Reduction of Superoxide Effected by Nickel-Containing Superoxide Dismutase Metallopeptide-Based Mimics. J. Phys. Chem. B, 2015, 119, 5453–5461. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme Name | Reaction |
|---|---|
| Hydrogenase |  |
| Methyl-coenzyme M reductase |  |
| CO dehydrogenase |  |
| Acetyl-CoA synthase |  |
| Acireductone dioxygenase |  |
| Quercetin 2,4-dioxygenase |  |
| Superoxide dismutase |  |
| Urease |  |
| Glyoxylase I |  |
| Lactate racemase |  |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siegbahn, P.E.M.; Chen, S.-L.; Liao, R.-Z. Theoretical Studies of Nickel-Dependent Enzymes. Inorganics 2019, 7, 95. https://doi.org/10.3390/inorganics7080095
Siegbahn PEM, Chen S-L, Liao R-Z. Theoretical Studies of Nickel-Dependent Enzymes. Inorganics. 2019; 7(8):95. https://doi.org/10.3390/inorganics7080095
Chicago/Turabian StyleSiegbahn, Per E. M., Shi-Lu Chen, and Rong-Zhen Liao. 2019. "Theoretical Studies of Nickel-Dependent Enzymes" Inorganics 7, no. 8: 95. https://doi.org/10.3390/inorganics7080095
APA StyleSiegbahn, P. E. M., Chen, S.-L., & Liao, R.-Z. (2019). Theoretical Studies of Nickel-Dependent Enzymes. Inorganics, 7(8), 95. https://doi.org/10.3390/inorganics7080095