Adding to the Family of Copper Complexes Featuring Borohydride Ligands Based on 2-Mercaptopyridyl Units

 , , and
, , and 
Abstract
1. Introduction
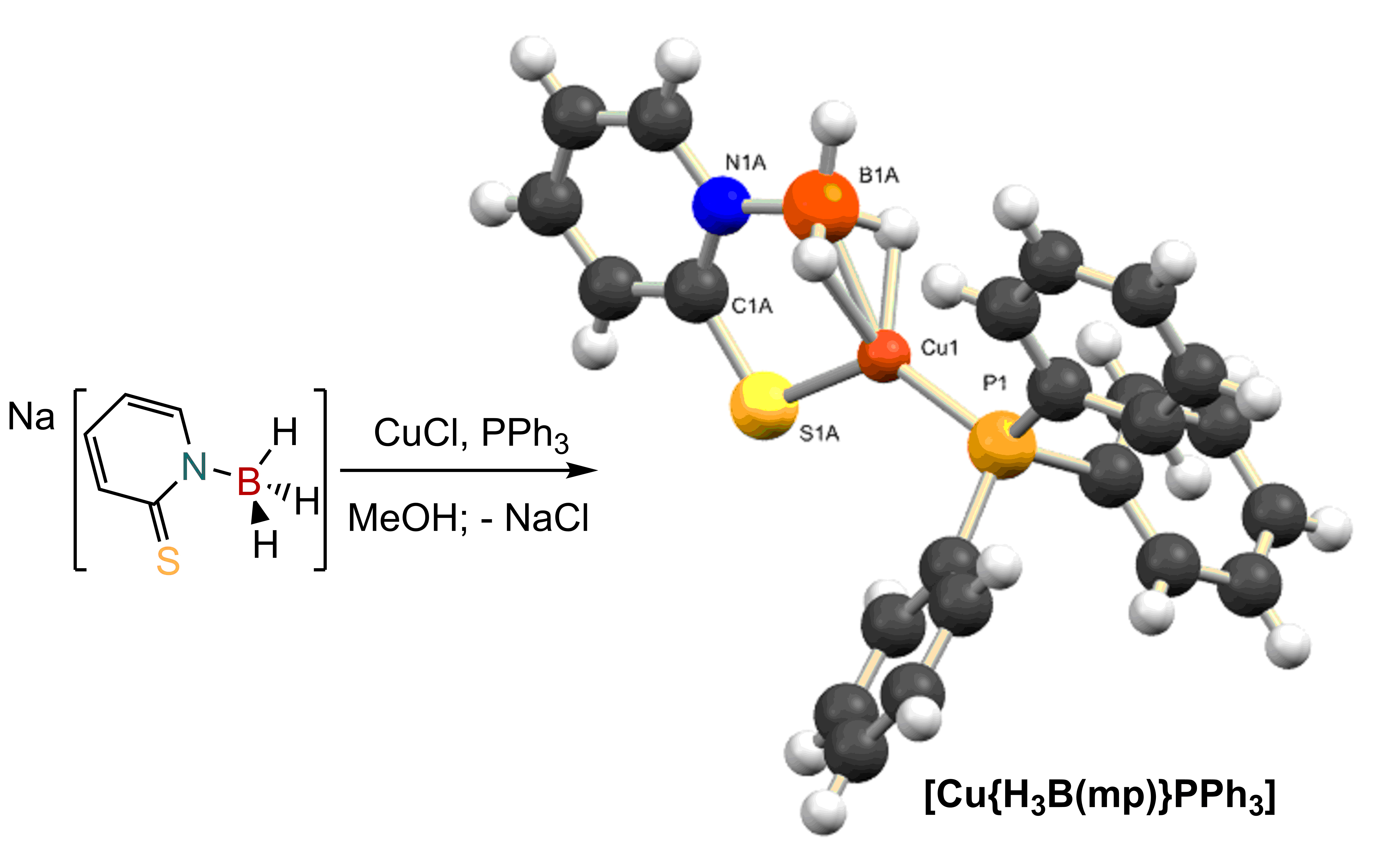
2. Results and Discussion
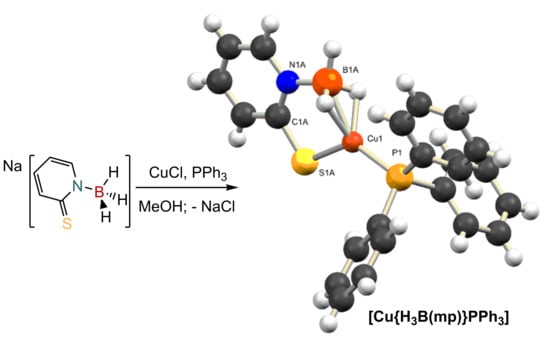
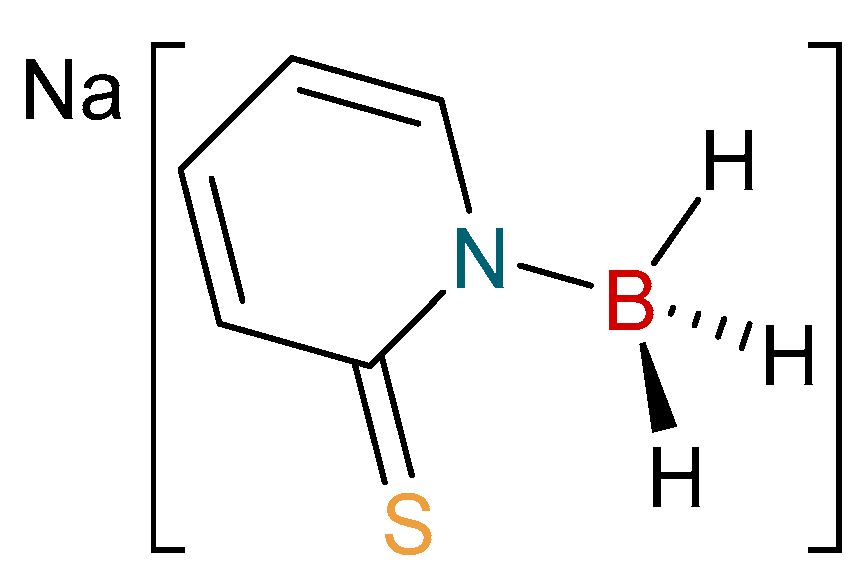
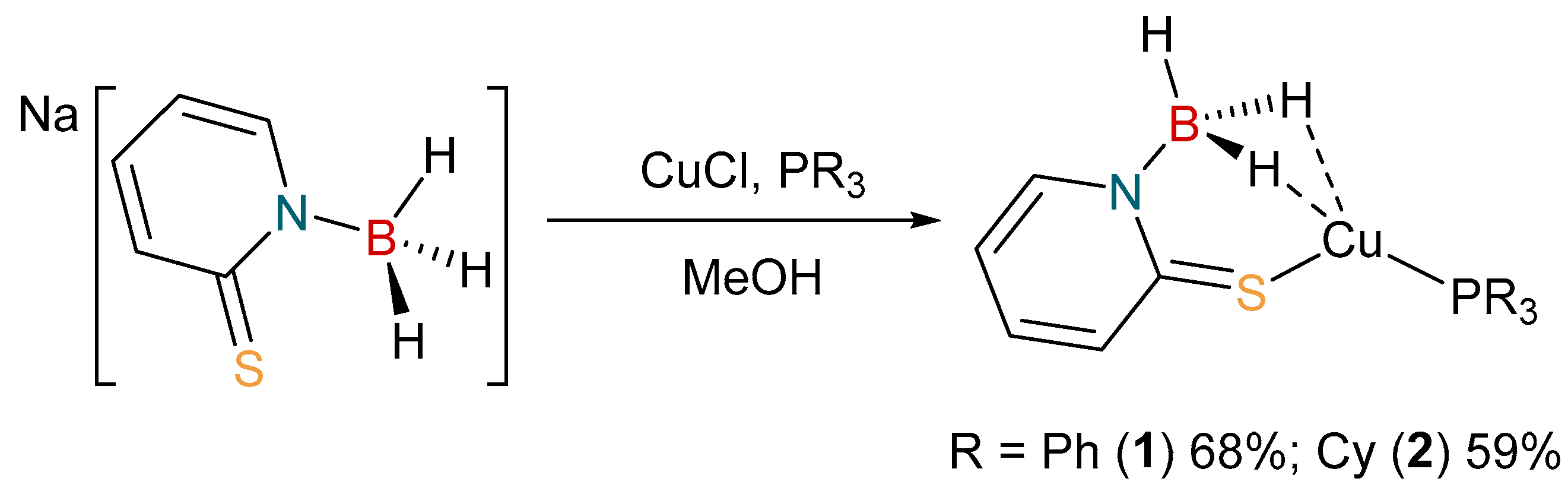
2.1. Synthesis and Characterization of Copper Complexes
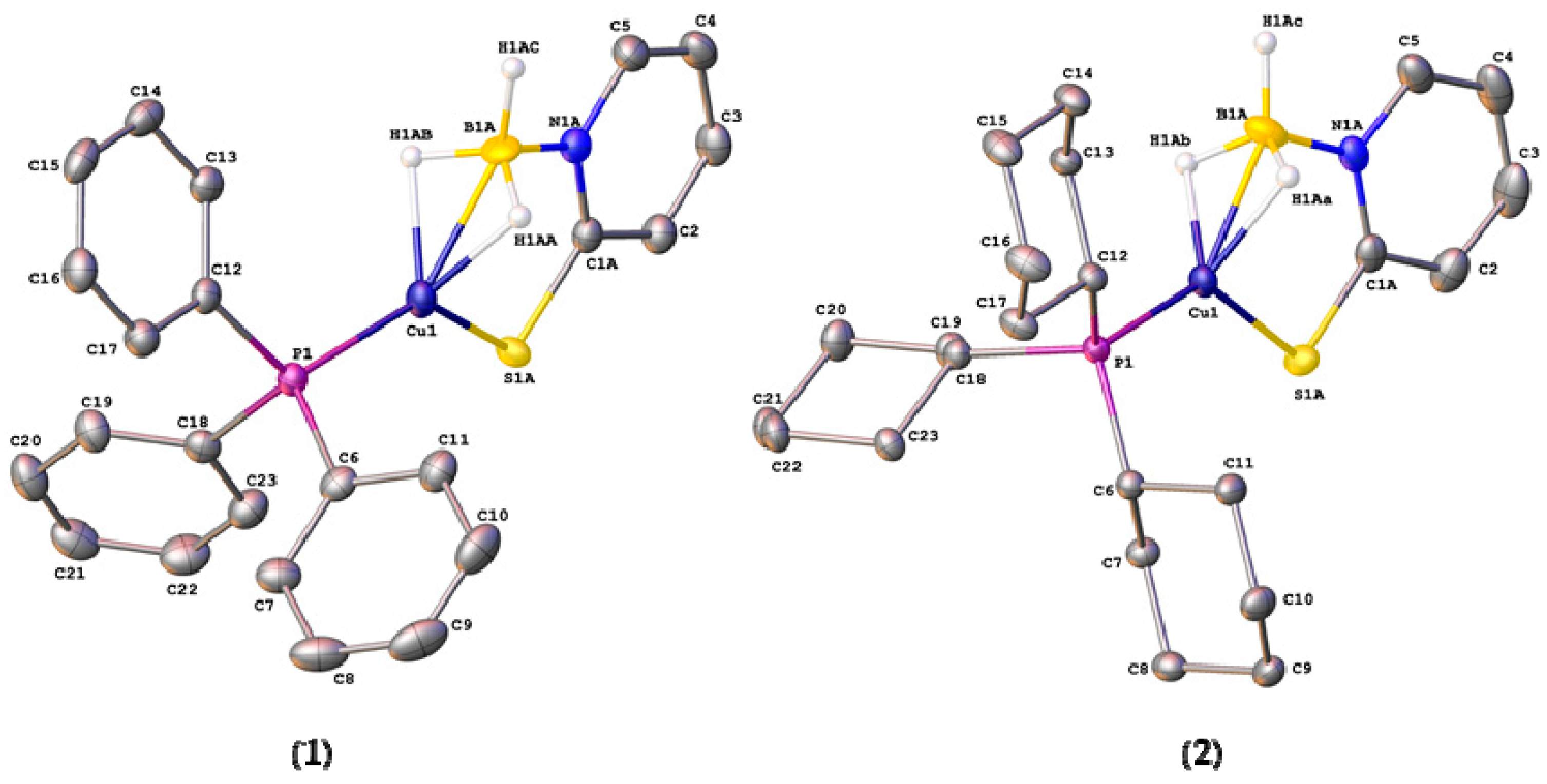
2.2. Structural Characterization of Copper Complexes
3. Materials and Methods
3.1. General Remarks
3.2. Synthesis of [Cu{H3B(mp)}(PPh3)]
3.3. Synthesis of [Cu{H3B(mp)}(PCy3)]
3.4. Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Marks, T.J.; Kolb, J.R. Covalent transition metal, lanthanide, and actinide tetrahydroborate complexes. Chem. Rev. 1977, 77, 263–293. [Google Scholar] [CrossRef]
- Bommer, J.C.; Morse, K.W. Slowing of the fluxional process in a diamagnetic copper(I) tetrahydroborate complex. Inorg. Chem. 1978, 17, 3708–3710. [Google Scholar] [CrossRef]
- Aqra, F.M.A.M. Bidentate bonding mode of tetrahydroborate and nitrite towards copper(II) in open-faced macrocyclic complexes. Trans. Met. Chem. 2004, 29, 921–924. [Google Scholar] [CrossRef]
- Golub, I.E.; Filippov, O.A.; Gutsul, E.I.; Belkova, N.V.; Epstein, L.M.; Rossin, A.; Peruzzini, M.; Shubina, E.S. Dimerization Mechanism of Bis(triphenylphosphine)copper(I) Tetrahydroborate: Proton Transfer via a Dihydrogen Bond. Inorg. Chem. 2012, 51, 6486–6497. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Lin, Z. Transition metal tetrahydroborato complexes: An orbital interaction analysis of their structure and bonding. Coord. Chem. Rev. 1996, 156, 139–162. [Google Scholar] [CrossRef]
- Lledos, A.; Duran, M.; Jean, Y.; Volatron, F. Ab initio Study of the coordination modes of tetrahydroborato ligands: The high-spin complex bis(phosphine)tris(tetrahydroborato)vanadium. Inorg. Chem. 1991, 30, 4440–4445. [Google Scholar] [CrossRef]
- Dias, H.V.R.; Lu, H.-L. Direct Synthesis of a Bis(pyrazolyl)boratocopper(I) Complex: Synthesis and Characterization of [H2B(3,5-(CF3)2Pz)2]Cu(PPh3)2 Displaying an Unusual Coordination Mode for a Poly(pyrazolyl)borate Ligand. Inorg. Chem. 2000, 39, 2246–2248. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Kuwata, S.; Ikariya, T. Synthesis and Reactivity of Tris(7-azaindolyl)boratoruthenium Complex. Comparison with Poly(methimazolyl)borate Analogue. Chem. Let. 2006, 35, 1224–1225. [Google Scholar] [CrossRef]
- Lenczyk, C.; Roy, D.K.; Ghosh, B.; Schwarzmann, J.; Phukan, A.K.; Braunschweig, H. First Bis(σ)-borane Complexes of Group 6 Transition Metals: Experimental and Theoretical Studies. Chem. Eur. J. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Trofimenko, S. Boron-Pyrazole Chemistry. J. Am. Chem. Soc. 1966, 88, 1842–1844. [Google Scholar] [CrossRef]
- Trofimenko, S. Scorpionates: The Coordination of Poly(pyrazolyl)borate Ligands; Imperial College Press: London, UK, 1999. [Google Scholar] [CrossRef]
- Trofimenko, S. Scorpionates: Genesis, milestones, prognosis. Polyhedron 2004, 23, 197–203. [Google Scholar] [CrossRef]
- Pettinari, C. Scorpionates II: Chelating Borate Ligands; Imperial College Press: London, UK, 2008. [Google Scholar] [CrossRef]
- Yap, G.P.A. A brief history of scorpionates. Acta Cryst. 2013, C69, 937–938. [Google Scholar] [CrossRef] [PubMed]
- Many special issues dedicated to the chemistry of scorpionate ligands has been published; see for example, Pettinari, C. Scorpionate Compounds. Eur. J. Inorg. Chem. 2016, 2016, 2209–2211. [CrossRef]
- Spicer, M.D.; Reglinski, J. Soft Scorpionate Ligands Based on Imidazole-2-thione Donors. Eur. J. Inorg. Chem. 2009, 1553–1574. [Google Scholar] [CrossRef]
- Hill, A.F.; Owen, G.R.; White, A.J.P.; Williams, D.J. The Sting of the Scorpion: A Metallaboratrane. Angew. Chem. Int. Ed. 1999, 38, 2759–2761. [Google Scholar] [CrossRef]
- Owen, G.R. Hydrogen atom storage upon Z-class borane ligand functions: An alternative approach to ligand cooperation. Chem. Soc. Rev. 2012, 41, 3535–3546. [Google Scholar] [CrossRef] [PubMed]
- Owen, G.R. Functional group migrations between boron and metal centres within transition metal–borane and –boryl complexes and cleavage of H–H, E–H and E–E′ bonds. Chem. Commun. 2016, 52, 10712–10726. [Google Scholar] [CrossRef] [PubMed]
- Bouhadir, G.; Bourissou, D. Complexes of ambiphilic ligands: Reactivity and catalytic applications. Chem. Soc. Rev. 2016, 45, 1065–1079. [Google Scholar] [CrossRef]
- Crossley, I.R.; Hill, A.F.; Willis, A.C. Unlocking the metallaboratrane cage: Reversible B–H activation in platinaboratranes. Dalton Trans. 2008, 201–203. [Google Scholar] [CrossRef]
- Neshat, A.; Shahsavari, H.R.; Mastrorilli, P.; Todisco, S.; Haghighi, M.G.; Notash, B. A Borane. A Borane Platinum Complex Undergoing Reversible Hydride Migration in Solution. Inorg. Chem. 2018, 57, 1398–1407. [Google Scholar] [CrossRef]
- Garner, M.; Reglinski, J.; Cassidy, I.; Spicer, M.D.; Kennedy, A.R. Hydrotris(methimazolyl)borate, a soft analogue of hydrotris(pyrazolyl)borate. Preparation and crystal structure of a novel zinc complex. Chem. Commun. 1996, 1975–1976. [Google Scholar] [CrossRef]
- Ma, C.; Hill, A.F. Methimazolyl based diptych bicyclo-[3.3.0]-ruthenaboratranes. Dalton Trans. 2019, 48, 1976–1992. [Google Scholar] [CrossRef] [PubMed]
- Foreman, M.R.S.-J.; Hill, A.F.; Ma, C.; Tshabang, N.; Whited, A.J.P. Synthesis and ligand substitution reactions of κ4-B,S,S′,S′′-ruthenaboratranes. Dalton Trans. 2019, 48, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F.; Schwich, T.; Xiong, Y. 5-Mercaptotetrazolyl-derived metallaboratranes. Dalton Trans. 2019, 48, 2367–2376. [Google Scholar] [CrossRef] [PubMed]
- Gomosta, S.; Ramalakshmi, R.; Arivazhagan, C.; Haridas, A.; Raghavendra, B.; Maheswari, K.; Roisnel, T.; Ghosh, S.Z. Synthesis, Structural Characterization, and Theoretical Studies of Silver(I) Complexes of Dihydrobis(2-mercapto-benzothiazolyl) Borate. Anorg. Allg. Chem. 2019, 645, 588–594. [Google Scholar] [CrossRef]
- Song, D.; Jia, W.L.; Wu, G.; Wang, S. Cu(I) and Zn(II) complexes of 7-azaindole-containing scorpionates: Structures, luminescence and fluxionality. Dalton Trans. 2005, 433–438. [Google Scholar] [CrossRef]
- Wagler, J.; Hill, A.F. 7-Azaindol-7-ylborate: A Novel Bidentate N^BH3 Chelating Ligand. Organometallics 2008, 27, 2350–2353. [Google Scholar] [CrossRef]
- Da Costa, R.C.; Rawe, B.W.; Tsoureas, N.; Haddow, M.F.; Sparkes, H.A.; Tizzard, G.J.; Coles, S.J.; Owen, G.R. Preparation and reactivity of rhodium and iridium complexes containing a methylborohydride based unit supported by two 7-azaindolyl heterocycles. Dalton Trans. 2018, 47, 11047–11057. [Google Scholar] [CrossRef]
- Tsoureas, N.; Hamilton, A.; Haddow, M.F.; Harvey, J.N.; Orpen, A.G.; Owen, G.R. Insight into the Hydrogen Migration Processes Involved in the Formation of Metal–Borane Complexes: Importance of the Third Arm of the Scorpionate Ligand. Organometallics 2013, 32, 2840–2856. [Google Scholar] [CrossRef]
- Holler, S.; Tüchler, M.; Belaj, F.; Veiros, L.F.; Kirchner, K.; Mösch-Zanetti, N.C. Thiopyridazine-Based Copper Boratrane Complexes Demonstrating the Z-type Nature of the Ligand. Inorg. Chem. 2016, 55, 4980–4991. [Google Scholar] [CrossRef]
- Tüchler, M.; Ramböck, M.; Glanzer, S.; Zangger, K.; Belaj, F.; Mösch-Zanetti, N.C. Mono- and Hexanuclear Zinc Halide Complexes with Soft Thiopyridazine Based Scorpionate Ligands. Inorganics 2019, 7, 24. [Google Scholar] [CrossRef]
- Maria, L.; Paulo, A.; Santos, I.C.; Santos, I.; Kurz, P.; Springler, B.; Alberto, R. Very Small and Soft Scorpionates: Water Stable Technetium Tricarbonyl Complexes Combining a Bis-agostic (k3-H, H, S) Binding Motif with Pendant and Integrated Bioactive Molecules. J. Am. Chem. Soc. 2006, 128, 14590–14598. [Google Scholar] [CrossRef] [PubMed]
- Videira, M.; Maria, L.; Paulo, A.; Santos, I.C.; Santos, I.; Vaz, P.D.; Calhorda, M.J. Mixed-Ligand Rhenium Tricarbonyl Complexes Anchored on a (κ2-H,S) Trihydro(mercaptoimidazolyl)borate: A Missing Binding Motif for Soft Scorpionates. Organometallics 2008, 27, 1334–1337. [Google Scholar] [CrossRef]
- Dyson, G.; Hamilton, A.; Mitchell, B.; Owen, G.R. A new family of flexible scorpionate ligands based on 2-mercaptopyridine. Dalton Trans. 2009, 6120–6126. [Google Scholar] [CrossRef] [PubMed]
- Iannetelli, A.; Tizzard, G.J.; Coles, S.J.; Owen, G.R. Sequential Migrations between Boron and Rhodium Centers: A Cooperative Process between Rhodium and a Monosubstituted Borohydride Unit. Inorg. Chem. 2018, 57, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Iannetelli, A.; Tizzard, G.J.; Coles, S.J.; Owen, G.R. Synthesis and Characterization of Platinum and Palladium Complexes Featuring a Rare Secondary Borane Pincer Motif. Organometallics 2018, 37, 2177–2187. [Google Scholar] [CrossRef]
- Zech, A.; Haddow, M.F.; Othman, H.; Owen, G.R. Utilizing the 8-Methoxycyclooct-4-en-1-ide Unit As a Hydrogen Atom Acceptor en Route to “Metal–Borane Pincers”. Organometallics 2012, 31, 6753–6760. [Google Scholar] [CrossRef]
- Anju, R.S.; Roy, D.K.; Mondal, B.; Yuvaraj, K.; Arivazhagan, C.; Saha, K.; Varghese, B.; Ghosh, S. Reactivity of Diruthenium and Dirhodium Analogues of Pentaborane(9): Agostic versus Boratrane Complexes. Angew. Chem. Int. Ed. 2014, 53, 2873–2877. [Google Scholar] [CrossRef]
- Roy, D.K.; Mondal, B.; Anju, R.S.; Ghosh, S. Chemistry of Diruthenium and Dirhodium Analogues of Pentaborane(9): Synthesis and Characterization of Metal N,S-Heterocyclic Carbene and B-Agostic Complexes. Chem. Eur. J. 2015, 21, 3640–3648. [Google Scholar] [CrossRef]
- Anju, R.S.; Mondal, B.; Saha, K.; Panja, S.; Varghese, B.; Ghosh, S. Hydroboration of Alkynes with Zwitterionic Ruthenium–Borate Complexes: Novel Vinylborane Complexes. Chem. Eur. J. 2015, 21, 11393–11400. [Google Scholar] [CrossRef]
- Ramalakshmi, R.; Saha, K.; Roy, D.K.; Varghese, B.; Phukan, A.K.; Ghosh, S. New Routes to a Series of σ-Borane/Borate Complexes of Molybdenum and Ruthenium. Chem. Eur. J. 2015, 21, 17191–17195. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.K.; Borthakur, R.; De, A.; Varghese, B.; Phukan, A.K.; Ghosh, S. Synthesis and Characterization of Bis(sigma)borate and Bis–zwitterionic Complexes of Rhodium and Iridium. Chem. Select 2016, 1, 3757–3761. [Google Scholar] [CrossRef]
- Saha, K.; Joseph, B.; Ramalakshmi, R.; Anju, R.S.; Varghese, B.; Ghosh, S. η4-HBCC-σ,π-Borataallyl Complexes of Ruthenium Comprising an Agostic Interaction. Chem. Eur. J. 2016, 22, 7871–7878. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Joseph, B.; Borthakur, R.; Ramalakshmi, R.; Roisnel, T.; Ghosh, S. Chemistry of ruthenium σ-borane complex, [Cp∗RuCO(μ-H)BH2L] (Cp∗ = η5-C5Me5; L = C7H4NS2) with terminal and internal alkynes: Structural characterization of vinyl hydroborate and vinyl complexes of ruthenium. Polyhedron 2017, 125, 246–252. [Google Scholar] [CrossRef]
- Nako, A.E.; White, A.J.P.; Crimmin, M.R. Bis(σ-B–H) complexes of copper(I): Precursors to a heterogeneous amine–borane dehydrogenation catalyst. Dalton Trans. 2015, 44, 12530–12534. [Google Scholar] [CrossRef] [PubMed]
- Hicken, A.; White, A.J.P.; Crimmin, M.R. Reversible Coordination of Boron–, Aluminum–, Zinc–, Magnesium–, and Calcium–Hydrogen Bonds to Bent {CuL2} Fragments: Heavy σ Complexes of the Lightest Coinage Metal. Inorg. Chem. 2017, 56, 8669–8682. [Google Scholar] [CrossRef] [PubMed]
- This signal was unambiguously confirmed via a COSY NMR experiment
- Coles, S.J.; Gale, P.A. Changing and challenging times for service crystallography. Chem. Sci. 2012, 3, 683. [Google Scholar] [CrossRef]
- CrysAlisPro Software System, Rigaku, V1.171.40.40a, Rigaku Oxford Diffraction, 2019
- Sheldrick, G.M. Crystal structure refinement with ShelXL. Acta Cryst. 2015, C27, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal structure determination. Acta Cryst. 2015, A71, 3. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound 1 | 11B{1H} NMR 2 | 31P{1H} NMR | 13C{1H} NMR C=S | 1H{11B} NMR 3 BHn | IR B–H 4 |
|---|---|---|---|---|---|
| Na[H3B(mp)] 5 | −14.1 (44) | - | 181.3 | 2.11 | 2307 |
| [Cu{H3B(mp)}(PPh3)] (1) | −13.9 (113) | 4.8 | 175.9 | 2.64 | 2439 (t)/2078 (κ2) |
| [Cu{H3B(mp)}(PCy3)] (2) | −13.4 (90) | 27.2 | 176.1 | 2.42 | 2448 (t)/2085 (κ2) |
| Na[H2B(mp)2] 6 | −3.7 (211) | - | 182.6 | 3.64 | 2438, 2370 |
| [Cu{H2B(mp)2}(PPh3)] (3) | 0.7 (265) | 1.7 | n.o. 7 | 4.12 | 2425 |
| [Cu{H2B(mp)2}(PCy3)] | −0.7 (248) | 19.0 | 178.2 | 3.99 | 2374 |
| K[HB(mp)3] 6 | 4.4 (560) | - | 182.5 | 4.83 | 2468 |
| [Cu{HB(mp)3}(PPh3)] | −0.1 (412) | −2.4 | 178.3 | n.o. 7 | 2458 |
| [Cu{HB(mp)3}(PCy3)] | −0.5 (331) | 17.4 | 181.0 | 5.86 | n.o. 7 |
| [Cu{H3B(mp)}PPh3] 1 | [Cu{H3B(mp)}PCy3] 2 | [Cu{H2B(mp)2}PPh3] 3 | |
|---|---|---|---|
| Cu(1)–P(1) | 2.1789(4) | 2.1876(4) | 2.216(3) |
| Cu(1)–B(1) | 2.113(17)/2.229(14) | 2.153(16)/2.10(3) | 2.7479(15) |
| Cu(1)–S(1) | 2.205(2)/2.221(4) | 2.2523(12)/2.296(12) | 2.255(4) and 2.248(4) |
| C(1)–S(1) | 1.7515(17)/1.722(2) | 1.7244(17)/1.751(13) | 1.707(14) and 1.708(14) |
| B(1)–N(1) | 1.551(8)/1.465(10) | 1.602(16)/1.61(2) | 1.592(2) and 1.583(18) |
| N(1)–C(1) | 1.3506(19)/1.3506(19) | 1.3550(19)/1.3550(19) | 1.3649(17) and 1.3648(19) |
| B(1)–H(1AA) | 1.17(2)/1.18(2) | 1.16(2)/1.16(2) | - |
| B(1)–H(1AB) | 1.16(2)/1.18(2) | 1.17(2)/1.15(2) | 1.090(18) (terminal) |
| B(1)–H(1AC) | 1.17(2)/1.17(2) | 1.14(2)/1.15(2) | 1.150(17) (bridging) |
| Cu(1)–H(1AA) | 1.75(3)/1.81(4) | 1.75(2)/1.68(8) | 1.832(17) |
| Cu(1)–H(1AB) | 1.81(3)/1.85(4) | 1.81(2)/1.82(8) | - |
| S(1)–Cu(1)–P(1) | 129.93(3)/134.69(5) | 129.93(3)/135.9(3) | 111.88(15) and 124.56(14) |
| S(1)–Cu(1)–B(1) | 89.2(2)/87.3(2) | 89.7(4)/90.2(5) | 82.29(3) and 80.27(3) |
| P(1)–Cu(1)–B(1) | 140.5(2)/137.5(3) | 140.3(4)/133.9(6) | 135.64(3) |
| Σangles around Cu 4 | 359.63/359.49 | 359.93/360.0 | 350.4 |
| C(1)–S(1)–Cu(1) | 99.53(9)/99.14(16) | 99.53(8)/96.2(5) | 106.49(5) and 109.83(5) |
| N(1)–B(1)–Cu(1) | 110.0(8)/108.7(7) | 107.0(8)/110.3(13) | 95.36 and 99.09 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goldsworthy, J.; Thomas, S.D.; Tizzard, G.J.; Coles, S.J.; Owen, G.R. Adding to the Family of Copper Complexes Featuring Borohydride Ligands Based on 2-Mercaptopyridyl Units. Inorganics 2019, 7, 93. https://doi.org/10.3390/inorganics7080093
Goldsworthy J, Thomas SD, Tizzard GJ, Coles SJ, Owen GR. Adding to the Family of Copper Complexes Featuring Borohydride Ligands Based on 2-Mercaptopyridyl Units. Inorganics. 2019; 7(8):93. https://doi.org/10.3390/inorganics7080093
Chicago/Turabian StyleGoldsworthy, Joseph, Simon D. Thomas, Graham J. Tizzard, Simon J. Coles, and Gareth R. Owen. 2019. "Adding to the Family of Copper Complexes Featuring Borohydride Ligands Based on 2-Mercaptopyridyl Units" Inorganics 7, no. 8: 93. https://doi.org/10.3390/inorganics7080093
APA StyleGoldsworthy, J., Thomas, S. D., Tizzard, G. J., Coles, S. J., & Owen, G. R. (2019). Adding to the Family of Copper Complexes Featuring Borohydride Ligands Based on 2-Mercaptopyridyl Units. Inorganics, 7(8), 93. https://doi.org/10.3390/inorganics7080093