Human Acireductone Dioxygenase (HsARD), Cancer and Human Health: Black Hat, White Hat or Gray?


Abstract
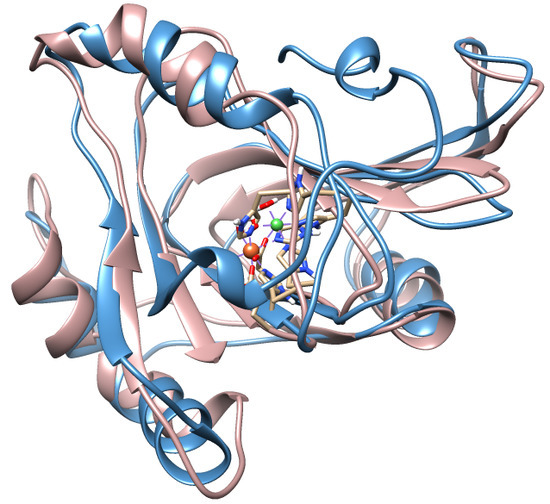
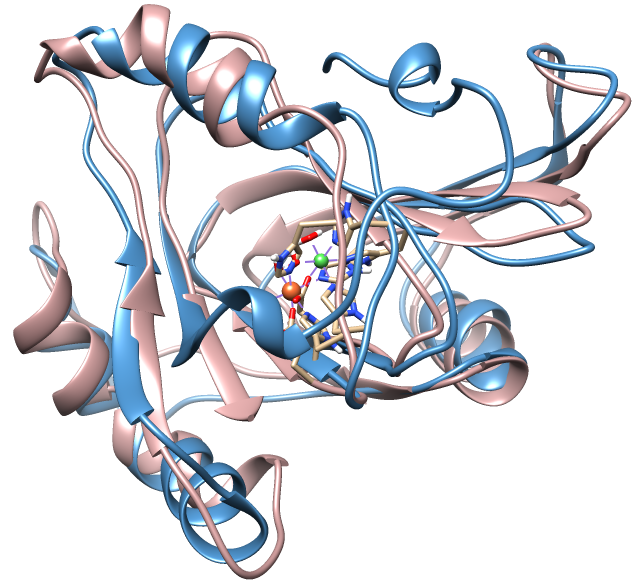
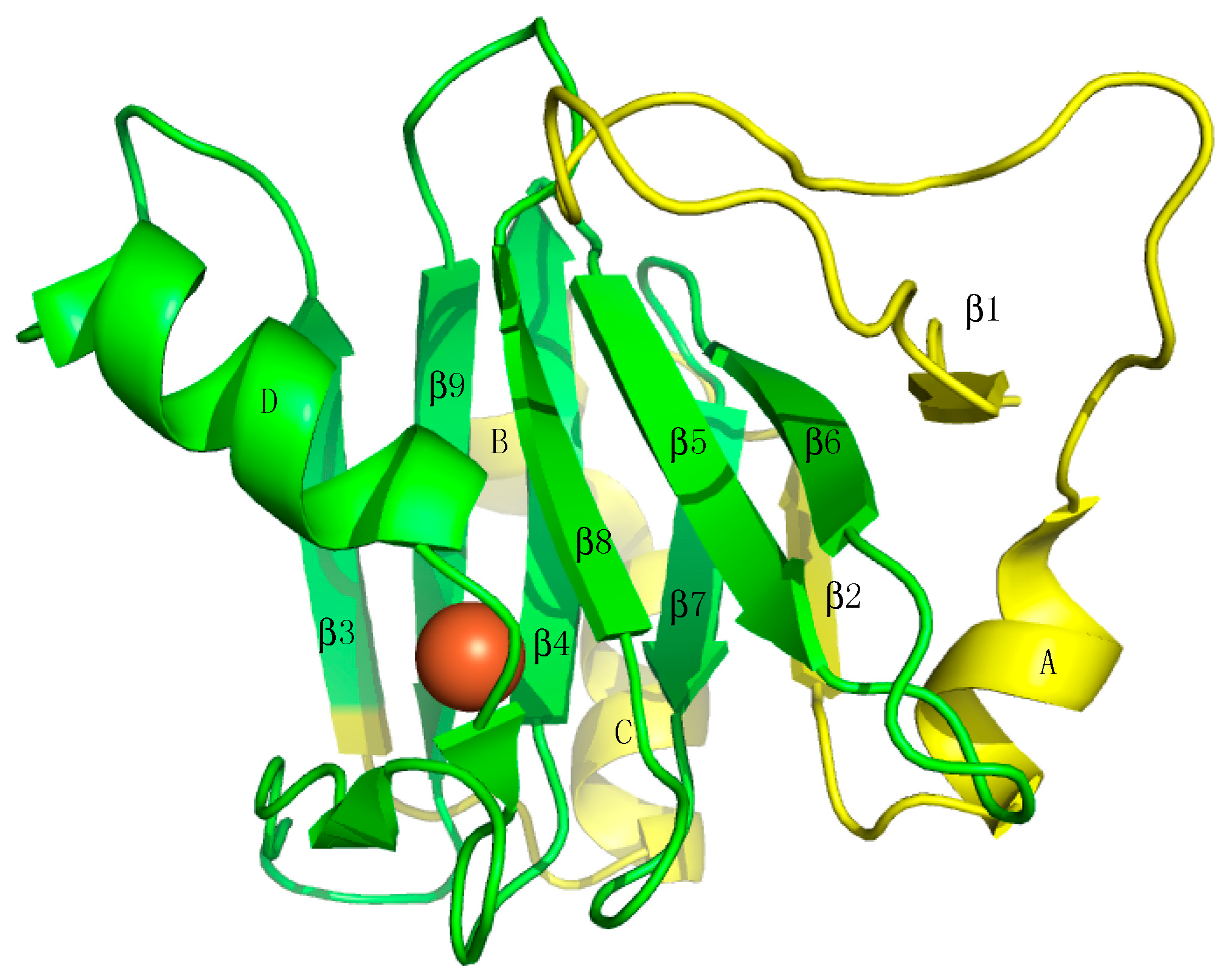
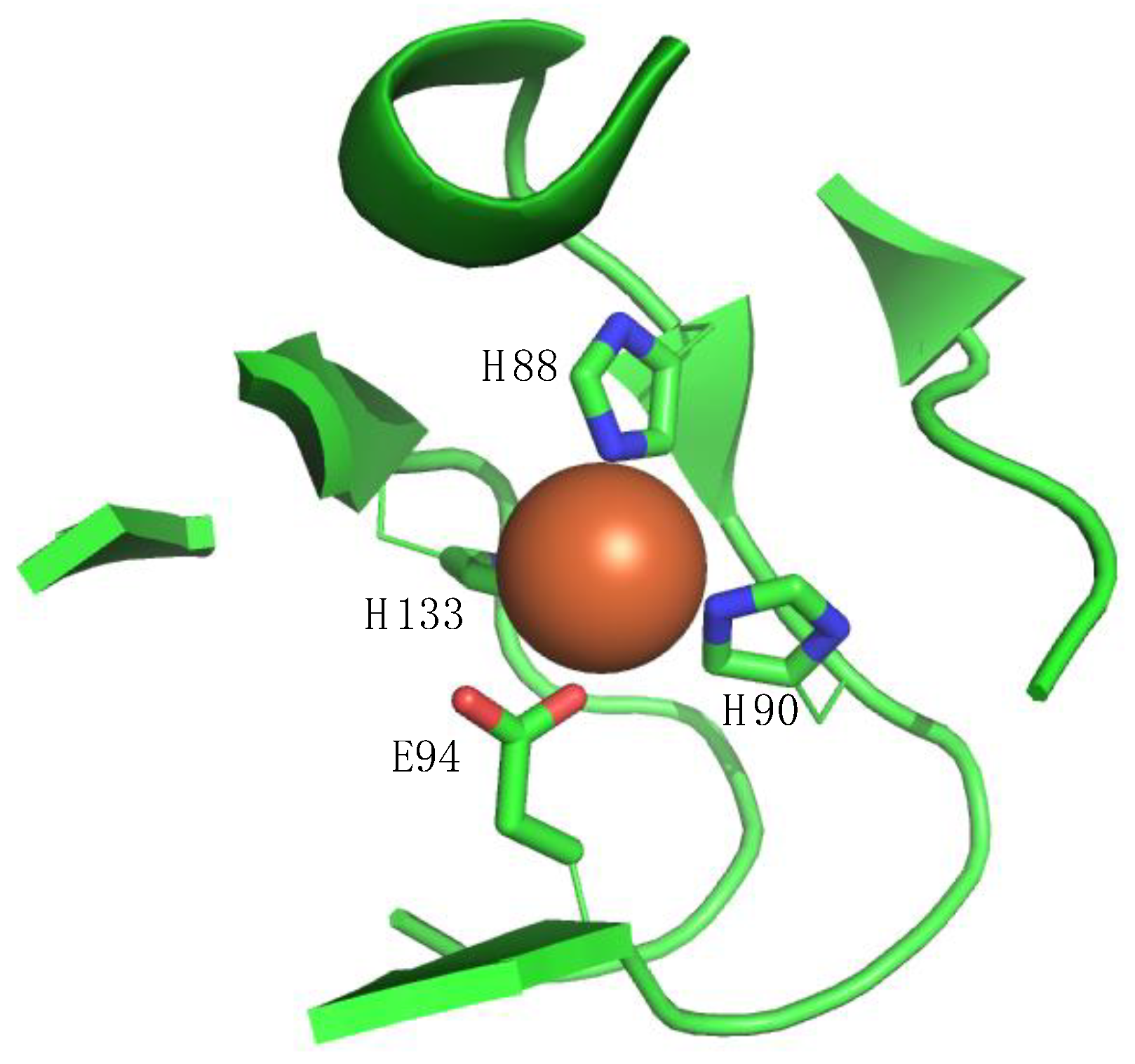
1. Introduction
2. ADI1 and ADI1GP
2.1. A Role for ADI1GP in Hepatitis C Virus (HCV) Infection
2.2. ADI1 in Hepatocellular Carcinoma (HCC)
2.3. ADI1GP Induces Apoptosis in Prostate Cancer Cell Lines
2.4. ADI1GP Regulation of Membrane-Type 1 Matrix Metalloproteinase (MT1-MMP)
2.5. ADI1GP/MT1-MMP Interaction Restricts Metastasis of Pancreatic Ductal Adenocarcinoma (PDAC)
3. Polyamines, MSP and Cancer
4. Carbon Monoxide as an Anti-Apoptotic Signal: A Potential off-MSP Role for ADI1GP in Carcinogenesis
5. Conclusions
- Does the metal-dependent on/off MSP divergence occur in human cell lines? If so, under what conditions? Is off-MSP chemistry observed only under certain circumstances (e.g., in transformed cells)?
- Is the methionine dependence of many cancer cell lines the result of off-MSP chemistry?
- Do different metals bound to the ADI1GP act as switches for regulation of cell processes such as division, migration, invasion, metastasis and apoptosis? Is it possible to distinguish normal cells from tumor cells based on the metal content of their ADI1GP?
- Does binding of metals other than Fe2+ change the interaction between ADI1 and MT1-MMP?
- A systems biology approach will be necessary to place all of these possibilities into a complex cellular context, but answering some of these questions will help to define the role(s) of ADI1GP and, particularly, HsARD in human health.
Author Contributions
Funding
Conflicts of Interest
References
- Dai, Y.; Wensink, O.C.; Abeles, R.H. One Protein, Two Enzymes. J. Biol. Chem. 1999, 274, 1193–1195. [Google Scholar] [CrossRef] [PubMed]
- Milaczewska, A.; Kot, E.; Amaya, J.A.; Makris, T.M.; Zajac, M.; Korecki, J.; Chumakov, A.; Trzewik, B.; Kedracka-Krok, S.; Minor, W.; et al. On the Structure and Reaction Mechanism of Human Acireductone Dioxygenase. Chem. A Eur. J. 2018, 24, 5225–5237. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Pochapsky, T.C.; Abeles, R.H. Mechanistic Studies of Two Dioxygenases in the Methionine Salvage Pathway of Klebsiella Pneumoniae. Biochemistry 2001, 40, 6379–6387. [Google Scholar] [CrossRef] [PubMed]
- Ju, T.T.; Goldsmith, R.B.; Chai, S.C.; Maroney, M.J.; Pochapsky, S.S.; Pochapsky, T.C. One Protein, Two Enzymes Revisited: A Structural Entropy Switch Interconverts the Two Isoforms of Acireductone Dioxygenase. J. Mol. Biol. 2006, 363, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.R.; Wagenpfeil, K.; Pochapsky, T.C.; Petsko, G.A.; Ringe, D. Metal-Dependent Function of a Mammalian Acireductone Dioxygenase. Biochemistry 2016, 55, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.R.; Pochapsky, T.C.; Petsko, G.A.; Ringe, D. Dual Chemistry Catalyzed by Human Acireductone Dioxygenase. Protein Eng. Des. Sel. 2017, 30, 109–206. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.R.; Pochapsky, T.C.; Ringe, D. The Metal Drives the Chemistry: Dual Functions of Acireductone Dioxygenase. Chem. Rev. 2017, 117, 10474–10501. [Google Scholar] [CrossRef] [PubMed]
- Najim, N.; Podmore, I.D.; McGown, A.; Estlin, E.J. Biochemical Changes and Cytotoxicity Associated with Methionine Depletion in Paediatric Central Nervous System Tumour Cell Lines. Anticancer Res. 2009, 29, 2971–2976. [Google Scholar] [PubMed]
- Kokkinakis, D.M.; Hoffman, R.M.; Frenkel, E.P.; Wick, J.B.; Han, Q.; Xu, M.; Tan, Y.; Schold, S.C. Synergy between Methionine Stress and Chemotherapy in the Treatment of Brain Tumor Xenografts in Athymic Mice. Cancer Res. 2001, 61, 4017–4023. [Google Scholar]
- Willmann, L.; Erbes, T.; Halbach, S.; Brummer, T.; Jäger, M.; Hirschfeld, M.; Fehm, T.; Neubauer, H.; Stickeler, E.; Kammerer, B. Exometabolom Analysis of Breast Cancer Cell Lines: Metabolic Signature. Sci. Rep. 2015, 5, 13374. [Google Scholar] [CrossRef]
- Kano, Y.; Sakamoto, S.; Kasahara, T.; Kusumoto, K.; Hida, K.; Suda, K.; Ozawa, K.; Miura, Y.; Takaku, F. Methionine Dependency of Cell Growth in Normal and Malignant Hematopoietic Cells. Cancer Res. 1982, 42, 3090–3092. [Google Scholar] [PubMed]
- Poirson-Bichat, F.; Gonçalves, R.A.B.; Miccoli, L.; Dutrillaux, B.; Poupon, M.F. Methionine Depletion Enhances the Antitumoral Efficacy of Cytotoxic Agents in Drug-Resistant Human Tumor Xenografts. Clin. Cancer Res. 2000, 6, 643–653. [Google Scholar] [PubMed]
- Lu, S.; Epner, D.E. Molecular Mechanisms of Cell Cycle Block by Methionine Restriction in Human Prostate Cancer Cells. Nutr. Cancer 2000, 38, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.-Y.; Herrera, H.; Groce, A.; Hoffman, R.M. Expression of the Biochemical Defect of Methionine Dependence in Fresh Patient Tumors in Primary Histoculture. Cancer Res. 1993, 53, 2479–2483. [Google Scholar] [PubMed]
- Mecham, J.O.; Rowitch, D.; Wallace, C.D.; Stern, P.H.; Hoffman, R.M. The Metabolic Defect of Methionine Dependence Occurs Frequently in Human Tumor Cell Lines. Biochem. Biophys. Res. Commun. 1983, 117, 429–434. [Google Scholar] [CrossRef]
- Sauter, M.; Lorbiecke, R.; OuYang, B.; Pochapsky, T.C.; Rzewuski, G. The Immediate-Early Ethylene Response Gene Osard1 Encodes an Acireductone Dioxygenase Involved in Recycling of the Ethylene Precursor S-Adenosylmethionine. Plant J. 2005, 44, 718–729. [Google Scholar] [CrossRef]
- Hirano, W.; Gotoh, I.; Uekita, T.; Seiki, M. Membrane-Type 1 Matrix Metalloproteinase Cytoplasmic Tail Binding Protein-1 (Mtcbp-1) Acts as an Eukaryotic Aci-Reductone Dioxygenase (Ard) in the Methionine Salvage Pathway. Genes Cells 2005, 10, 565–574. [Google Scholar] [CrossRef]
- Deshpande, A.R.; Wagenpfeil, K.; Pochapsky, T.C.; Petsko, G.; Ringe, D. Metal Drives Chemistry: Dual-Function of Acireductone Dioxygenase Enzymes. FASEB J. 2017, 31, 1. [Google Scholar] [CrossRef]
- Yeh, C.-T.; Lai, H.-Y.; Chen, T.-C.; Chu, C.-M.; Liaw, Y.-F. Identification of a Hepatic Factor Capable of Supporting Hepatitis C Virus Replication in a Nonpermissive Cell Line. J. Virol. 2001, 75, 11017–11024. [Google Scholar] [CrossRef]
- Yeh, C.-T.; Lai, H.-Y.; Yeh, Y.-J.; Cheng, J.-C. Hepatitis C Virus Infection in Mouse Hepatoma Cells Co-Expressing Human Cd81 and Sip-L. Biochem. Biophys. Res. Commun. 2008, 372, 157–161. [Google Scholar] [CrossRef]
- Cheng, J.C.; Yeh, Y.J.; Pai, L.M.; Chang, M.L.; Yeh, C.T. 293 Cells over-Expressing Human ADI1 and Cd81 Are Permissive for Serum-Derived Hepatitis C Virus Infection. J. Med. Virol. 2009, 81, 1560–1568. [Google Scholar] [CrossRef]
- Oram, S.W.; Ai, J.; Pagani, G.M.; Hitchens, M.R.; Stern, J.A.; Eggener, S.; Pins, M.; Xiao, W.; Cai, X.; Haleem, R.; et al. Expression and Function of the Human Androgen-Responsive Gene ADI1 in Prostate Cancer. Neoplasia 2007, 9, 643–651. [Google Scholar] [CrossRef][Green Version]
- Chang, M.-L.; Huang, Y.-H.; Cheng, J.-C.; Yeh, C.-T. Interaction between Hepatic Membrane Type 1 Matrix Metalloproteinase and Acireductone Dioxygenase 1 Regulates Hepatitis C Virus Infection. J. Viral Hepat. 2016, 23, 256–266. [Google Scholar] [CrossRef]
- Chu, Y.D.; Lai, H.Y.; Pai, L.M.; Huang, Y.H.; Lin, Y.H.; Liang, K.H.; Yeh, C.T. The Methionine Salvage Pathway-Involving ADI1 Inhibits Hepatoma Growth by Epigenetically Altering Genes Expression Via Elevating S-Adenosylmethionine. Cell Death Dis. 2019, 10, 240. [Google Scholar] [CrossRef]
- Miyamori, H.; Takino, T.; Kobayashi, Y.; Tokai, H.; Itoh, Y.; Seiki, M.; Sato, H. Claudin Promotes Activation of Pro-Matrix Metalloproteinase-2 Mediated by Membrane-Type Matrix Metalloproteinases. J. Biol. Chem. 2001, 276, 28204–28211. [Google Scholar] [CrossRef]
- Seiki, M. The Cell Surface: The Stage for Matrix Metalloproteinase Regulation of Migration. Curr. Opin. Cell Biol. 2002, 14, 624–632. [Google Scholar] [CrossRef]
- Chai, S.C.; Ju, T.T.; Dang, M.; Goldsmith, R.B.; Maroney, M.J.; Pochapsky, T.C. Characterization of Metal Binding in the Active Sites of Acireductone Dioxygenase Isoforms from Klebsiella Atcc 8724. Biochemistry 2008, 47, 2428–2438. [Google Scholar] [CrossRef]
- Williams, T.M.; Lisanti, M.P. Caveolin-1 in Oncogenic Transformation, Cancer, and Metastasis. Am. J. Physiol. Cell Physiol. 2005, 288, C494–C506. [Google Scholar] [CrossRef]
- Liu, P.; Rudick, M.; Anderson, R.G.W. Multiple Functions of Caveolin-1. J. Biol. Chem. 2002, 277, 41295–41298. [Google Scholar] [CrossRef]
- Wiechen, K.; Diatchenko, L.; Agoulnik, A.; Scharff, K.M.; Schober, H.; Arlt, K.; Zhumabayeva, B.; Siebert, P.D.; Dietel, M.; Schäfer, R.; et al. Caveolin-1 Is Down-Regulated in Human Ovarian Carcinoma and Acts as a Candidate Tumor Suppressor Gene. Am. J. Pathol. 2001, 159, 1635–1643. [Google Scholar] [CrossRef]
- Oram, S.; Jiang, F.; Cai, X.Y.; Haleem, R.; Dincer, Z.; Wang, Z. Identification and Characterization of an Androgen-Responsive Gene Encoding an Aci-Reductone Dioxygenase-Like Protein in the Rat Prostate. Endocrinology 2004, 145, 1933–1942. [Google Scholar] [CrossRef][Green Version]
- Lu, S.C.; Mato, J.M. S-Adenosylmethionine in Cell Growth, Apoptosis and Liver Cancer. J. Gastroenterol. Hepatol. 2008, 23, S73–S77. [Google Scholar] [CrossRef]
- Ansorena, E.; García-Trevijano, E.R.; Martínez-Chantar, M.L.; Huang, Z.; Chen, L.; Mato, J.M.; Iraburu, M.; Lu, S.C.; Avila, M.A. S-Adenosylmethionine and Methylthioadenosine Are Antiapoptotic in Cultured Rat Hepatocytes but Proapoptotic in Human Hepatoma Cells. Hepatology 2002, 35, 274–280. [Google Scholar] [CrossRef]
- Tang, B.; Kadariya, Y.; Murphy, M.E.; Kruger, W.D. The Methionine Salvage Pathway Compound 4-Methylthio-2-Oxobutanate Causes Apoptosis Independent of Down-Regulation of Ornithine Decarboxylase. Biochem. Pharmacol. 2006, 72, 806–815. [Google Scholar] [CrossRef]
- Avila, M.A.; García-Trevijano, E.R.; Lu, S.C.; Corrales, F.J.; Mato, J.M. Methylthioadenosine. Int. J. Biochem. Cell Biol. 2004, 36, 2125–2130. [Google Scholar] [CrossRef]
- Pegg, A.E. Regulation of Ornithine Decarboxylase. J. Biol. Chem. 2006, 281, 14529–14532. [Google Scholar] [CrossRef]
- Uekita, T.; Gotoh, I.; Kinoshita, T.; Itoh, Y.; Sato, H.; Shiomi, T.; Okada, Y.; Seiki, M. Membrane-Type 1 Matrix Metalloproteinase Cytoplasmic Tail-Binding Protein-1 Is a New Member of the Cupin Superfamily—A Possible Multifunctional Protein Acting as an Invasion Suppressor Down-Regulated in Tumors. J. Biol. Chem. 2004, 279, 12734–12743. [Google Scholar] [CrossRef]
- Pratt, J.; Iddir, M.; Bourgault, S.; Annabi, B. Evidence of Mtcbp-1 Interaction with the Cytoplasmic Domain of Mt1-Mmp: Implications in the Autophagy Cell Index of High-Grade Glioblastoma. Mol. Carcinog. 2016, 55, 148–160. [Google Scholar] [CrossRef]
- Seiki, M.; Koshikawa, N.; Yana, I. Role of Pericellular Proteolysis by Membrane-Type 1 Matrix Metalloproteinase in Cancer Invasion and Angiogenesis. Cancer Metastasis Rev. 2003, 22, 129–143. [Google Scholar] [CrossRef]
- Sato, H.; Takino, T.; Okada, Y.; Cao, J.; Shinagawa, A.; Yamamoto, E.; Seiki, M. A Matrix Metalloproteinase Expressed on the Surface of Invasive Tumour Cells. Nature 1994, 370, 61. [Google Scholar] [CrossRef]
- Gotoh, I.; Uekita, T.; Seiki, M. Regulated Nucleo-Cytoplasmic Shuttling of Human Aci-Reductone Dioxygenase (HADI1) and Its Potential Role in Mrna Processing. Genes Cells 2007, 12, 105–117. [Google Scholar] [CrossRef]
- Qiang, L.; Cao, H.; Chen, J.; Weller, S.G.; Krueger, E.W.; Zhang, L.; Razidlo, G.L.; McNiven, M.A. Pancreatic Tumor Cell Metastasis Is Restricted by Mt1-Mmp Binding Protein Mtcbp-1. J. Cell Biol. 2019, 218, 317–332. [Google Scholar] [CrossRef]
- Ridley, A.J. Life at the Leading Edge. Cell 2011, 145, 1012–1022. [Google Scholar] [CrossRef]
- Linder, S. The Matrix Corroded: Podosomes and Invadopodia in Extracellular Matrix Degradation. Trends Cell Biol. 2007, 17, 107–117. [Google Scholar] [CrossRef]
- McNiven, M.A.; Kim, L.; Krueger, E.W.; Orth, J.D.; Cao, H.; Wong, T.W. Regulated Interactions between Dynamin and the Actin-Binding Protein Cortactin Modulate Cell Shape. J. Cell Biol. 2000, 151, 187–198. [Google Scholar] [CrossRef]
- Bae, D.H.; Lane, D.J.R.; Jansson, P.J.; Richardson, D.R. The Old and New Biochemistry of Polyamines. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2053–2068. [Google Scholar] [CrossRef]
- Gerner, E.W.; Meyskens, F.L., Jr. Polyamines and Cancer: Old Molecules, New Understanding. Nat. Rev. Cancer 2004, 4, 781–792. [Google Scholar] [CrossRef]
- Nowotarski, S.L.; Woster, P.M.; Casero, R.A. Polyamines and Cancer: Implications for Chemotherapy and Chemoprevention. Expert Rev. Mol. Med. 2013, 15, e3. [Google Scholar] [CrossRef]
- Cervelli, M.; Pietropaoli, S.; Signore, F.; Amendola, R.; Mariottini, P. Polyamines Metabolism and Breast Cancer: State of the Art and Perspectives. Breast Cancer Res. Treat. 2014, 148, 233–248. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Day, C.P. S-Adenosylmethionine (Same) Therapy in Liver Disease: A Review of Current Evidence and Clinical Utility. J. Hepatol. 2012, 57, 1097–1109. [Google Scholar] [CrossRef]
- Lieber, C.S.; Packer, L. S-Adenosylmethionine: Molecular, Biological, and Clinical Aspects—An Introduction. Am. J. Clin. Nutr. 2002, 76, 1148S–1150S. [Google Scholar] [CrossRef]
- Martínez-Chantar, M.L.; García-Trevijano, E.R.; Latasa, M.U.; Pérez-Mato, I.; del Pino, M.M.S.; Corrales, F.J.; Avila, M.A.; Mato, J.M. Importance of a Deficiency in S-Adenosyl-l-Methionine Synthesis in the Pathogenesis of Liver Injury. Am. J. Clin. Nutr. 2002, 76, 1177S–1182S. [Google Scholar] [CrossRef]
- Mato, J.M.; Lu, S.C. Role of S-Adenosyl-l-Methionine in Liver Health and Injury. Hepatology 2007, 45, 1306–1312. [Google Scholar] [CrossRef]
- Lieber, C.S. S-Adenosyl-l-Methionine: Its Role in the Treatment of Liver Disorders. Am. J. Clin. Nutr. 2002, 76, 1183S–1187S. [Google Scholar] [CrossRef]
- Finkelstein, J.D. The Metabolism of Homocysteine: Pathways and Regulation. Eur. J. Pediatr. 1998, 157, S40–S44. [Google Scholar] [CrossRef]
- Subhi, A.L.; Diegelman, P.; Porter, C.W.; Tang, B.; Lu, Z.J.; Markham, G.D.; Kruger, W.D. Methylthioadenosine Phosphorylase Regulates Ornithine Decarboxylase by Production of Downstream Metabolites. J. Biol. Chem. 2003, 278, 49868–49873. [Google Scholar] [CrossRef]
- Williams-Ashman, H.G.; Seidenfeld, J.; Galletti, P. Trends in the Biochemical Pharmacology of 5’-Deoxy-5’-Methylthioadenosine. Biochem. Pharmacol. 1982, 31, 277–288. [Google Scholar] [CrossRef]
- Nobori, T.; Takabayashi, K.; Tran, P.; Orvis, L.; Batova, A.; Yu, A.L.; Carson, D.A. Genomic Cloning of Methylthioadenosine Phosphorylase: A Purine Metabolic Enzyme Deficient in Multiple Different Cancers. Proc. Natl. Acad. Sci. USA 1996, 93, 6203–6208. [Google Scholar] [CrossRef]
- Pegg, A.E. Polyamine Metabolism and Its Importance in Neoplastic Growth and a Target for Chemotherapy. Cancer Res. 1988, 48, 759–774. [Google Scholar]
- Su, L.; Yang, K.; Li, S.; Liu, C.; Han, J.G.; Zhang, Y.; Xu, G.Z. Enolase-Phosphatase 1 as a Novel Potential Malignant Glioma Indicator Promotes Cell Proliferation and Migration. Oncol. Rep. 2018, 40, 2233–2241. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, T.; Yang, K.; Xu, J.; Ren, L.J.; Li, W.P.; Liu, W.L. Cerebral Microvascular Endothelial Cell Apoptosis after Ischemia: Role of Enolase-Phosphatase 1 Activation and Aci-Reductone Dioxygenase 1 Translocation. Front. Mol. Neurosci. 2016, 9, 79. [Google Scholar] [CrossRef]
- Almeida, A.S.; Figueiredo-Pereira, C.; Vieira, H.L.A. Carbon Monoxide and Mitochondria—Modulation of Cell Metabolism, Redox Response and Cell Death. Front. Physiol. 2015, 6, 33. [Google Scholar] [CrossRef]
- Bakhautdin, B.; Das, D.; Mandal, P.; Roychowdhury, S.; Danner, J.; Bush, K.; Pollard, K.; Kaspar, J.W.; Li, W.; Salomon, R.G.; et al. Protective Role of Ho-1 and Carbon Monoxide in Ethanol-Induced Hepatocyte Cell Death and Liver Injury in Mice. J. Hepatol. 2014, 61, 1029–1037. [Google Scholar] [CrossRef]
- Rochette, L.; Cottin, Y.; Zeller, M.; Vergely, C. Carbon Monoxide: Mechanisms of Action and Potential Clinical Implications. Pharmacol. Ther. 2013, 137, 133–135. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Citation | Type of Cancer | Cell Lines | Met Dependence Ccomplete, ++, Partial, +, Independent, − |
|---|---|---|---|
| Najim et al. (2009) [8] | Central nervous system | Daoy (medulloblastoma) | ++ |
| D-54 (glioma) | ++ | ||
| Kokkinakis et al. (2001) [9] | Brain | D-54 | ++ |
| SWB77 (glioblastoma) | ++ | ||
| Daoy | ++ | ||
| Willmann et al. (2015) [10] | Breast cancer | epithelial cell line MCF-10A | ++ |
| Kano et al. (1982) [11] | Leukemia | Raji (Burkitt) | ++ |
| BALL (B-cell) | ++ | ||
| TALL (T-cell) | ++ | ||
| MOLT-3 (T-cell) | − | ||
| MOLT 4B (T-cell) | ++ | ||
| HL60 (promyelocytic) | ++ | ||
| K562 (chronic myelogenous leukemia in blastic crisis) | ++ | ||
| Poirson-Bichat et al. (2000) [12] | Colon, lung, glioma | TC71-MA (colon) | ++ |
| SCLC6 (small cell lung) | ++ | ||
| SNB19 (glioma) | ++ | ||
| Lu et al. (2000) [13] | Prostate | LNCaP (lymph node metastasis) | − |
| PC-3 (distant metastasis) | ++ | ||
| DU 145(distant metastasis) | + | ||
| Guo et al. (1993) [14] | Prostate, lung, fibrosarcoma | PC3 | ++ |
| SKLU-I (lung carcinoma) | ++ | ||
| HT 1080 (fibrosarcoma) | ++ | ||
| Mecham et al. (1983) [15] | Bladder, breast, cervical, colon, kidney, lung, prostate, fibrosarcoma, osteogenic sarcoma, glioblastoma, neuroblastoma | EJ (bladder) | ++ |
| J82 (bladder) | ++ | ||
| SK-BR-2-II (breast) | ++ | ||
| MCF-7 (breast) | ++ | ||
| HeLa (cervical) | ++ | ||
| SK-CO-1 (colon) | ++ | ||
| A498 (kidney) | ++ | ||
| A2182 (lung) | ++ | ||
| PC-3 | ++ | ||
| 8387 (fibrosarcoma) | ++ | ||
| HT-1080 | ++ | ||
| HOS (osteogenic sarcoma) | ++ | ||
| Human neurological tumors | ++ | ||
| A172 (glioblastoma) | ++ | ||
| SK-N-SH (neuroblastoma) | ++ |
| Types of Cancer | Tissue/Cell Type | ADI1GP Expression | Metastatic/Apoptotic | Mechanism |
|---|---|---|---|---|
| Hepatocellular carcinoma [24] | Tissue, J7 and Huh7 | Down regulated by CAV1, overexpressed | No information | Not proposed |
| Prostate cancer [22,31] | Tissue, LNCaP and PC3 | Observed in epithelial cells, little/no expression in stromal cells. Expression may be Mib regulated, Gleason gr. 3 prostate tumor tissue < ADI1GP than benign tissue. | Apoptotic | Associated with metal binding |
| Pancreatic ductal adenocarcinoma [42] | DanG, BxPC3, and Panc-1 | Overexpressed | Metastatic | ADI1GP MT1-MMP interaction |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Pochapsky, T.C. Human Acireductone Dioxygenase (HsARD), Cancer and Human Health: Black Hat, White Hat or Gray? Inorganics 2019, 7, 101. https://doi.org/10.3390/inorganics7080101
Liu X, Pochapsky TC. Human Acireductone Dioxygenase (HsARD), Cancer and Human Health: Black Hat, White Hat or Gray? Inorganics. 2019; 7(8):101. https://doi.org/10.3390/inorganics7080101
Chicago/Turabian StyleLiu, Xinyue, and Thomas C. Pochapsky. 2019. "Human Acireductone Dioxygenase (HsARD), Cancer and Human Health: Black Hat, White Hat or Gray?" Inorganics 7, no. 8: 101. https://doi.org/10.3390/inorganics7080101
APA StyleLiu, X., & Pochapsky, T. C. (2019). Human Acireductone Dioxygenase (HsARD), Cancer and Human Health: Black Hat, White Hat or Gray? Inorganics, 7(8), 101. https://doi.org/10.3390/inorganics7080101