Understanding Factors that Control the Structural (Dis)Assembly of Sulphur-Bridged Bimetallic Sites


Abstract
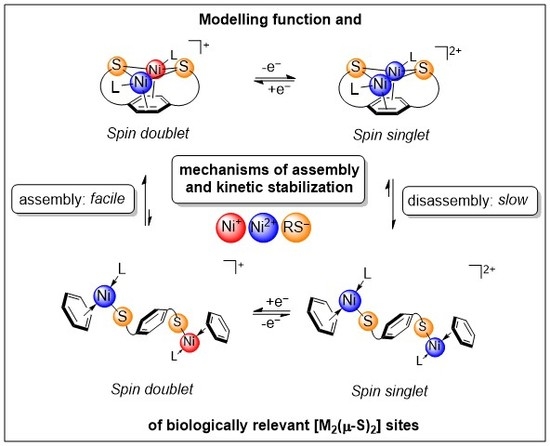
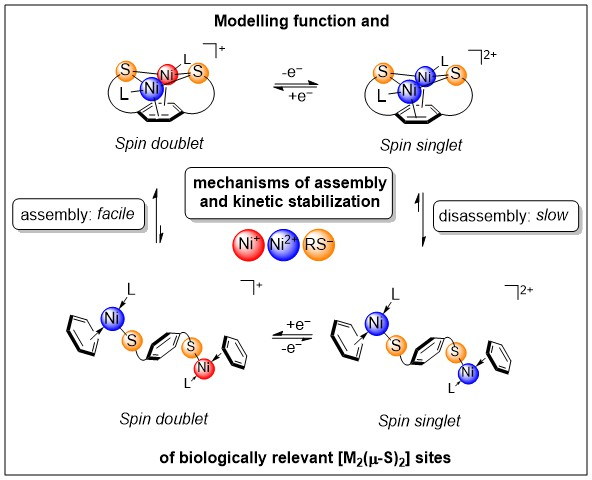{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
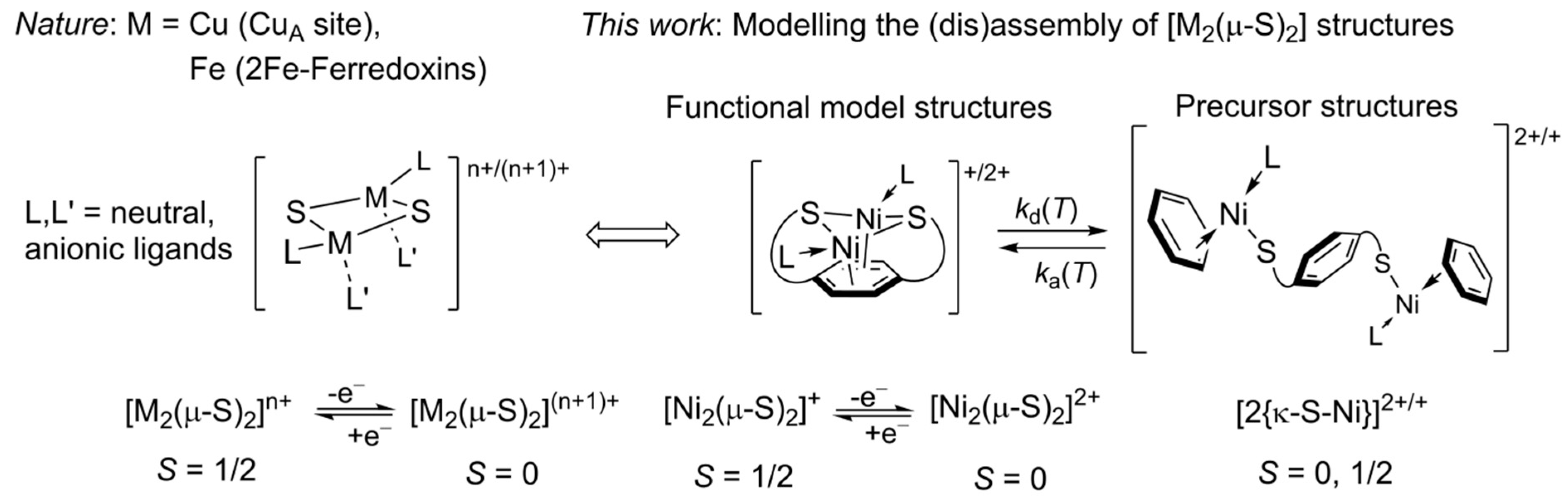
2. Results
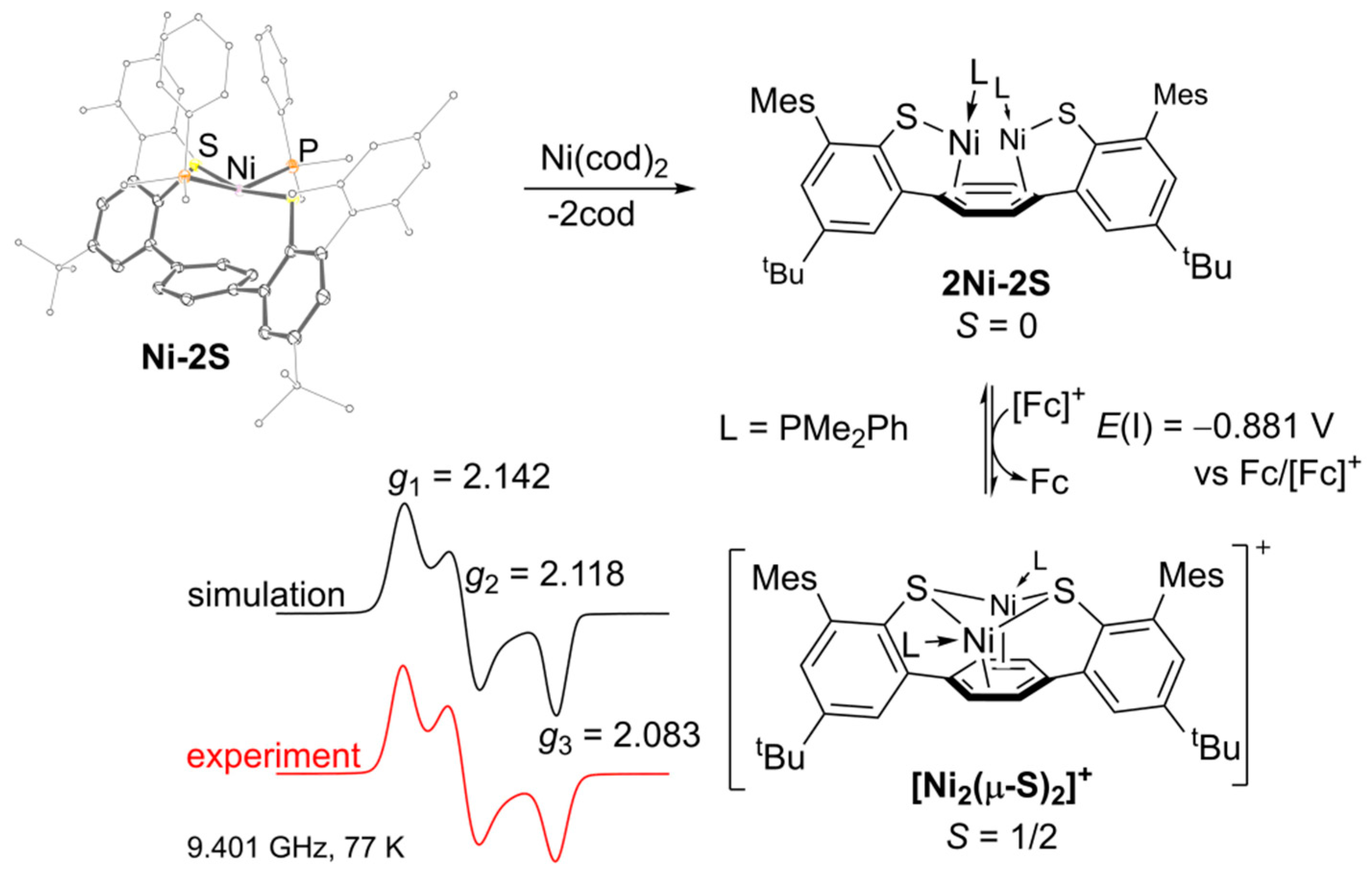
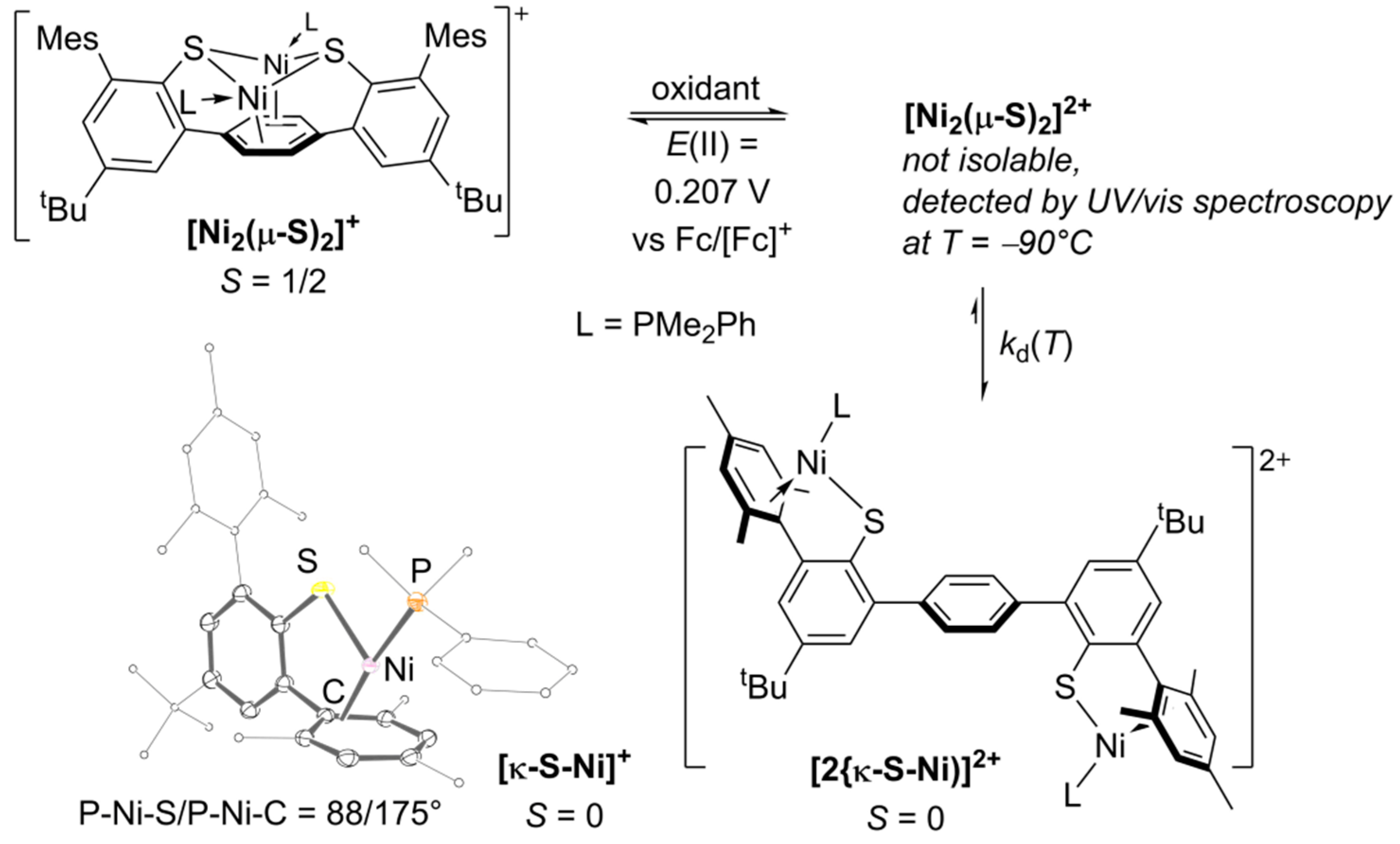
2.1. Preparation and Characterization of Bimetallic Compounds
2.2. Quantitative UV–VIS Spectroscopic Study of Oxidation-Induced Disassembly of [Ni2(µ-S)2]+
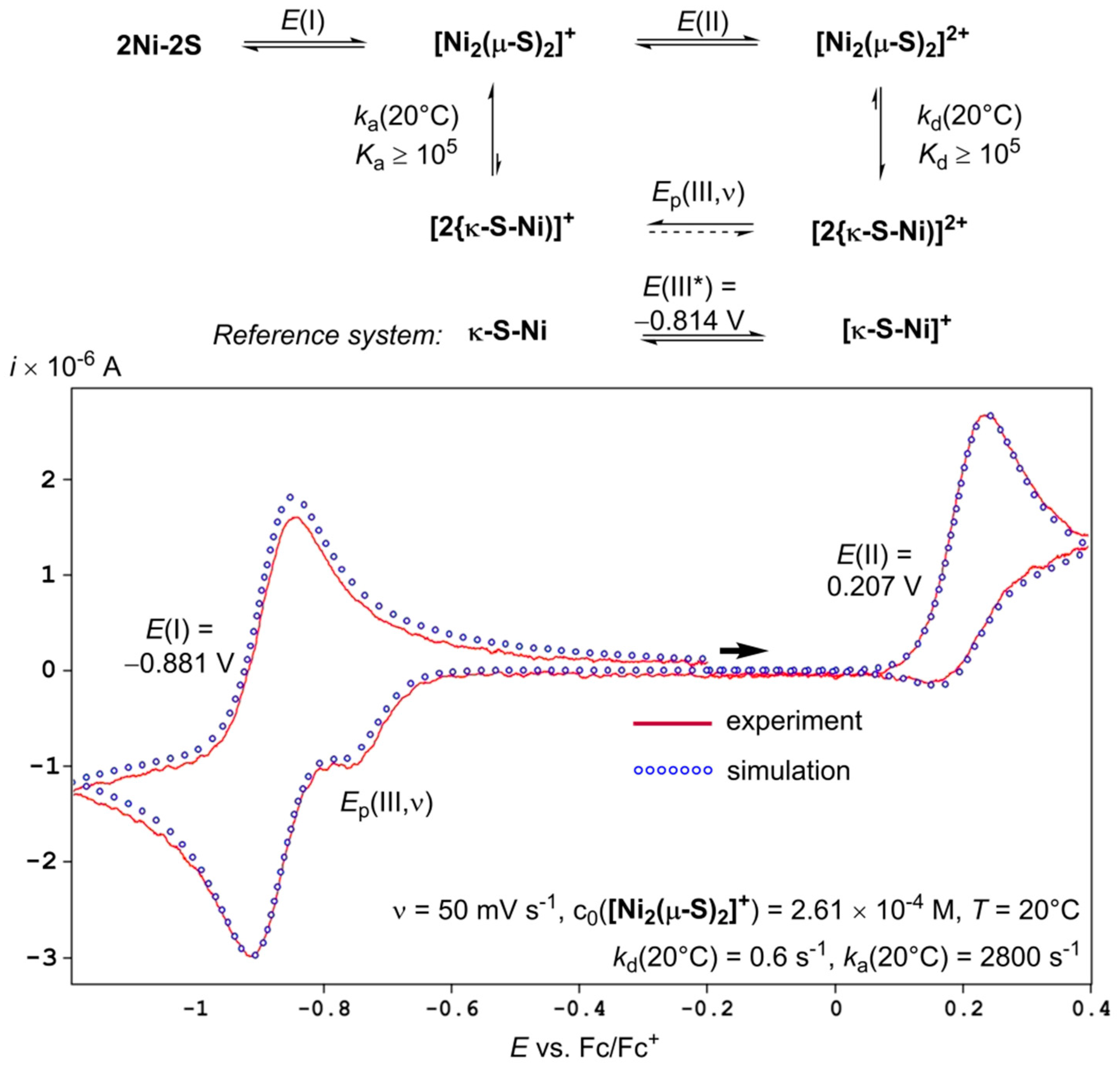
2.3. Quantitative Electrochemical Study of Rate Constants ka and kd for (dis)Assembly of [Ni2(µ-S)2]+
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Solomon, E.I.; Heppner, D.E.; Johnston, E.M.; Ginsbach, J.W.; Cirera, J.; Qayyum, M.; Kieber-Emmons, M.T.; Kjaergaard, C.H.; Hadt, R.G.; Tian, L. Copper Active Sites in Biology. Chem. Rev. 2014, 114, 3659–3853. [Google Scholar] [CrossRef]
- Solomon, E.I.; Xie, X.; Dey, A. Mixed valent sites in biological electron transfer. Chem. Soc. Rev. 2008, 37, 623–638. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C.; Dean, D.R.; Smith, A.D.; Johnson, M.K. Structure, Function, and Formation of Biological Iron–Sulfur Clusters. Annu. Rev. Biochem. 2005, 74, 247–281. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.Q.; Bill, E.; Trautwein, A.X.; Winkler, H.; Kostikas, A.; Papaefthymiou, V.; Simopoulos, A.; Beardwood, P.; Gibson, J.F. Exchange interactions, charge delocalization, and spin relaxation in a mixed-valence di-iron complex studied by Mössbauer spectroscopy. J. Phys. Chem. 1993, 99, 6421–6428. [Google Scholar] [CrossRef]
- Glaser, T.; Hedman, B.; Hodgson, K.O.; Solomon, E.I. Ligand K-Edge X-ray Absorption Spectroscopy: A Direct Probe of Ligand–Metal Covalency. Acc. Chem. Res. 2000, 33, 859–868. [Google Scholar] [CrossRef]
- Wilson, T.D.; Savelieff, M.G.; Nilges, M.J.; Marshall, N.M.; Lu, Y. Kinetics of Copper Incorporation into a Biosynthetic Purple CuA Azurin: Characterization of Red, Blue, and a New Intermediate Species. J. Am. Chem. Soc. 2011, 133, 20778–20792. [Google Scholar] [CrossRef]
- Chacón, K.N.; Blackburn, N.J. Stable Cu(II) and Cu(I) Mononuclear Intermediates in the Assembly of the CuA Center of Thermus thermophilus Cytochrome Oxidase. J. Am. Chem. Soc. 2012, 134, 16401–16412. [Google Scholar] [CrossRef]
- Ghosh, M.K.; Basak, P.; Mazumdar, S. Mechanism of Copper Incorporation in Subunit II of Cytochrome c Oxidase from Thermus thermophilus: Identification of Intermediate Species. Biochemistry 2013, 52, 4620–4635. [Google Scholar] [CrossRef]
- Chakraborty, S.; Polen, M.J.; Chacón, K.N.; Wilson, T.D.; Yu, Y.; Reed, J.; Nilges, M.J.; Blackburn, N.J.; Lu, Y. Binuclear CuA Formation in Biosynthetic Models of CuA in Azurin Proceeds via a Novel Cu(Cys)2His Mononuclear Copper Intermediate. Biochemistry 2015, 54, 6071–6081. [Google Scholar] [CrossRef]
- Dance, I.G.; Guerney, P.J.; Rae, A.D.; Scudder, M.L. Planar bridging thiolate in (Ph3P)2Cu(μ-SPh)2Cu(PPh3)2. Inorg. Chem. 1983, 22, 2883–2887. [Google Scholar] [CrossRef]
- Houser, R.P.; Young, V.G.; Tolman, W.B. A Thiolate-Bridged, Fully Delocalized Mixed-Valence Dicopper(I,II) Complex That Models the CuA Biological Electron-Transfer Site. J. Am. Chem. Soc. 1996, 118, 2101–2102. [Google Scholar] [CrossRef]
- Thomas, A.M.; Lin, B.-L.; Wasinger, E.C.; Stack, T.D.P. Ligand Noninnocence of Thiolate/Disulfide in Dinuclear Copper Complexes: Solvent-Dependent Redox Isomerization and Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2013, 135, 18912–18919. [Google Scholar] [CrossRef]
- Ording-Wenker, E.C.M.; van der Plas, M.; Siegler, M.A.; Bonnet, S.; Bickelhaupt, F.M.; Fonseca Guerra, C.; Bouwman, E. Thermodynamics of the CuII μ-Thiolate and CuI Disulfide Equilibrium: A Combined Experimental and Theoretical Study. Inorg. Chem. 2014, 53, 8494–8504. [Google Scholar] [CrossRef]
- Boniecki, M.T.; Freibert, S.A.; Mühlenhoff, U.; Lill, R.; Cygler, M. Structure and functional dynamics of the mitochondrial Fe/S cluster synthesis complex. Nat. Commun. 2017, 8, 1287. [Google Scholar] [CrossRef]
- Bonfio, C.; Valer, L.; Scintilla, S.; Shah, S.; Evans, D.J.; Jin, L.; Szostak, J.W.; Sasselov, D.D.; Sutherland, J.D.; Mansy, S.S. UV-light-driven prebiotic synthesis of iron–sulfur clusters. Nat. Chem. 2017, 9, 1229. [Google Scholar] [CrossRef]
- Hagen, K.S.; Reynolds, J.G.; Holm, R.H. Definition of reaction sequences resulting in self-assembly of [Fe4S4(SR)4]2− clusters from simple reactants. J. Am. Chem. Soc. 1981, 103, 4054–4063. [Google Scholar] [CrossRef]
- Do, Y.; Simhon, E.D.; Holm, R.H. Improved syntheses of tetrachlorodi-μ-sulfidodiferrate(2−) ([Fe2S2Cl4]2−) and hexachloro-μ-oxodiferrate(2−) ([Fe2OCl6]2−) and oxo/sulfido ligand substitution by use of silylsulfide reagents. Inorg. Chem. 1983, 22, 3809–3812. [Google Scholar] [CrossRef]
- Beinert, H.; Holm, R.H.; Münck, E. Iron–Sulfur Clusters: Nature’s Modular, Multipurpose Structures. Science 1997, 277, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Venkateswara Rao, P.; Holm, R.H. Synthetic Analogues of the Active Sites of Iron−Sulfur Proteins. Chem. Rev. 2004, 104, 527–560. [Google Scholar] [CrossRef] [PubMed]
- Ballmann, J.; Albers, A.; Demeshko, S.; Dechert, S.; Bill, E.; Bothe, E.; Ryde, U.; Meyer, F. A Synthetic Analogue of Rieske-Type [2Fe–2S] Clusters. Angew. Chem. Int. Ed. 2008, 47, 9537–9541. [Google Scholar] [CrossRef] [PubMed]
- Ballmann, J.; Sun, X.; Dechert, S.; Schneider, B.; Meyer, F. A convenient ligand exchange pathway to [2Fe–2S] ferredoxin analogues. Dalton Trans. 2009, 4908–4917. [Google Scholar] [CrossRef]
- Saouma, C.T.; Kaminsky, W.; Mayer, J.M. Protonation and Concerted Proton–Electron Transfer Reactivity of a Bis-Benzimidazolate Ligated [2Fe–2S] Model for Rieske Clusters. J. Am. Chem. Soc. 2012, 134, 7293–7296. [Google Scholar] [CrossRef]
- Bergner, M.; Roy, L.; Dechert, S.; Neese, F.; Ye, S.; Meyer, F. Ligand Rearrangements at Fe/S Cofactors: Slow Isomerization of a Biomimetic [2Fe–2S] Cluster. Angew. Chem. Int. Ed. 2017, 56, 4882–4886. [Google Scholar] [CrossRef] [PubMed]
- Koch, F.; Berkefeld, A.; Speiser, B.; Schubert, H. Mechanistic Aspects of Redox-Induced Assembly and Disassembly of S-Bridged [2M–2S] Structures. Chem. Eur. J. 2017, 23, 16681–16690. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, R.S.; Shain, I. Theory of Stationary Electrode Polarography. Single Scan and Cyclic Methods Applied to Reversible, Irreversible, and Kinetic Systems. Anal. Chem. 1964, 36, 706–723. [Google Scholar] [CrossRef]
- Mews, N.M.; Berkefeld, A.; Hörner, G.; Schubert, H. Controlling Near-Infrared Chromophore Electronic Properties through Metal–Ligand Orbital Alignment. J. Am. Chem. Soc. 2017, 139, 2808–2815. [Google Scholar] [CrossRef]
- Koch, F.; Schubert, H.; Sirsch, P.; Berkefeld, A. Binuclear complexes of Ni(I) from 4-terphenyldithiophenol. Dalton Trans. 2015, 44, 13315–13324. [Google Scholar] [CrossRef]
- Gollas, B.; Krauß, B.; Speiser, B.; Stahl, H. Design of a Single-Unit Haber-Luggin Capillary/Dual Reference-Electrode System. Curr. Sep. 1994, 13, 42–44. [Google Scholar]
- Kuwana, T.; Bublitz, D.E.; Hoh, G. Chronopotentiometric Studies on the Oxidation of Ferrocene, Ruthenocene, Osmocene and Some of their Derivatives1. J. Am. Chem. Soc. 1960, 82, 5811–5817. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alrefai, R.; Eggenweiler, H.; Schubert, H.; Berkefeld, A. Understanding Factors that Control the Structural (Dis)Assembly of Sulphur-Bridged Bimetallic Sites. Inorganics 2019, 7, 42. https://doi.org/10.3390/inorganics7040042
Alrefai R, Eggenweiler H, Schubert H, Berkefeld A. Understanding Factors that Control the Structural (Dis)Assembly of Sulphur-Bridged Bimetallic Sites. Inorganics. 2019; 7(4):42. https://doi.org/10.3390/inorganics7040042
Chicago/Turabian StyleAlrefai, Riyadh, Henri Eggenweiler, Hartmut Schubert, and Andreas Berkefeld. 2019. "Understanding Factors that Control the Structural (Dis)Assembly of Sulphur-Bridged Bimetallic Sites" Inorganics 7, no. 4: 42. https://doi.org/10.3390/inorganics7040042
APA StyleAlrefai, R., Eggenweiler, H., Schubert, H., & Berkefeld, A. (2019). Understanding Factors that Control the Structural (Dis)Assembly of Sulphur-Bridged Bimetallic Sites. Inorganics, 7(4), 42. https://doi.org/10.3390/inorganics7040042