Abstract
The catalytic potential of linked redox centres is exemplified by the catalytic site of [FeFe]-hydrogenases, which feature a diiron subsite linked by a cysteinyl S atom to a 4Fe4S cube. The investigation of systems possessing similarly-linked redox sites is important because it provides a context for understanding the biological system and the rational design of abiological catalysts. The structural, electrochemical and spectroscopic properties of Fe2(CO)5(CH3C(CH2S)2CH2SPhNO2, I-bzNO2 and the aniline analogue, I-bzNH2, are described and IR spectroelectrochemical studies have allowed investigation of the reduction products and their reactions with CO and protons. These measurements have allowed identification of the nitrobenzenyl radical anion, quantification of the shifts of the (CO) bands on ligand-based reduction compared with NO2/NH2 exchange and protonation of the pendent ligand. The strength of thioether coordination is related to the electronic effects, where competitive binding studies with CO show that CO/thioether exchange can be initiated by redox processes of the pendent ligand. Stoichiometric multi electron/proton transfer reactions of I-bzNO2 localised on nitrobenzene reductions occur at mild potentials and a metal-centred reduction in the presence of protons does not lead to significant electrocatalytic proton reduction.
1. Introduction
Dithiolato-bridged diiron carbonyl compounds have important structural and functional similarities with the catalytic site of [FeFe]-hydrogenases [1,2,3,4,5,6] and this has driven detailed investigations into the chemistry of this class of compound [7,8,9,10,11,12]. Critical to the functioning of the catalytic centre, or H-cluster, is the transfer of electrons between the diiron subsite and a 4Fe4S cube which is linked through a bridging S atom from a cysteinyl residue. It has been demonstrated that tripodal bridging ligand systems can be exploited to provide the diiron-bridging dithiolato S atoms together with a thioether which is able to bind, reversibly, to one of the Fe atoms [13,14]. We have previously shown that such systems can be used to generate mixed carbonyl, cyano complexes which are excellent spectroscopic models of the CO-inhibited forms of the H-cluster [15,16] and have allowed the preparation of a diiron subsite with S-atom linking to a 4Fe4S cube—mimicking key characteristics of the full H-cluster [17]. An important extension of this work is the exploration of the impact of a redox-active group bound to the linking S-atom on the chemistry of the system [18,19,20,21]. Interaction between the diiron and S-linked redox groups can be studied by exploiting the sensitivity of the ν(CO) bands to the electronic structure of the metal to which the CO group is bound. Further, cases where the remote redox centre has similarly sensitive reporter bands allow reliable assignment of the character of the redox process and interpretation of the chemistry which follows oxidation or reduction. Nitrobenzene presents an ideal redox partner for such studies. The compound has a well-defined one-electron reduction at mild potentials and the nitro group gives strong NO2 stretching bands in the IR spectrum with wavenumbers which are sensitive to the local environment. The interaction between the linked redox sites can be explored through the examination of the impact of altering the redox state of a proximal nitrobenzene active group on the {2Fe3S}-carbonyl subsite, to which it is attached by S-ligation (I-bzNO2, Scheme 1).
Scheme 1.
Structures of Fe2(CO)5(CH3C(CH2S)2CH2SPhNO2, I-bzNO2 (left) and the aniline analogue, I-bzNO2 (right).
In this investigation we set out to (i) establish whether the primary reduction of I-bzNO2 is localised on the nitrobenzene group, (ii) explore the impact of the pendent redox group on reduction of the diiron subsite by comparison of the reduction chemistry of I-bzNO2 and the related aniline adduct I-bzNH2 and (iii) examine the reduction chemistry of I-bzNO2 under reducing conditions in the presence of a non-coordinating acid, 2,6-lutidinium (LutH+).
2. Results
2.1. Electrochemistry of I-bzNO2 and I-bzNH2
The nitrobenzene complex, I-bzNO2, shows two well-defined reduction processes and a more complicated reoxidation wave (Figure 1). The primary electron-transfer process at E°’ = −0.85 V versus Ag/AgCl is a diffusion-controlled, single-electron, reversible reduction; thus the peak separation ΔE = 75 mV (293 K) is close to that expected for a one-electron process; the peak current, ipreduction, is linearly dependent on the square-root of the scan-rate, ν1/2, and the peak current ratio, ipoxidation/ipreduction, is close to unity. The second partly reversible reduction (Ep = −1.21 V) is similar to that of related complexes with different pendent thioethers [13], but the more positive potential and high reversibility of the primary electron-transfer step suggests that it is associated with the pendent nitrobenzene group. This conclusion is supported by the similarity of the cyclic voltammetric response of nitrobenzene and the uncoordinated tripodal ligand (Figure S1).
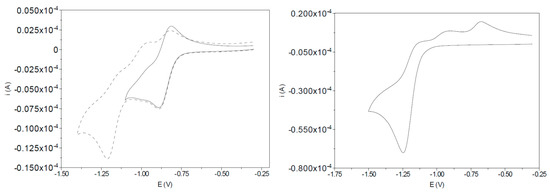
Figure 1.
Cyclic voltammogram of (left) I-bzNO2 (2 mM, 0.2 M [NBu4][BF4] in CH3CN, 0.1 V·s−1) and (right) I-bzNH2 (7 mM, 0.2 M [NBu4][BF4] in CH3CN, 0.1 V·s−1). The cyclic voltammograms of nitrobenzene and CH3C(CH2S)2CH2S(C6H4-p-NO2) recorded under the same experimental conditions are given in Figure S1 and show a single reversible reduction process at potentials near −1.0 V.
The metal-based reduction of Fe2(μ-SCH2)2C(CH3)SR(CO)5 compounds [13] exhibit limited reversibility with the potential of peak cathodic current (Epc) sensitive to the donor properties of the R group. The similarity of the Epc values for the diiron-based reduction of I-bzNO2 and I-bzNH2 suggests that the S-bound reduced nitrobenzene and aniline groups have similar electronic interaction with the diiron centre. For both complexes weak re-oxidation features are evident in the anodic scan.
The addition of a proton source to the reaction mixture leads to significant increases in current response consistent with multielectron reduction. The nitrobenzene-localised reduction of I-bzNO2 is shifted to more positive potentials and current increase and loss of reversibility is very similar to that observed for nitrobenzene (Figure S2) indicative of the replacement of the one-electron reversible process by a 6 electron/6 proton reaction at high acid concentration. The reaction product, I-bzNH2, may then undergo metal-based electron/proton reactions where acid-dependent voltammetry of I-bzNH2 reveal shifts of the onset reduction potential and marked increases in current consistent with electrocatalytic proton reduction (Figure 2). The shift of the reduction potential to more positive potentials with increasing acid concentration is indicative of coupled electron proton reactions and the sigmoidal character of the onset current may indicate that the initial electron-proton product has limited electrocatalytic activity. This detail of the electrochemical response is explored with the aid of spectroelectrochemical methods in Section 2.6.
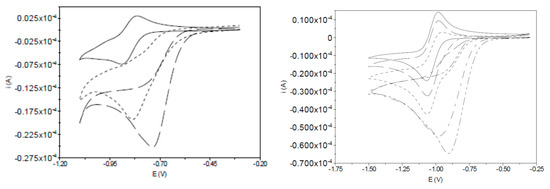
Figure 2.
Cyclic voltammograms of the primary reduction processes of (left) I-bzNO2 (2 mM) in the absence and in the presence of acetic acid (0.2 M [NBu4][BF4] in CH3CN, 0.1 V·s−1) and (right) I-bzNH2 (7 mM, 0.2 M [NBu4][BF4] in CH3CN, 0.1 V·s−1) in absence of acid (―) and in presence of 1 (– – –), 5 (―――) and 10 equivalents (― - ― -) of 2,6-lutidinium, LutH+). The voltammetry of LutH+ under equivalent conditions is given in Figure S3.
A compound with diiron subsites and nitrobenzene components has been reported by Sun and coworkers, [Fe2(CO)6{(µ-SCH2)2N(C6H4-p-NO2)}] (Fe2-adt(bzNO2)) [22]. In that case the reduction processes associated with the diiron centres was attributed to the first reduction wave and the reduction of nitrobenzene was not explicitly considered. Addition of HOAc to solutions of Fe2-adt(bzNO2) were observed to give an increased current response which was attributed to electrocatalytic proton reduction [22].
2.2. EPR Specroscopy of the In-Situ Generated Reduction Product of I-bzNO2
Despite the electrochemical reversibility of the primary reduction process of I-bzNO2, the one-electron reduced product was unstable under anaerobic conditions and it was necessary to employ in-situ methods of sample generation in order to obtain EPR spectra. Application of potentials sufficient to generate a significant cathodic current were accompanied by the appearance of weak, but highly resolved spectra (Figure 3). Simulation of the spectra are consistent with a radical centred on the pendent nitrobenzene group with a g value of 2.00543 and hyperfine coupling dominated by a single N (aN = 24.43 MHz) and inequivalence of the coupling constants to the four protons of the substituted-nitrobenzene ring (aH = 10.3, 8.4, 3.6 and 3.4 MHz). The inequivalence of the nitrobenzene protons is accounted for by hindered conformational change of the pendent nitrobenzene group. Weaker hyperfine coupling, to the methylene protons of the linker (aH = 1.4 MHz), are also evident in the spectra. While the conformation of the thioether-bound form of the complex renders the methylene protons inequivalent, the simulation shown in Figure 3 was calculated using a single coupling constant for these protons. Support for coupling of the methylene protons is based on DFT calculations showing a small population of the SOMO on the methyl group and reported EPR spectra of reduced 1-(methylthio)-4-nitrobenzene in DMSO [23]. The hyperfine coupling constants aN, a2,6, a3,5 and aR have values corresponding to 26.7, 9.46, 3.24, 0.64 MHz. The transient character of the reduced product was confirmed by the observed loss of the EPR product within ca. 1 min of switching the potentiostat to open circuit.
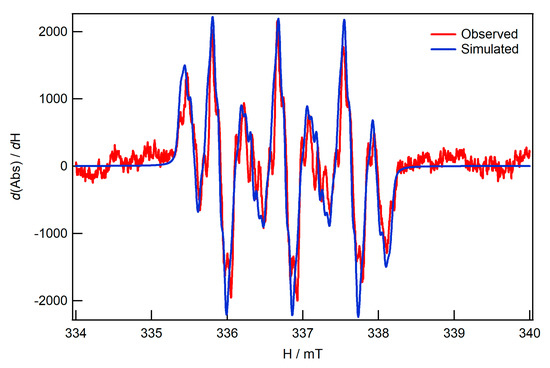
Figure 3.
X-band EPR spectra of in-situ reduced I-bzNO2 and simulated spectrum. The simulated spectrum was calculated using the EasySpin suite of routines [24]. Details of the simulation parameters are given in Table S1.
Assignment of the EPR spectrum to [I-bzNO2]− is based on the g value and the good agreement between the relative magnitudes of the hyperfine coupling constants for [I-bzNO2]− and those of the radical anion of 1-(methylthio)-4-nitrobenzene. The inequivalence of the hyperfine coupling constants of the four protons of the substituted nitrobenzene ring is indicative of the bridging thioether remaining in the closed, bound, form for the reduced species. The barrier to conformational change of the substituted nitrobenzene group of I-bzNO2 has been estimated from NMR spectroscopy (Table S2). The coalescence temperature of the ortho and meta protons is 200 K and the temperature-dependence of the NMR spectra can be simulated with a ΔG‡ of 38–40 kJ·mol−1. If the coordinate bond between the Fe and the thioether is lost for the EPR-active species, it is expected that the ortho and meta protons will become equivalent on the EPR timescale as is observed for substituted nitrobenzene radicals [23].
Given the assignment of the EPR spectrum to [I-bzNO2], the magnitude of the hyperfine coupling constants can be used to assess the localization of charge on the diiron and nitrobenzene fragments of the molecule. Notwithstanding the small sensitivity of the hyperfine coupling constants of the anion of 1-(methylthio)-4-nitrobenzene to the solvent [23], the similarity of the magnitude of the values obtained for [I-bzNO2]− suggests that reduction is strongly localised on the nitrobenzene fragment. This observation is consistent with arguments based on the electrochemical measurements discussed in the previous section.
2.3. Infrared Spectroelectrochemistry (IR-SEC) of the Reduction of I-bzNH2
The reduction chemistry of the diiron subsite, free of complications associated with the redox chemistry of the pendent nitrobenzene moiety, is most conveniently explored using the aniline analogue, I-bzNH2. IR-SEC methods have been developed which allow collection of spectra from samples maintained in a strictly anaerobic environment and under elevated pressures of selected gases [25]. This allows the collection of IR spectra through the redox process, allowing the identification of longer-lived (>ca. 2 s) intermediate products. The spectra are presented in differential absorption format and are calculated using as reference the spectrum of the thin film of solution trapped between the working electrode and the IR-transmitting window at a resting potential of the system (usually 0 V) immediately before application of a reducing potential. Reduction at −1.2 V leads to at least two products which are evident either immediately following application of a reducing potential (Figure 4a) or at longer times (Figure 4b). The product bands (1910, 1890 and 1830 cm−1) are in a spectral region consistent with CO terminally-bound to electron-rich iron centres. The broad band profile is insufficiently distinct to provide significant insight into the possible structures of the reduced species, however the absence of bands near 1700 cm−1 would suggest that CO-bridged species are not formed in significant concentration. This behaviour contrasts with the chemistry of reduced dithiolato-bridged hexacarbonyl complexes [26,27,28]. Re-oxidation of the sample at 0 V leads to ca. 20% recovery of the starting material. The residual bands in the spectrum have a band profile significantly different from the starting complex (Figure 4c).
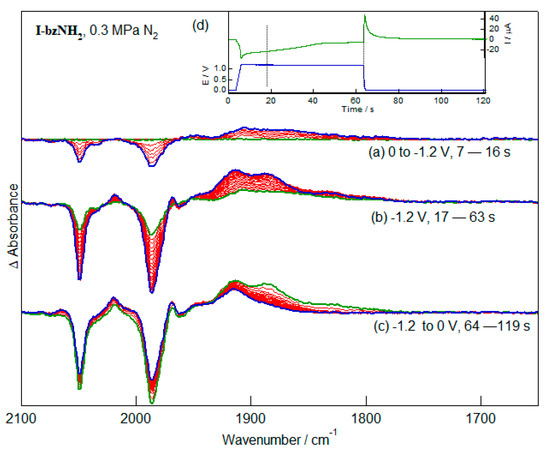
Figure 4.
IR-SEC measurements of the reduction (a,b) and re-oxidation (c) of I-bzNH2. The applied potential and current response of the cell during the IR-SEC experiment is shown in (d). For this, and subsequent, figures the initial spectrum of each block is shown in green and the final spectrum in blue.
2.4. IR-SEC of the Reduction of I-bzNO2
The study of the interaction between the diiron-centred and nitrobenzene-centred redox processes is facilitated by the strong IR chromophores sensitive to the electron richness of the respective centres. As suggested by the partial-reversibility of the electrochemistry (Figure 1), the reduced compound is unstable and interrogation of these species requires rapid collection of IR spectra during reduction. IR-SEC measurements recorded following reduction at potentials between the first and second reduction processes of I-bzNO2 and reoxidation cycles are shown in Figure 5. Experiments have been conducted using both acetonitrile and dichloromethane as solvent. The spectra recorded in dichloromethane give a better-defined separation between the two reduction processes and additionally are less affected by solvent absorption in the 1200 to 1600 cm−1 region. This allows better characterisation of the spectral changes in the NO2 stretching region.
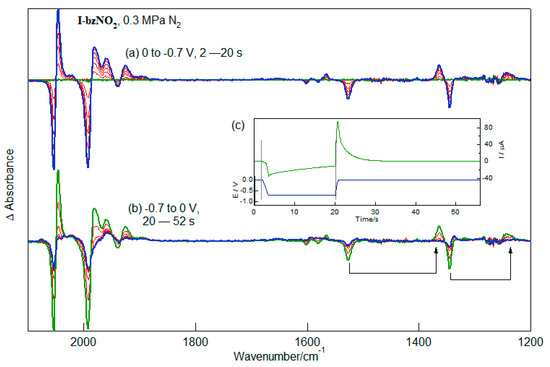
Figure 5.
IR-SEC recorded during (a) reduction and (b) re-oxidation of I-bzNO2 in CH2Cl2 (2 mM, 0.2 M TBAPF6). The applied potential and current response of the cell during the IR-SEC experiment is shown in (c).
The spectral changes in the ν(CO) region following reduction of I-bzNO2 at mild potentials are complex but diagnostic of a small spectral shift of the band profile (Figure 5a). In the early stages of the reduction the pattern of ν(CO) bands is retained, consistent with the retention of the geometry about the diiron centre. The complete removal of the bands due to I-bzNO2 and the small wavenumber shift of the ν(CO) bands is more accurately determined with reference to the single beam spectra which are available as Figure S4. The 5–6 cm−1 shift of the highest-wavenumber band to lower wavenumber may be compared with the 71 cm−1 shift of the highest wavenumber ν(CO) band of Fe2(μS(CH2)3S)(CO)6 following metal-centred one-electron reduction [29,30]. As the reduction proceeds there is a growth of lower-wavenumber ν(CO) bands (1958, 1923 and 1892 cm−1) which are due to a byproduct which is not reoxidised at 0 V. The lower wavenumber region (1200–1650 cm−1) has symmetric and antisymmetric stretches of the nitro group together with ring vibrations of the substituted benzene. Since reduction leads to population of π antibonding orbitals it is likely that the ring stretching vibrations (1601 and 1580 cm−1) shift to lower wavenumbers, with the growth band near 1566 cm−1 a plausible candidate. This implies that the NO stretching vibrations are significantly shifted following reduction of I-bzNO2, with the asymmetric and symmetric stretches shifting from 1527 and 1345 cm−1 to 1364 and 1243 cm−1. The large wavenumber shifts of the NO stretching vibrations implies significant involvement of the nitro group in the SOMO, a conclusion consistent with the large hyperfine coupling constant for the N atom of [I-bzNO2]−. It is noted that the higher noise level in the IR spectra between 1250 and 1280 cm−1 is associated with strong solvent absorption and this distorts the apparent wavenumber and profile of the lower-wavenumber growth band (1243 cm−1). The application of a re-oxidising potential leads to the rapid depletion of the growth bands with concomitant recovery of ca. 75% recovery of the starting complex (Figure 5b).
Reduction of the diiron subsite of I-bzNO2 can be achieved by application of more reducing potentials, where sequential reduction of the nitrobenzene and diiron fragments is shown in Figure 6. After generation of [I-bzNO2]− further reduction leads to shifts of the ν(CO) bands, together with the aryl and NO2 stretching bands (Figure 6b). The ν(CO) bands give a group of broad, unresolved bands centred about 1913, 1885 and 1822 cm−1 which are not experimentally distinguishable in form and wavenumber from those obtained following diiron-based reduction of I-bzNH2 (Figure 4). Both the complexity of the band profile and variations in the time-dependence of the increases in absorbance at different wavenumbers suggest that a mixture of products is formed. The current response for the second reduction is at least twice that of the 1-electron nitrobenzene-localised reaction, this being consistent with a 2-electron reduction at the diiron centre (Figure 6e), possibly with further rearrangement and reduction processes. Re-oxidation at −0.7 V leads to loss of the lower wavenumber ν(CO) bands (at 1913, 1885 and 1822 cm−1) with some recovery of the starting complex (Figure 6c) and another species can be oxidised at 0 V (Figure 6d) but less than 10% of the starting complex is recovered in these experiments. The markedly different pattern of ν(CO) bands indicates a significant change in the geometry of the diiron pentacarbonyl core.
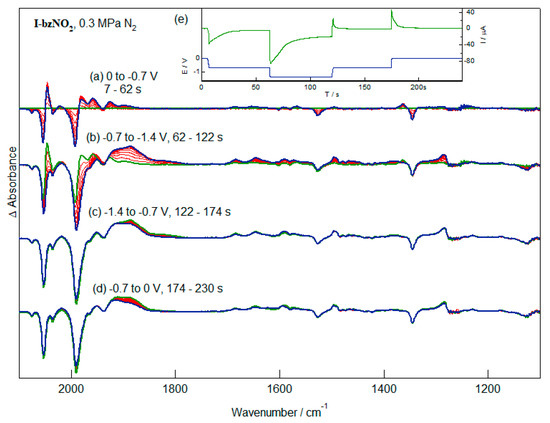
Figure 6.
IR-SEC measurements of the first (a) and second (b) reduction processes of I-bzNO2 followed by reoxidation at mild (c) and more forcing (d) conditions. The applied potential and current response of the cell during the IR-SEC experiment is shown in (e).
More rapid generation of the reduced species by stepping the potential directly into the second reduction results in transient formation of [I-bzNO2]− with a subsequent band profile analogous to that of the final spectrum of Figure 6b. Re-oxidation in these experiments lead to similarly poor recovery of the starting material. These experiments show that the metal centred reduction of I-bzNO2 is both electrochemically and chemically irreversible. Most likely this proceeds through a two-electron reduction of the diiron centre with rearrangement and/or ligand dissociation.
2.5. Redox-Dependent Competitive Binding of CO to I-bzNO2 and I-bzNH2
The products of diiron-centred reduction of I-bzNO2 and I-bzNH2 differ from those obtained following reduction of the diiron hexacarbonyl analogues, where this is most marked by an absence of bands in the region between 1700 and 1800 cm−1, those typical of a bridging CO moiety. Previous studies have indicated that the binding of the pendent S group is sensitive to oxidation state and a likely cause of the differing reaction chemistry is associated with the lability of the linking S atom. The relative strength of interaction between the Fe atom and the bridging S atom can be deduced from competition studies involving an alternative ligand. In this regard CO is an ideal ligand as it binds strongly to the Fe centre to give a hexacarbonyl species with a well-defined spectral profile. Measurements for a series of pendent thioethers are summarised in Table 1 of ref. [31]. There is a clear relationship between the electron-withdrawing character of the substituent and the equilibrium constant for thioether replacement by CO with I-bzNO2 having a value ca. 30 times larger than that of I-bzNH2. If it is assumed that the free energies of the hexacarbonyl species are approximately the same, then the Keq values will reflect the relative stability of the S-bound form. The strongest interaction is in the cases where the substitute is most electron rich.
The addition of CO to the gas mixture in the IR-SEC cell containing I-bzNH2 gives a mixture of pentacarbonyl and hexacarbonyl species. Reduction leads to depletion of the bands of both species, with the loss of the hexacarbonyl species slightly preferred over I-bzNH2. It is important to note that among the products generated during the reduction reaction is a species giving rise to a weak band near 1720 cm−1 (Figure 7a). The wavenumber of this band is consistent with its assignment to a bridging CO group. Application of increasingly positive potentials to the solution result in oxidation first of the species with ν(CO) bands between 1800 and 1900 cm−1 (Figure 7b), then of species with higher-wavenumber ν(CO) bands (Figure 7c) before oxidation of the species with the bridging CO group (Figure 7d). Similarly high potentials were required to oxidise the most stable CO-bridged species obtained after reduction of Fe2(μ-(SCH2)2CH2)(CO)6 [29].
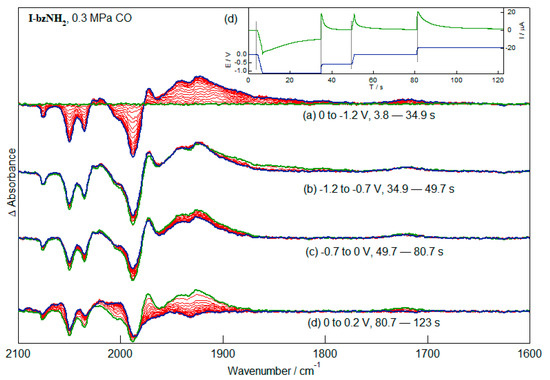
Figure 7.
IR-SEC of I-bzNH2 (2 mM, CH3CN, 0.2 M TBAPF6) with CO (ca. 0.3 MPa). The spectra show the reduction (a) and re-oxidation at increasingly positive potentials (b–d). The applied potential and current response of the cell during the IR-SEC experiment is shown in (e).
The impact of a change in the electron richness of the pendent group on the equilibrium constant for CO displacement of the bridging S atom is well illustrated by experiments of I-bzNO2 in CH2Cl2 conducted under elevated pressure of CO (Figure 8). The application of mild reducing potentials leads to depletion bands of the nitrobenzene and, at a slower initial rate, the diiron hexacarbonyl fragment. Notably, the growth ν(CO) bands have a different pattern from those of I-bzNH2 obtained in related experiments (Figure 7) and the wavenumbers of the bands (2048, 1983, 1964 and 1930 cm−1) are in good agreement with the spectral bands of [I-bzNO2]− (Figure 5) after taking into account the different depletion bands in the two experiments. In addition to the bands due to the terminally-bound CO groups, there is a weak band at 1690 cm−1 which appears to be associated with the initial product. The reduction products of the reaction are highly dependent on the final potential and experiments conducted with a more reducing potential give more complex spectra with further reduction of the diiron core. The spectral features obtained following reduction of CO-saturated CH2Cl2 solutions of I-bzNO2 are consistent with the initial species in the form of the S-dissociated hexacarbonyl species and the initial one-electron reduction product having re-coordination of the bridging S-atom. The explanation of the 1690 cm−1 band remains open. The very low wavenumber of the band is inconsistent with its assignment to a bridging CO group as judged by the electron richness of the diiron centre based on the wavenumbers of the terminally-bound ν(CO) modes.
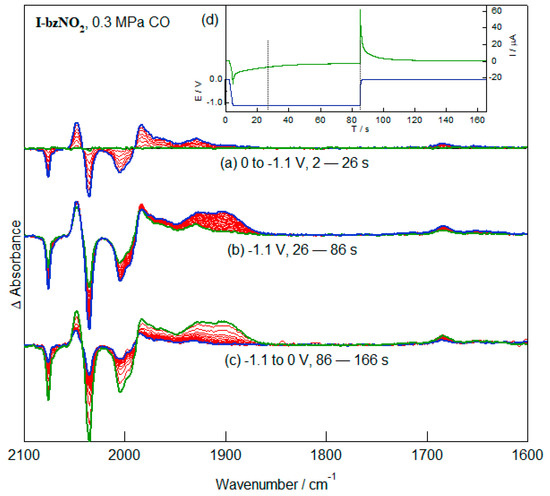
Figure 8.
IR-SEC spectra recorded during (a) and (b) the reduction of I-bzNO2 (2 mM, 0.2 M TBAPF6) in CH2Cl2 saturated with CO (0.3 MPa) and (c) re-oxidation. The time between spectra was 2.68 s. The applied potential and current response of the cell during the IR-SEC experiment is shown in (d).
2.6. Reduction of I-bzNO2 in the Presence of Lutidinium, HLut+
The thiolato-bridged diiron carbonyl compounds are well known catalysts of proton reduction, albeit at high thermodynamic cost. The reduction of I-bzNO2 in the presence of an excess of HLut+ at potentials sufficiently positive to avoid reduction of the diiron centre are shown in Figure 9a. The selection of HLut+ as acid is based on the non-coordinating character of the conjugate base, Lut. The ν(CO) spectra show differential absorption spectra similar to those obtained in experiments conducted in the absence of acid (Figure 6a) and these indicate that there is a small shift of the ν(CO) band profile to lower wavenumbers. In the absence of acid conversion of the pendent nitrobenzenethe group to the radical anion is associated with a 5.5 cm−1 shift of the two intense ν(CO) bands. In these experiments a shift of 3.5 cm−1 is observed. While small, this difference is significant considering the 2.5 cm−1 difference in wavenumbers of the corresponding bands of I-bzNO2 and I-bzNH2. Under the conditions of the experiment reduction of the pendent nitrobenzene group will be a 6e, 6H+ process leading to formation of I-bzNH2 which may then engage in protonation equilibria giving [I-bzNH3]+. The reaction of the nitro group is indicated by the depletion of the NO2 stretching bands without the appearance of the corresponding bands of the radical anion (Figure 6a). Consumption of the acid during reduction is indicated by depletion bands of LutH+ (1655, 1627, 1238 cm−1) and growth bands due to Lut (1655, 1627, 1238 cm−1). If the potential is returned to 0 V at this stage of the reaction, there is no recovery of the starting material. The slow rate of depletion of the starting complex relative to that observed in experiments conducted in the absence of acid (Figure 8) is due to the slow rate of ion diffusion in the thin layer geometry of the IR-SEC cell. This, of course, leads to more pronounced effects for the multi-electron process.
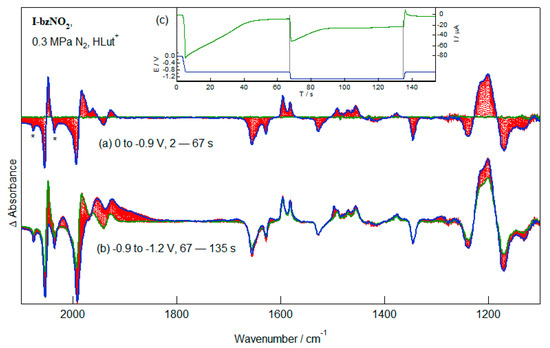
Figure 9.
IR-SEC spectra recorded during the reduction of I-bzNO2 in the presence of HLut+ at (a) −0.9 V and (b) −1.3 V. Weak ν(CO) depletion bands (denoted by an asterisk) are due a small concentration of the hexacarbonyl species. Re-oxidation at 0 V after the initial reduction at −0.9 V does not lead to any significant spectral changes (Figure S5).
If, after initial reduction, a potential is applied sufficiently to reduce the diiron centre then the ν(CO) bands of the initial product are rapidly replaced by broad, lower-wavenumber bands centred about 1950 cm−1 (Figure 9b). Reduction of the diiron centre is associated with additional, though limited, loss of LutH+. If the initial conversion of the nitrobenzene fragment of I-bzNO2 to [I-bzNH3]+, a net 6e, 7H+ process, then the spectral changes are consistent with the further consumption of 2–3 protons/complex. Neither the spectral response nor the current response during reduction is consistent with there being significant electrocatalytic proton reduction.
The sensitivity of the protonation chemistry of the reduced forms of I-bzNO2 is of interest both because the starting complex has CO substitution of the bridging S atom (Figure 8) and because it is well known that CO is known to inhibit electrocatalytic proton reduction, both of the enzyme and of different of the model compounds. As is evident from the depletion bands in the ν(CO) region, mixtures of I-bzNO2 and LutH+ in CH2Cl2 under elevated pressures of CO are in the open hexacarbonyl form (Figure 10). Reduction of the pendent nitrobenzene fragment proceeds with product bands indistinguishable from those obtained in experiments conducted in the absence of CO (Figure 10a). In this case, reduction of the nitrobenzene fragment and its conversion to the aniline analogue is accompanied by coordination of the bridging S atom and elimination of CO. The stoichiometry of LutH+ consumption appears to be similar irrespective of whether the starting I-bzNO2 complex is in the closed pentacarbonyl or open hexacarbonyl forms. As expected for the [I-bzNH3]+ product, no spectral changes are associated with the application of a re-oxidising potential (Figure 10b). Application of potentials sufficient to reduce the diiron core do not lead to significant electrocatalysis.
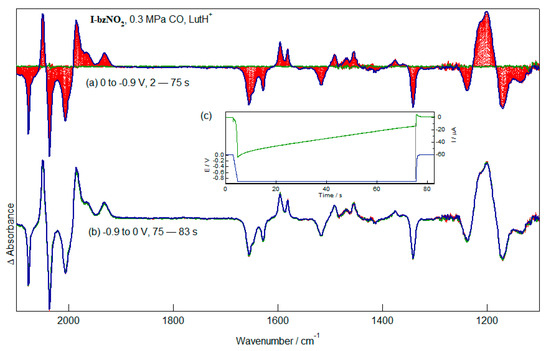
Figure 10.
IR-SEC spectra recorded during (a) reduction and (b) re-oxidation of solutions of I-bzNO2 (2 mM, CH2Cl2, 0.2 M TBAPF6) in CH2Cl2 with LutH+ (ca. 50 mM) and saturated with CO (0.3 MPa).
3. Discussion
Details of the redox chemistry of I-bzNO2 and I-bzNH2 and the interactions between the redox-accessible species with CO and LutH+ have been explored using a combination of electrochemical and in-situ spectroscopic methods. A summary of the chemistry is given in Scheme 2. Identification of the species derived from I-bzNO2 and I-bzNH2 is based primarily on the ν(CO) spectra where this is supplemented by their bands due to the NO2 stretching modes and EPR spectra of the one-electron reduced form of I-bzNO2. These studies have allowed unambiguous assignment of the first reduction of I-bzNO2 to a ligand centred process. This leads to an increase in the electron richness of the bridging S atom and this is reflected by a 5–6 cm−1 shift of the ν(CO) band profile to lower wavenumbers. The I-bzNH2 compound has a ν(CO) band profile intermediate between that of I-bzNO2 and [I-bzNO2]−. The small resultant shift in the wavenumbers of the ν(CO) bands reflect subtle changes in the electronic structure of the diiron core which can be translated into dramatic changes in the relative stability of different reaction products. This is illustrated by competitive binding of CO and the pendent thioether. The equilibrium constant for the reaction of the thioether-bound form of the 2Fe3S pentacarbonyl complexes with CO to give the substitution product with CO replacing the thioether strongly favours the pentacarbonyl, thioether-bound form for I-bzNO2 but strongly favours the hexacarbonyl, thioether dissociated form for [I-bzNO2]−. The distribution of products formed following metal-based reduction is expected to be similarly sensitive to the electronic structure of the core and this may be critical in determining the productivity of this general class of compound in catalytic reactions at strongly reducing potentials.

Scheme 2.
Summary of the chemical and redox interconversions of I-bzNO2 and I-bzNH2. The metal or ligand-based character of the redox reactions are emphasised using red text. The spectra of different of the species have been obtained by subtraction, and the wavenumbers for the bands in the subtraction spectra are given. In some cases, the absence of a good reference spectrum leads to distortions of the baseline and the introduction of features due to water vapour. Note that the initial spectra obtained following reduction of I-bzNH2 under CO have a band profile and wavenumber consistent with the generation of [I-(bzNH2)CO]− in chemistry analogous to that of Fe2(μ-S(CH2)3S)(CO)6 [29].
The reduction of I-bzNO2 in the presence of LutH+ reveals a well-defined 6 electron, 6 or 7 proton reaction with formation mostly of [I-bzNH3]+, where the shift of the band profile by ca. 4 cm−1 is approximately midway between that of I-bzNO2 and I-bzNH2 (ca. 2.5 cm−1). After conversion of I-bzNO2 to [I-bzNH3]+ further reduction of the diiron subsite leads to further consumption of protons (2–3 equivalents) in a reaction which is unlikely to be catalytic. In this case the increases in current response with the addition of acid to I-bzNO2 involves stoichiometric consumptions of protons unless the applied potential is greater than that required for reduction of the diiron subsite. In this case the sigmoidal current response observed for reduction of I-bzNH2 with acid (Figure 2) is most likely due to stoichiometric reaction between protons and the initial reduction products. More reducing potentials are needed to obtain an electrocatalytic response. It is interesting to note that despite the different starting species, the reaction products obtained following reduction of I-bzNO2 in the presence of acid are the same for reactions conducted under elevated pressures of N2 or CO. Clearly the reduction of the nitrobenzene fragment to protonated aniline proceeds similarly for the thioether bound, pentacarbonyl and thioether dissociated, hexacarbonyl forms and the equilibrium for the protonated aniline adduct lies strongly in favour of the thioether bound, pentacarbonyl form. The reaction is rapid, at least on the timescale of the IR-SEC experiments as no additional intermediate species were observed.
The behaviour of I-bzNO2 on reduction in the presence of protons provides a more solid basis for the interpretation of the voltammetric response of Fe2-adt(bzNO2) [22]. In light of the voltammetry of nitrobenzene and thioether-linked nitrobenzene ligands examined herein, together with the markedly different reversibility of the primary reduction process of the complex relative to that of the aniline adduct, Fe-adt(bzNH2) (Figure 2 of ref. [22]), are more consistent with the first wave being dominated by reduction at the nitrobenzene subsite. Further, it is significant that the current associated with the first reduction process saturates at low proton concentrations. It is more likely that stoichiometric electron/proton reactions following reduction of nitrobenzene account for the purported electrocatalysis at milder potentials. At more reducing potentials, the assignment of any proton-dependent current increases to electrocatalysis becomes problematic owing to formation of species unrelated to the starting complex or heterogeneous electron transfer from insoluble reduction products [32]. While, perhaps, the interpretation of the electrochemistry of Fe2-adt(bzNO2) is in error, resolution of the details of the chemistry of reactive systems such as the dithiolato-bridged carbonyl compounds clearly requires the combination of techniques.
The observation of stoichiometric, as opposed to electrocatalytic proton reduction following metal-based reduction of I-bzNO2 in excess HLut+ is significant as it suggests that electrocatalytic proton reduction of weak acids requires either the application of more strongly reducing potentials or reorganization of the parent complex. In this case the relationship between the catalytic species and the initial diiron compound is ill-defined. Similar comments apply in relation to studies of the purported electrocatalytic behaviour of a broader range of dithiolato-bridged diiron compounds at strongly reducing potentials, a situation which applies frequently when acetic acid is the proton source in non-aqueous solvents [33,34,35,36,37]. In this contribution we highlight the importance of monitoring both the current response from the system and the extent of reduction of the pendent redox centre and the diiron centre of this important class of compound.
4. Materials and Methods
4.1. General
All manipulations were performed under inert atmosphere of N2 using the Schlenk technique. Solvents were dried and distilled under N2 following usual procedures (Na for toluene and hexane, Na/benzophenone for tetrahydrofuran and CaH2 for acetonitrile). Chemical compounds were purchased from Aldrich (St. Louis, MO, USA) and were used as supplied without further purification. Micro-analysis was performed by Medac Ltd. (Egham, Surrey, UK). NMR spectroscopy was recorded on a JEOL Lambda 400 MHz (Tokyo, Japan) and FT-IR on a Shimadzu FTIR-8300 (Kyoto, Japan). A Bruker ECS 106 X-band spectrometer (Billerican, MA, USA) was used to collect EPR spectra and a Wilmad WG-810 electrolytic cell (Vineland, NJ, USA) assembly allowed in situ electrochemical generation of reduced compounds. EPR spectra were simulated with the aid of the EasySpin subroutines [24] which operate within a MatLab environment.
4.2. Electrochemistry
Cyclic voltammetry experiments were controlled using an Autolab PGSTAT 30 (Metrohm, Herisau, Switzerland) and digital simulations were performed using Digisim version 3.0 software (BASi, West Lafayette, IN, USA). Experiments were carried out in a three-compartment glass cell with a vitreous carbon disk as working electrode (1 mm2), a carbon strip as counting electrode and a Ag/AgCl reference electrode (Ag/AgCl, CH2Cl2, 0.45 M [NBu4][BF4], 0.05 M [NBu4]Cl). All electrodes were cleaned before use and the working electrode was polished with 9 μm alumina particles on polishing cloth. All potentials measured are referred against Ag/AgCl ({Fc+/Fc} = 0.52 V in CH3CN). The supporting electrolyte (0.2 M [NBu4][BF4]) was purged with N2 before experiments. Experiments in acidic conditions were carried out using 2,6-lutidinium (tetrafluoroborate as counter-cation) prepared in diethyl ether from 2,6-lutidine and HBF4.
4.3. Spectroelectrochemistry
Spectroelectrochemical (SEC) experiments were conducted using a purpose built cell previously described [25]. All experiments employed a 3 mm diameter vitreous carbon working, silver pseudo-reference and platinum foil counter electrodes. The potentials of the SEC experiments are uncorrected and given relative to the silver pseudo-reference electrode. Solutions for SEC analysis were prepared under strictly anaerobic conditions either through the agency of a Vacuum Atmospheres glove box or using standard Schlenk techniques. The applied potential was controlled using a PAR model 362 potentiostat (Princeton Applied Research, Oak Ridge, TN, USA). A Powerlab 4/20 interface (ADInstruments, Dunedin, New Zealand) using EChem V1.5.2 or Chart V4.12 provided a means of setting the applied potential and monitoring the potential and current response during SEC experiments. IR spectra were obtained using a Bio-Rad FT175C FTIR equipped with a Ge/KBr beamsplitter and narrow band MCT detector (Hercules, CA, USA). Spectral subtraction and curve fitting were performed using either in-house programs or Grams/32 AI software (Galactic Industries Corporation, Salem, NH, USA). Final plots were generated using Igor Pro (version 5.04B, Wavemetrics, Lake Oswego, OR, USA).
4.4. Synthesis
CH3C(CH2S)2CH2SC6H4-p-NO2, 4-((4-nitrophenylthio)methyl)-4-methyl-1,2-dithiolane: NaH (60% oil suspension, 0.33 g, 8.2 mmol) was washed with hexane and then dissolved in THF (50 cm3) under a dinitrogen atmosphere. CH3C(CH2S)2CH2SH (1.00 g, 6.0 mmol) was dissolved in THF (5 cm3), added to the solution of NaH and stirred at room temperature. 4-bromo-nitrobenzene (1.49 g, 7.4 mmol) was dissolved in THF (5 cm3), added to the mixture and stirred at 60 °C overnight. Excess of sodium hydride was quenched by ammonium chloride-saturated water and the product was extracted with dichloromethane. The organic phase was dried (MgSO4) and purified by flash chromatography (hexane/chloroform 4:1) to give a yellow solid (0.86 g, 3.0 mmol, 50%). 1H-NMR (400 MHz; solvent CDCl3; standard SiMe4) 1.39 (3H, s, CH3), 2.97 (2H, d, J 11.5 Hz, CH2SS), 3.17 (2H, d, J 11.5 Hz, CH2SS), 3.28 (2H, s, CH2SPh), 7.41 (2H, d, J 8.8 Hz, Ph), 8.13 (2H, d, J 9.0 Hz, Ph); Elemental analysis for C11H13NO2S3: Calcd.: C 45.97; H 4.56; N 4.87; S 33.45; Found: C 45.77; H 4.59; N 4.86; S 32.30.
Fe2(CO)5(CH3C(CH2S)2CH2SC6H4-p-NO2) (I-bzNO2): [Fe3(CO)12] (0.97 g, 1.9 mmol) was dissolved in toluene (50 cm3). CH3C(CH2S)2CH2SC6H4-p-NO2 (0.46 g, 1.6 mmol) was added to the solution. The dark green mixture turned red brown when it was heated at 90 °C for 90 min. The solvent was removed, and the compound was purified by flash chromatography under dinitrogen (diethyl ether/hexane 4:1) to give a red-brown powder (0.28 g, 0.52 mmol, 32%). IR: νmax/cm−1 (CO) 1933, 1988 and 2052 cm−1 (acetonitrile); 1H-NMR (400 MHz; solvent CDCl3; standard SiMe4) 0.95 (3H, s, CH3), 1.74 (2H, d, J 13.9 Hz, CH2SFe), 2.32 (2H, d, J 13.9 Hz, CH2SFe), 2.44 (2H, s, CCH2SPh), 7.95 (2H, d, J 8.8 Hz, Ph), 8.33 (2H, d, J 8.8 Hz, Ph); Elemental analysis for Fe2C16H13S3O7N: Calcd.: C 35.64; H 2.43; N 2.60; S 17.84; Found: C 35.74; H 2.50; N 2.67; S 17.84.
CH3C(CH2SH)2CH2SC6H4-p-NH2, 2-((4-aminophenylthio)methyl)-2-methylpropane-1,3-dithiol: Pd/C (12 mg) and NaBH4 (0.39 g, 10.3 mmol) were suspended in 2 M NaOH (20 cm3). CH3C(CH2S)2CH2SC6H4-p-NO2 (0.28 g, 0.97 mmol) was added and the mixture stirred at 90 °C overnight. The excess of NaBH4 was quenched with 2 M HCl and the solution was neutralised with 2 M NaOH. The compound was then extracted with chloroform, dried over magnesium sulphate and purified by flash chromatography (chloroform/ethyl acetate 1:1) to give a yellow solid (0.24 g, 0.92 mmol, 95%). 1H-NMR (400 MHz; solvent CDCl3; standard SiMe4) 1.04 (3H, s, CH3), 1.18 (2H, t, J 9.0 Hz, 2 × SH), 1.54 (2H, s, CCH2SPh), 2.61 (4H, d, J 9.2 Hz, 2 × CH2SH), 3.71 (2H, br s, PhNH2), 6.61 (2H, d, J 8.8 Hz, Ph), 7.26 (2H, d, J 8.8 Hz, Ph).
Fe2(CO)5(CH3C(CH2S)2CH2S C6H4-p-NO2) (I-bzNH2): [Fe3(CO)12] (0.56 g, 1.1 mmol) was dissolved in toluene (20 cm3) and stirred under nitrogen. CH3C(CH2SH)2CH2SPhNH2 (0.24 g, 0.92 mmol) was added to the solution. The dark green mixture turned red brown when it was heated at 90 °C for 90 min. The solvent was removed and the compound was purified by flash chromatography under dinitrogen (diethyl ether/hexane 1:1) to give a red-brown powder (0.26 g, 0.51 mmol, 55%). IR: νmax/cm−1 (CO) 1928, 1983 and 2048 cm−1 (acetonitrile); 1H-NMR (400 MHz; solvent CDCl3; standard SiMe4) 0.83 (3H, s, CH3), 1.66 (2H, d, J 13.7 Hz, CH2SPh), 2.25 (4H, d, J 13.4 Hz, 2 × CH2SFe), 3.95 (2H, s, NH2), 7.95 (2H, d, J 8.8 Hz, Ph), 8.33 (2H, d, J 8.8 Hz, Ph); Elemental analysis for Fe2C16H15S3O5N: Calcd.: C 37.74; H 2.97; N 2.75; S 18.89; Found: C 37.76; H 2.99; N 2.66; S 18.94.
4.5. X-Ray Structure of I-bzNH2
CCDC 1898467 contains the supplementary crystallographic data for I-bzNH2. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk).
Crystals were orange-brown blocks. A single crystal of size 0.40 × 0.29 × 0.24 mm (corresponding to the c, a and b axes) was selected, mounted on a glass fiber and coated in silicone grease for photographic examination and the data collection. Diffraction data were measured on an Enraf-Nonius CAD4 diffractometer with monochromated Mo Kα radiation. Cell parameters were refined using least-squares methods from the settings of 25 reflections in the range 10 to 11 degrees in theta. Each reflection was centred in 4 orientations. 3517 unique reflections were collected to theta of 25°, with 2632 having intensities greater than 2σI. 3 reflections were chosen to monitor any decay in intensities (measured every 10,000 s) and any change in orientation (measured every 300 reflections). There was slight decay of the crystal with an overall decrease in intensities of 1.11%. Diffractometer data showed a monoclinic crystal in the space group P21/c (equivalent to no. 14) with cell dimensions a = 8.797(3) Å, b = 12.528(4) Å, c = 18.181(4) Å, β = 94.30(2) Å and V = 1998.0(10) Å3. The data were corrected for Lorentz-polarisation effects, for decay of the intensities, for absorption by semi-empirical psi-scan methods, and for negative intensities by Bayesian statistical methods. The structure was solved using direct methods in the SHELXS-97 program and refined on F2 using full-matrix least-squares procedures in SHELXL-97. The refinement process showed one [Fe2(CO)5{CH3C(CH2S)CH2SC6H4-p-NH2}] molecule in the unit cell. All non-hydrogen atoms were refined with anisotropic thermal parameters. Hydrogen coordinates were included in idealised positions and subsequently refined freely. All hydrogen isotropic thermal parameters were refined freely. Convergence was reached with R1 = 0.043 and wR2 = 0.066 for all data weighted w = 1/[σ2(Fo2) + (0.029P)2] where P = (Fo2 + 2Fc2)/3; for the 2632 observed data R1 = 0.027 and wR2 = 0.059. In the final difference map the residual electron density was 0.30 e−/Å3 for the largest peak, which was close to an S atom.
5. Conclusions
A combination of electrochemical and spectroelectrochemical measurements have shown that reduction of the nitrobenzene fragment of I-bzNO2 occurs at potentials more positive than that of its diiron core. Based on shifts of the ν(CO)-band profile the impact of nitrobenzene reduction on the diiron core is approximately 8% of that for a metal-centred reduction. At mild potentials electrochemically-generated [I-bzNO2]− reacts rapidly with HLut+ to give I-bzNH2 and [I-bzNH3]+.
Competitive binding experiments between CO and the pendant thioether of the oxidised and reduced forms of I-bzNO2 and the neutral and protonated forms of I-bzNH2 show that thioether displacement is favoured when more electron withdrawing groups are constituents of the thioether. Nitrobenzene reduction or conversion to aniline shifts the equilibrium between the open, thioether dissociated, and closed forms. These results demonstrate that redox reactions of a remote site can be used to drive ligand dissociation and binding at the diiron centre.
Considering the many reports of electrocatalytic proton reduction of diiron compounds in the presence of weak acids, it is perhaps surprising that reduction of I-bzNO2 in the presence of excess HLut+ at potentials sufficient to reduce the diiron core does not result in catalytic proton reduction. Generally, the purported electrocatalytic behaviour is based only on the current response of the system where the form and phase of the catalytic species is not defined. This study highlights the value of spectroelectrochemical techniques when elucidating the chemistry of reactive, and reacting, systems.
Supplementary Materials
The following are available online at https://www.mdpi.com/2304-6740/7/3/37/s1, Figure S1 Cyclic voltammogram of (— —) nitrobenzene (2 mM) and (—) CH3C(CH2S)2CH2S(C6H4-p-NO2) (2 mM) (0.2 M [NBu4][BF4] in acetonitrile, 0.1 V·s−1). Figure S2. Cyclic voltammograms of the primary reduction processes of nitrobenzene (2 mM, 0.2 M [NBu4][BF4] in acetonitrile, 0.1 V·s−1) in the absence and in the presence of acetic acid: 0 (—), 1 (— —), 5 (---), 15 (– - – -) and 30 equivalents (— - - —). Figure S3. Cyclic voltammogram of 2,6-lutidinium tetrafluoroborate (0.4 M, 0.2 M [NBu4][BF4] in acetonitrile, 0.1 V·s−1). Figure S4. IR-SEC single beam spectra recorded during (top) reduction and (bottom) re-oxidation of I-bzNO2 in CH2Cl2 (2 mM, 0.2 M TBAPF6). For this, and subsequent, figures the initial spectrum of each block is shown in green and the final spectrum in blue. Figure S5. IR-SEC spectra recorded during (a) the reduction and (b) re-oxidation of I-bzNO2 in the presence of HLut+. Table S1. EasySpin EPR simulation parameters for [I-bzNO2]−. Table S2. Determination of the free energy activation (ΔG‡) parameter in CDCl3; Tc is the coalescence temperature; Δνab is determined from the slow limit spectra. Cif and checkcif files.
Author Contributions
Conceptualization, C.J.P. and S.P.B.; methodology, S.P.B.; formal analysis, S.P.B. and C.J.P.; investigation, C.T., S.J.B. and S.A.F.; resources, C.J.P. and S.P.B.; data curation, S.P.B.; writing—original draft preparation, S.P.B. and C.J.P.; writing—review and editing, S.P.B., C.J.P. and C.T.; supervision, C.J.P. and S.P.B.; funding acquisition, C.J.P. and S.P.B.
Funding
This research was funded by the Australian Research Council, grant number DP0344556, the John Innes Centre and the BBSRC.
Acknowledgments
We thank David Hughes for the determination of the crystal structure of I-bzNH2. SPB thanks Alcoa, Australia for the donation of the glove box used at the University of Melbourne and for the expert assistance provided by the mechanical workshop of the School of Chemistry, University of Melbourne.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rauchfuss, T.B. Diiron azadithiolates as models for the [FeFe]-hydrogenase active site and paradigm for the role of the second coordination sphere. Acc. Chem. Res. 2015, 48, 2107–2116. [Google Scholar] [CrossRef] [PubMed]
- Best, S.P. Spectroelectrochemistry of hydrogenase enzymes and related compounds. Coord. Chem. Rev. 2005, 249, 1536–1554. [Google Scholar] [CrossRef]
- Lubitz, W.; Ogata, H.; Ruediger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef] [PubMed]
- Capon, J.-F.; Gloaguen, F.; Petillon, F.Y.; Schollhammer, P.; Talarmin, J. On the electrochemistry of diiron dithiolate complexes related to the active site of the [FeFe]H2ase. C. R. Chim. 2008, 11, 842–851. [Google Scholar] [CrossRef]
- Wright, J.A.; Pickett, C.J. Towards a functional model of the [FeFe]-hydrogenase: Dihydrogen oxidation. ChemCatChem 2012, 4, 1723–1724. [Google Scholar] [CrossRef]
- Xu, T.; Chen, D.; Hu, X. Hydrogen-activating models of hydrogenases. Coord. Chem. Rev. 2015, 303, 32–41. [Google Scholar] [CrossRef]
- Tard, C.; Pickett, C.J. Structural and functional analogues of the active sites of the [Fe]-, [NiFe]-, and [FeFe]-hydrogenases. Chem. Rev. 2009, 109, 2245–2274. [Google Scholar] [CrossRef]
- Apfel, U.-P.; Petillon, F.Y.; Schollhammer, P.; Talarmin, J.; Weigand, W. [FeFe] hydrogenase models: An overview. In Bioinspired Catalysis: Metal-Sulfur Complexes; Weigand, W., Schollhammer, P., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; pp. 79–103. [Google Scholar]
- Capon, J.-F.; Gloaguen, F.; Schollhammer, P.; Talarmin, J. Catalysis of the electrochemical H2 evolution by di-iron sub-site models. Coord. Chem. Rev. 2005, 249, 1664–1676. [Google Scholar] [CrossRef]
- Schilter, D.; Camara, J.M.; Huynh, M.T.; Hammes-Schiffer, S.; Rauchfuss, T.B. Hydrogenase enzymes and their synthetic models: The role of metal hydrides. Chem. Rev. 2016, 116, 8693–8749. [Google Scholar] [CrossRef]
- Lansing, J.C.; Manor, B.C.; Rauchfuss, T.B. Hydrogenase Models; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2014; pp. 1–21. [Google Scholar]
- Wittkamp, F.; Senger, M.; Stripp, S.T.; Apfel, U.P. [FeFe]-hydrogenases: Recent developments and future perspectives. Chem. Commun. 2018, 54, 5934–5942. [Google Scholar] [CrossRef]
- Razavet, M.; Davies, S.C.; Hughes, D.L.; Barclay, J.E.; Evans, D.J.; Fairhurst, S.A.; Liu, X.; Pickett, C.J. All-iron hydrogenase: Synthesis, structure and properties of {2Fe3S}-assemblies related to the di-iron sub-site of the h-cluster. Dalton Trans. 2003, 586–595. [Google Scholar] [CrossRef]
- Jablonskyte, A.; Wright, J.A.; Fairhurst, S.A.; Webster, L.R.; Pickett, C.J. [FeFe] hydrogenase: Protonation of {2Fe3S} systems and formation of super-reduced hydride states. Angew. Chem. Int. Ed. 2014, 53, 10143–10146. [Google Scholar] [CrossRef]
- Razavet, M.; Borg, S.J.; George, S.J.; Best, S.P.; Fairhurst, S.A.; Pickett, C.J. Transient ftir spectroelectrochemical and stopped-flow detection of a mixed valence {Fe(I)–Fe(II)} bridging carbonyl intermediate with structural elements and spectroscopic characteristics of the di-iron sub-site of all-iron hydrogenase. Chem. Commun. 2002, 700–701. [Google Scholar] [CrossRef]
- George, S.J.; Cui, Z.; Razavet, M.; Pickett, C.J. The di-iron subsite of all-iron hydrogenase: Mechanism of cyanation of a synthetic {2Fe3S}—Carbonyl assembly. Chem. Eur. J. 2002, 8, 4037–4046. [Google Scholar] [CrossRef]
- Tard, C.; Liu, X.; Ibrahim, S.K.; Bruschi, M.; De Gioia, L.; Davies, S.C.; Yang, X.; Wang, L.-S.; Sawers, G.; Pickett, C.J. Synthesis of the H-cluster framework of iron-only hydrogenase. Nature 2005, 433, 610–613. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Rahaman, A.; Holt, K.B.; Nordlander, E.; Richmond, M.G.; Kabir, S.E.; Hogarth, G. Hydrogenase biomimetics with redox-active ligands: Electrocatalytic proton reduction by [Fe2(CO)4(κ2-diamine)(μ-edt)] (diamine = 2,2′-bipy, 1,10-phen). Polyhedron 2016, 116, 127–135. [Google Scholar] [CrossRef]
- Lansing, J.C.; Camara, J.M.; Gray, D.E.; Rauchfuss, T.B. Hydrogen production catalyzed by bidirectional, biomimetic models of the [FeFe]-hydrogenase active site. Organometallics 2014, 33, 5897–5906. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Groy, T.L.; Jones, A.K. Biomimetic model for [FeFe]-hydrogenase: Asymmetrically disubstituted diiron complex with a redox-active 2,2′-bipyridyl ligand. Dalton Trans. 2013, 42, 3843–3853. [Google Scholar] [CrossRef] [PubMed]
- Camara, J.M.; Rauchfuss, T.B. Combining acid-base, redox and substrate binding functionalities to give a complete model for the [FeFe]-hydrogenase. Nat. Chem. 2012, 4, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.B.; Wang, M.; Shi, Z.; Cui, H.G.; Dong, W.B.; Chen, J.S.; Akermark, B.; Sun, L.C. Synthesis, structures and electrochemical properties of nitro- and amino-functionalized diiron azadithiolates as active site models of fe-only hydrogenases. Chem.-Eur. J. 2004, 10, 4474–4479. [Google Scholar] [CrossRef] [PubMed]
- Alberti, A.; Martelli, G.; Pedulli, G.F. Radical anions and nitroxides from alkylthio-, alkylsulfinyl-, and alkylsulfonyl nitrobenzenes. J. Chem. Soc. Perkin Trans. 2 1977, 1252–1255. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. Easyspin, a comprehensive software package for spectral simulation and analysis in epr. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Borg, S.J.; Best, S.P. Spectroelectrochemical cell for the study of interactions between redox-activated species and moderate pressures of gaseous substrates. J. Electroanal. Chem. 2002, 535, 57–64. [Google Scholar] [CrossRef]
- Borg, S.J.; Tye, J.W.; Hall, M.B.; Best, S.P. Assignment of molecular structures to the electrochemical reduction products of diiron compounds related to [Fe–Fe] hydrogenase: A combined experimental and density functional theory study. Inorg. Chem. 2007, 46, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Borg, S.J.; Bondin, M.I.; Best, S.P.; Razavet, M.; Liu, X.; Pickett, C.J. Electrocatalytic proton reduction by dithiolate-bridged diiron carbonyl complexes: A connection to the H-cluster? Biochem. Soc. Trans. 2005, 33, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Pickett, C.J.; Vincent, K.A.; Ibrahim, S.K.; Gormal, C.A.; Smith, B.E.; Fairhurst, S.A.; Best, S.P. Synergic binding of carbon monoxide and cyanide to the femo cofactor of nitrogenase: Relic chemistry of an ancient enzyme? Chem. Eur. J. 2004, 10, 4770–4776. [Google Scholar] [CrossRef] [PubMed]
- Borg, S.J.; Behrsing, T.; Best, S.P.; Razavet, M.; Liu, X.; Pickett, C.J. Electron transfer at a dithiolate-bridged diiron assembly: Electrocatalytic hydrogen evolution. J. Am. Chem. Soc. 2004, 126, 16988–16999. [Google Scholar] [CrossRef]
- Wang, S.; Aster, A.; Mirmohades, M.; Lomoth, R.; Hammarstroem, L. Structural and kinetic studies of intermediates of a biomimetic diiron proton-reduction catalyst. Inorg. Chem. 2018, 57, 768–776. [Google Scholar] [CrossRef]
- Xu, F.; Tard, C.; Wang, X.; Ibrahim, S.K.; Hughes, D.L.; Zhong, W.; Zeng, X.; Luo, Q.; Liu, X.; Pickett, C.J. Controlling carbon monoxide binding at di-iron units related to the iron-only hydrogenase sub-site. Chem. Commun. 2008, 606–608. [Google Scholar] [CrossRef]
- Borg, S.J.; Ibrahim, S.K.; Pickett, C.J.; Best, S.P. Electrocatalysis of hydrogen evolution by synthetic diiron units using weak acids as the proton source: Pathways of doubtful relevance to enzymic catalysis by the diiron subsite of [FeFe] hydrogenase. C. R. Chim. 2008, 11, 852–860. [Google Scholar] [CrossRef]
- Zhao, P.-H.; Hu, M.-Y.; Li, J.-R.; Ma, Z.-Y.; Wang, Y.-Z.; He, J.; Li, Y.-L.; Liu, X.-F. Influence of dithiolate bridges on the structures and electrocatalytic performance of small bite-angle PNP-chelated diiron complexes Fe2(μ-xdt)(CO)4{κ2-(Ph2P)2NR} related to [FeFe]-hydrogenases. Organometallics 2019, 38, 385–394. [Google Scholar] [CrossRef]
- Zhao, P.-H.; Ma, Z.-Y.; Hu, M.-Y.; He, J.; Wang, Y.-Z.; Jing, X.-B.; Chen, H.-Y.; Wang, Z.; Li, Y.-L. PNP-chelated and -bridged diiron dithiolate complexes Fe2(μ-pdt)(CO)4{(Ph2P)2NR} together with related monophosphine complexes for the [2Fe]H subsite of [FeFe]-hydrogenases: Preparation, structure, and electrocatalysis. Organometallics 2018, 37, 1280–1290. [Google Scholar] [CrossRef]
- Durgaprasad, G.; Das, S.K. Modeling the active site of [FeFe]-hydrogenase: Electro-catalytic hydrogen evolution from acetic acid catalysed by [Fe2(μ-L)(CO)6] and [Fe2(μ-L(CO)5(PPh3)] (L = pyrazine-2,3-dithiolate, quinoxaline-2,3-dithiolate and pyrido[2,3-b]pyrazine-2,3-dithiolate). J. Chem. Sci. 2015, 127, 295–305. [Google Scholar] [CrossRef]
- Song, L.-C.; Cao, M.; Du, Z.-Q.; Feng, Z.-H.; Ma, Z.; Song, H.-B. CO substitution reactions of diiron complexes [{(μ-SCH2)2x}Fe2(CO)6] and [{(μ-SeCH2)2x}Fe2(CO)6] (x = O, CH2) with Ph2PCl/Me3NO to give Ph2PCl−, Ph2PNMe2−, and Ph2PP(:O)Ph2-substituted complexes related to [FeFe] hydrogenases. Eur. J. Inorg. Chem. 2014, 2014, 1886–1895. [Google Scholar] [CrossRef]
- Harb, M.K.; Daraosheh, A.; Goerls, H.; Smith, E.R.; Meyer, G.J.; Swenson, M.T.; Sakamoto, T.; Glass, R.S.; Lichtenberger, D.L.; Evans, D.H.; et al. Effects of alkane linker length and chalcogen character in [FeFe]-hydrogenase inspired compounds. Heteroat. Chem. 2014, 25, 592–606. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).