Bond Forming Reactions Involving Isocyanides at Diiron Complexes




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
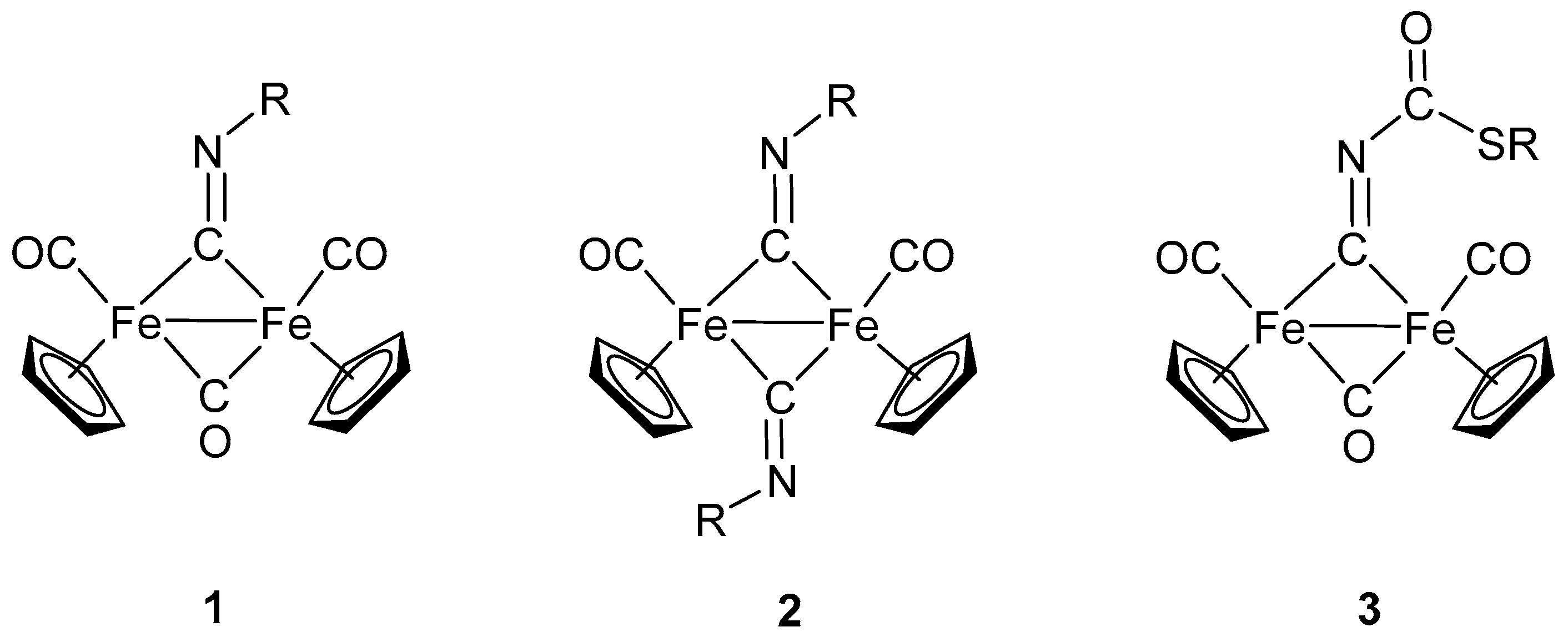
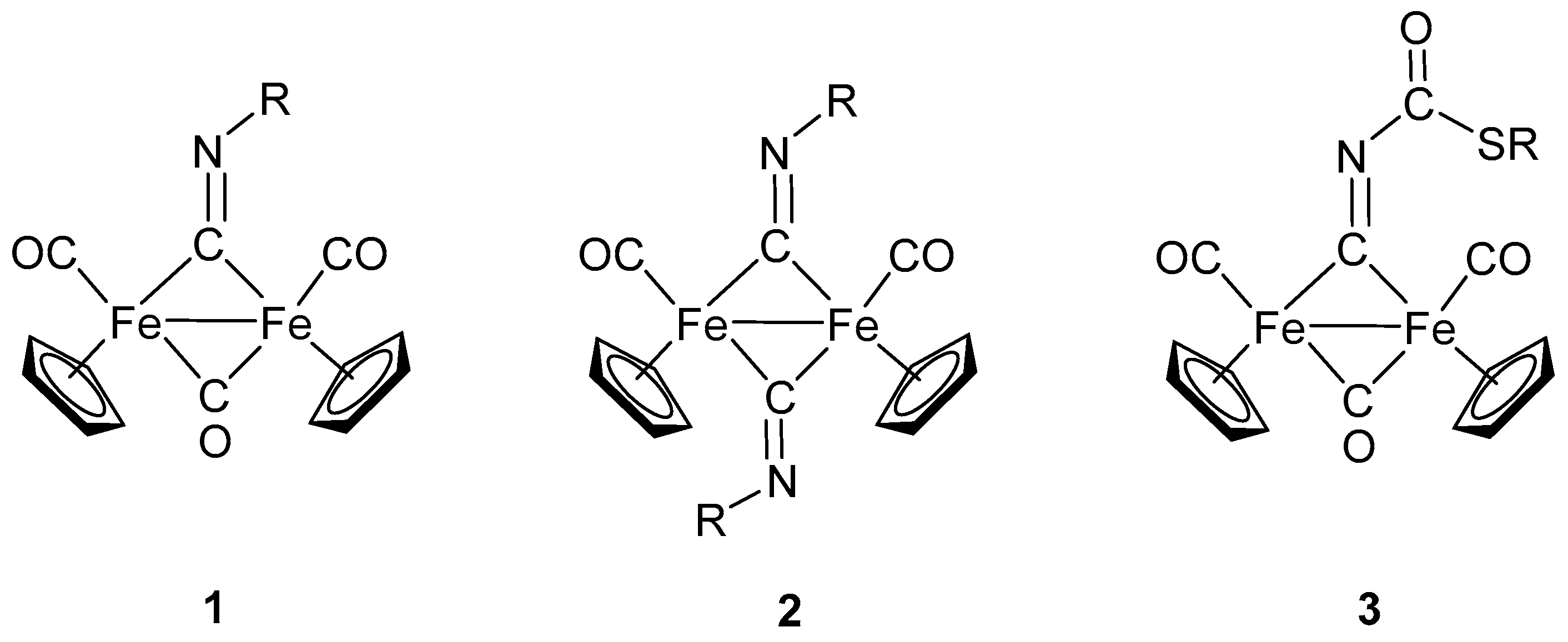
2. Diiron Complexes with Isocyanide Ligands
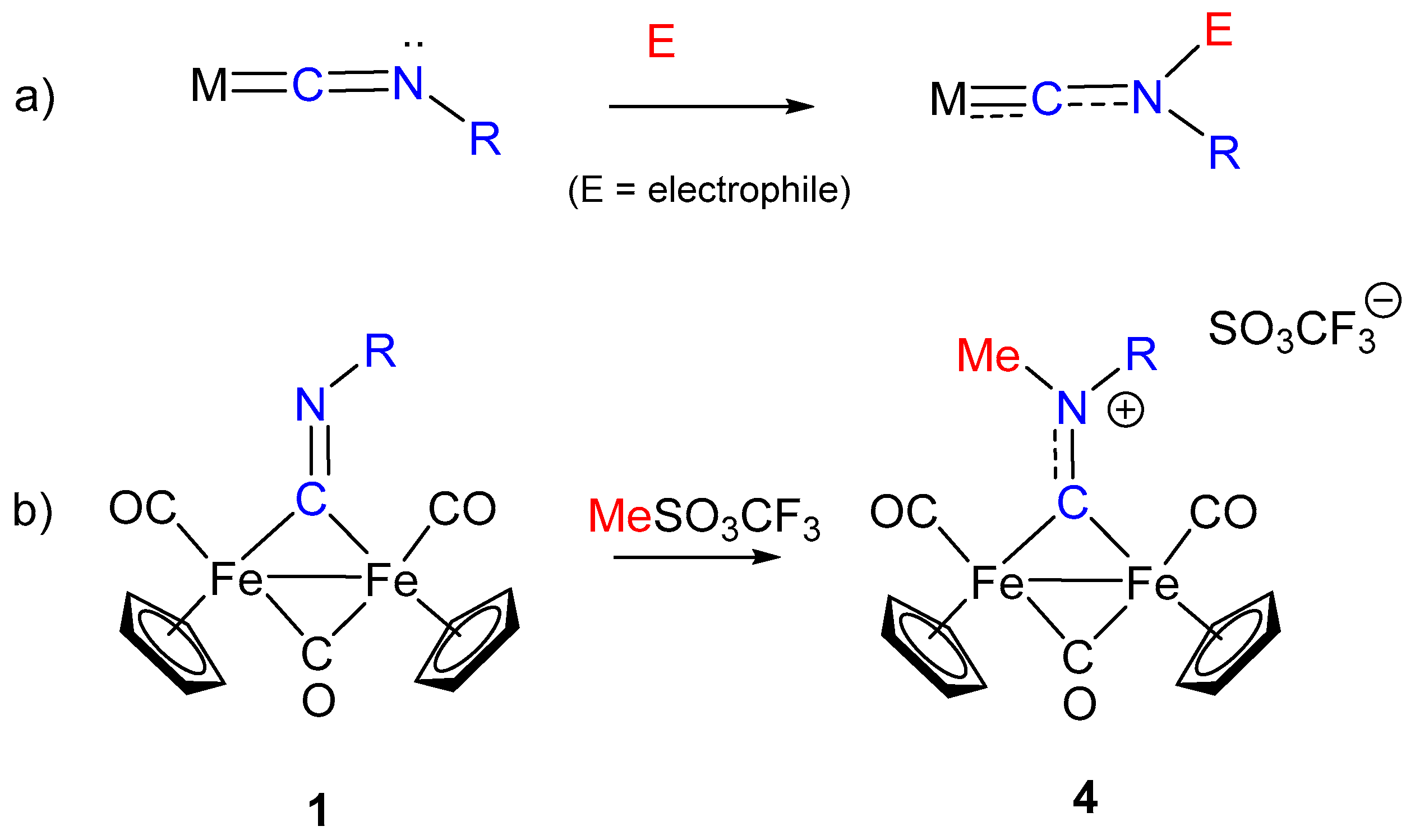
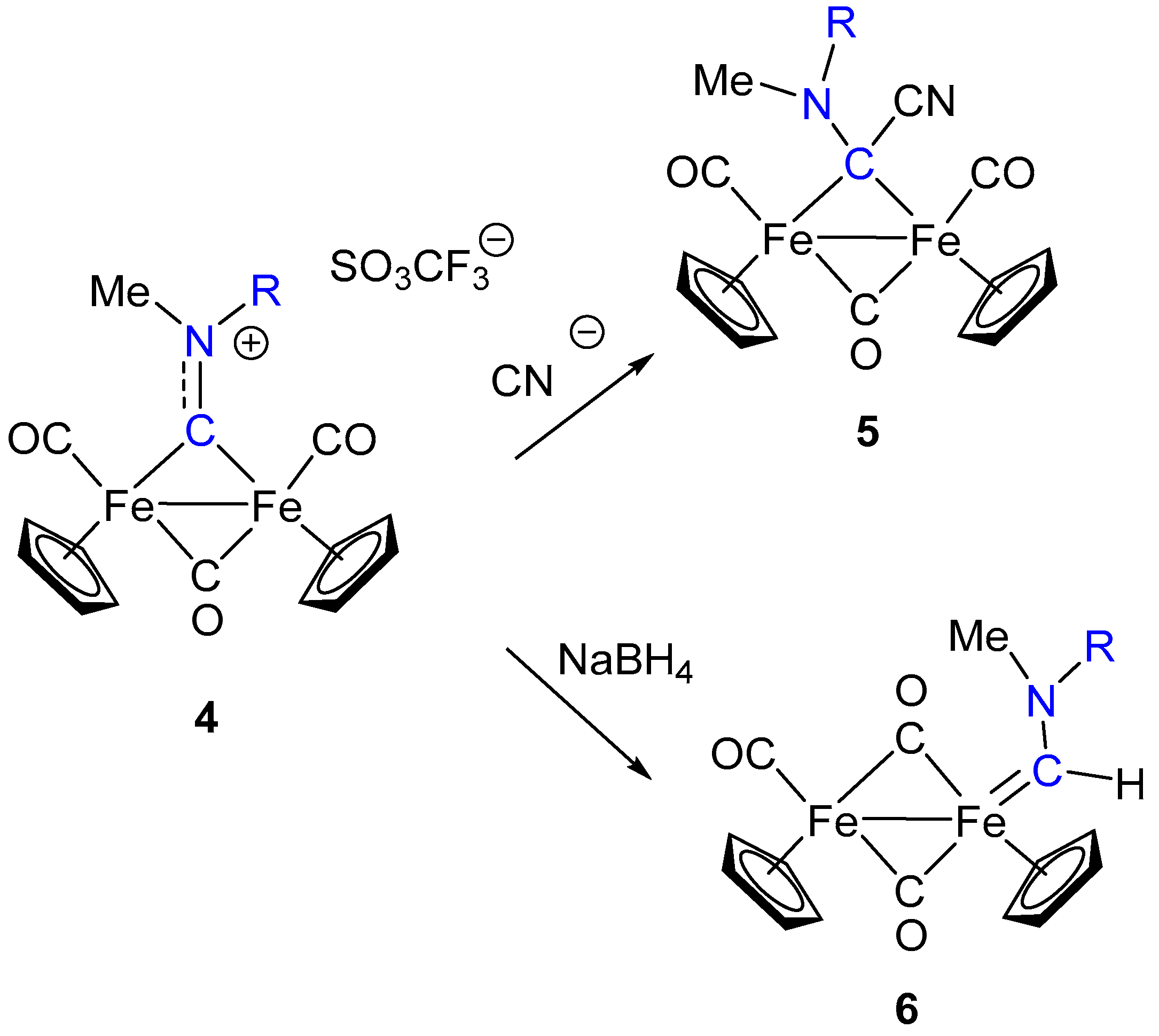
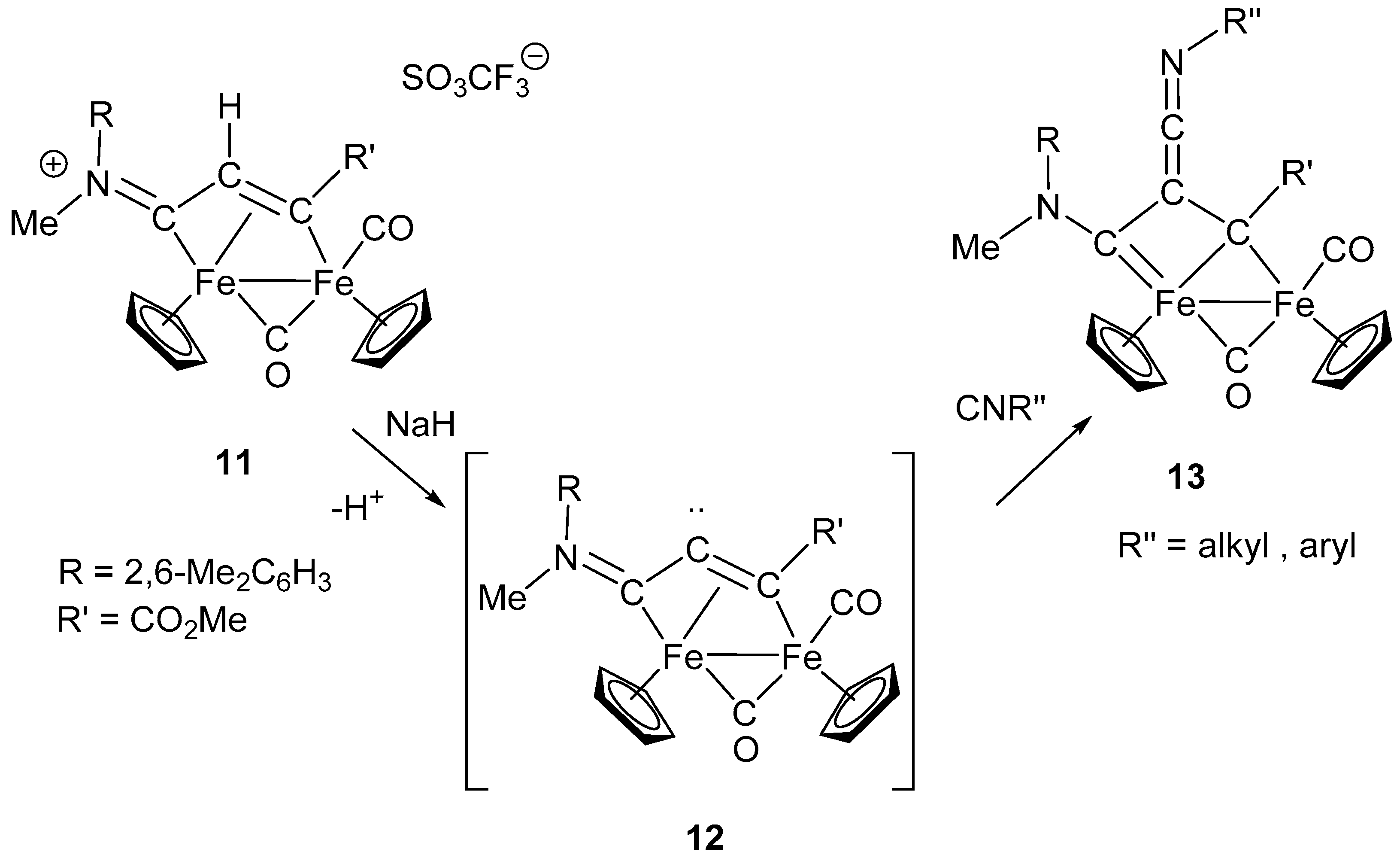
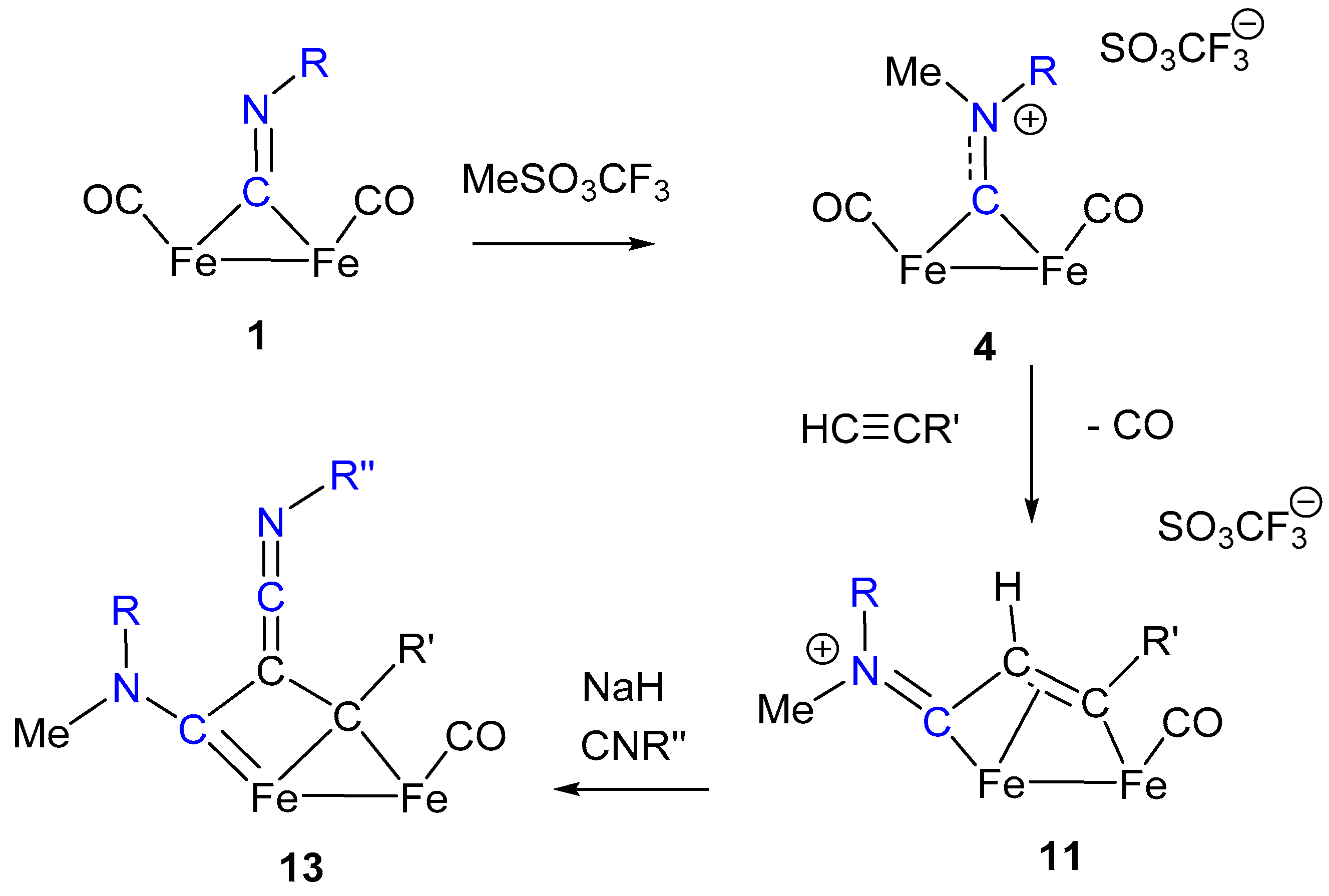
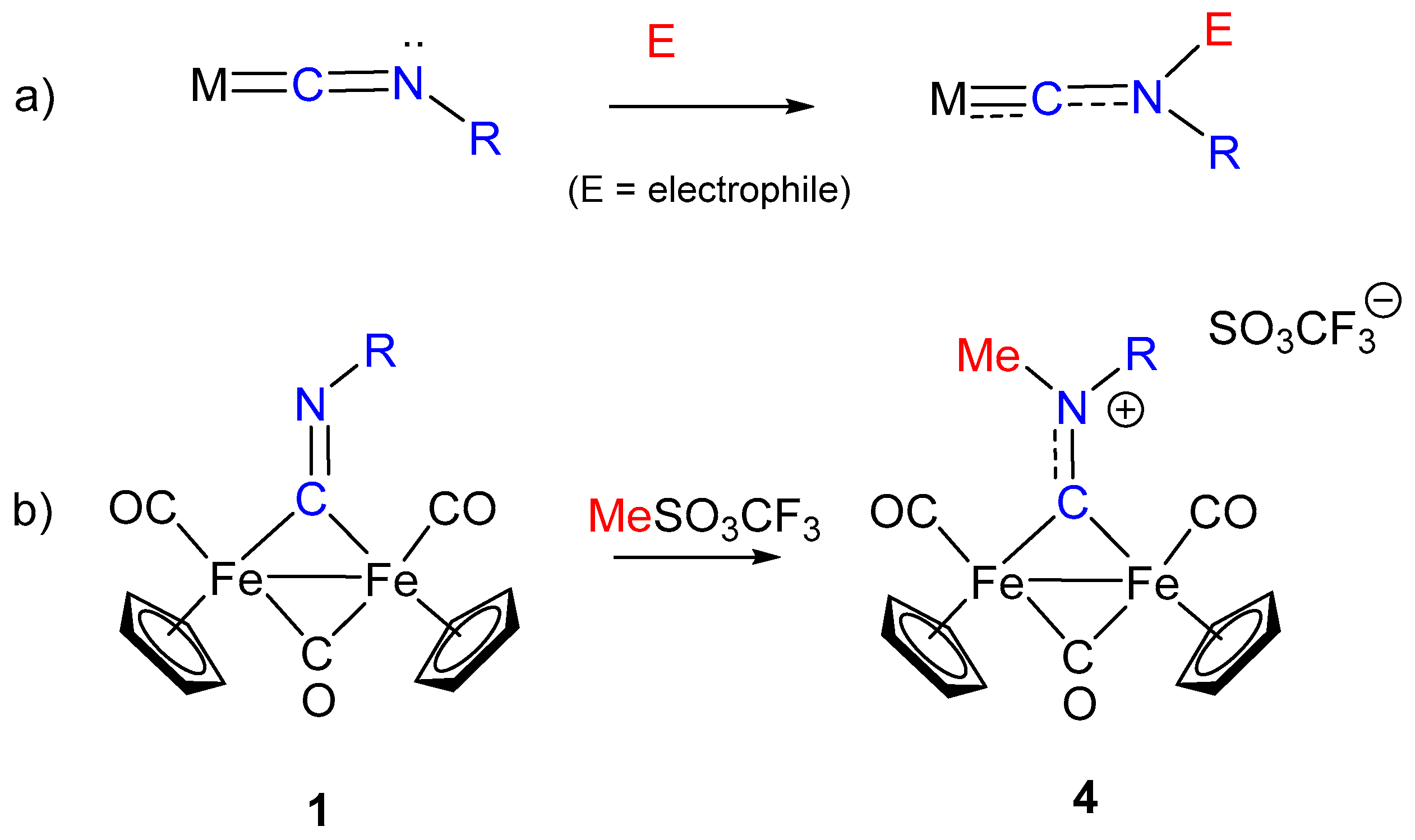
3. Electrophilic Addition at Bridging CNR as Effective Route to Bridging Aminoalkylidyne
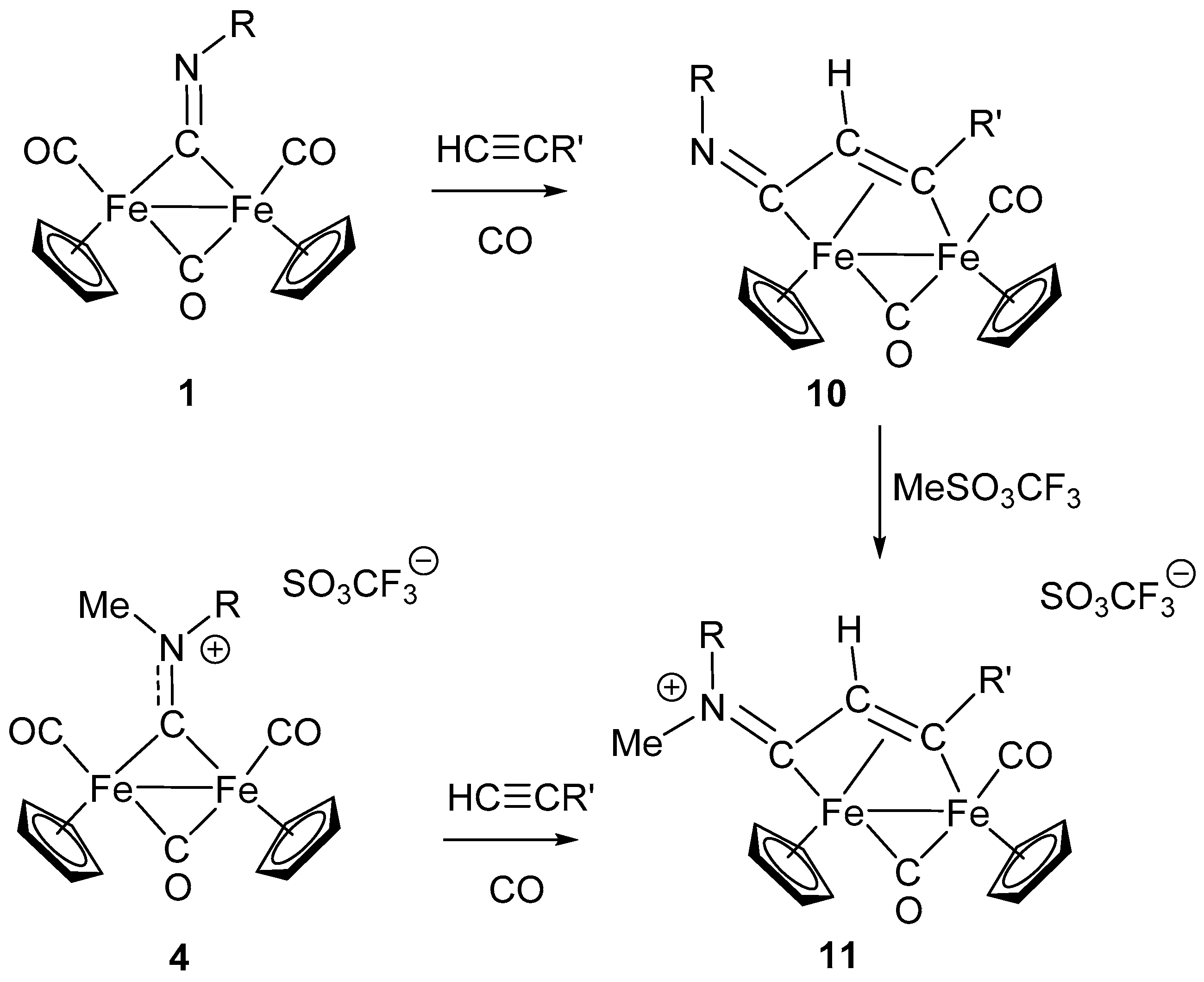
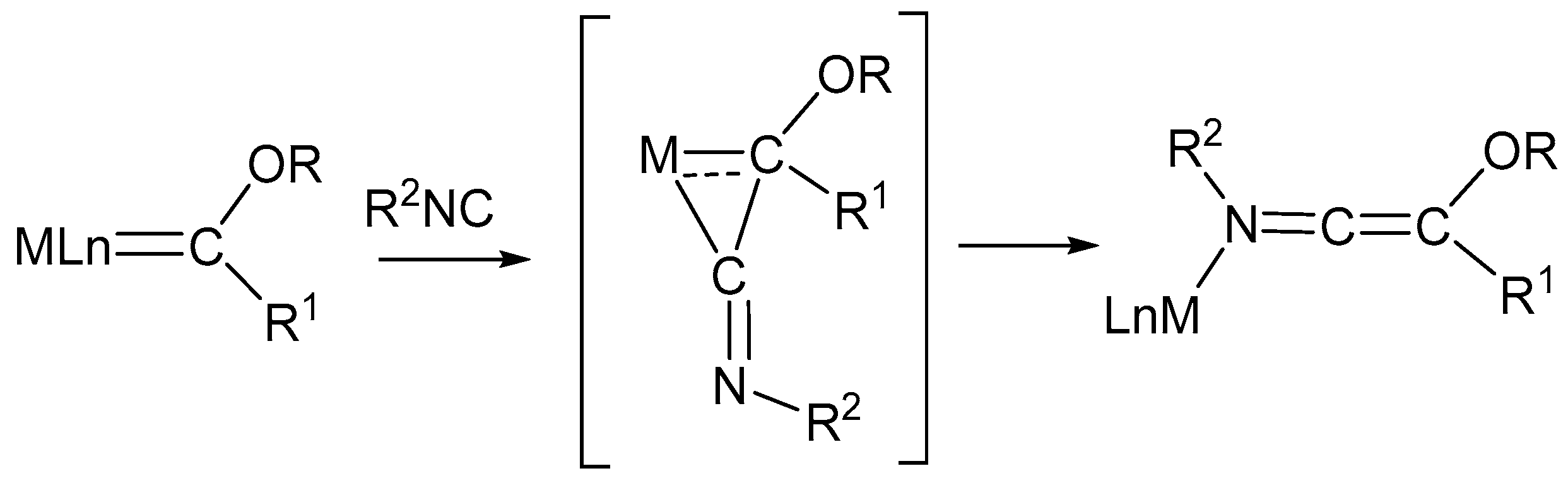
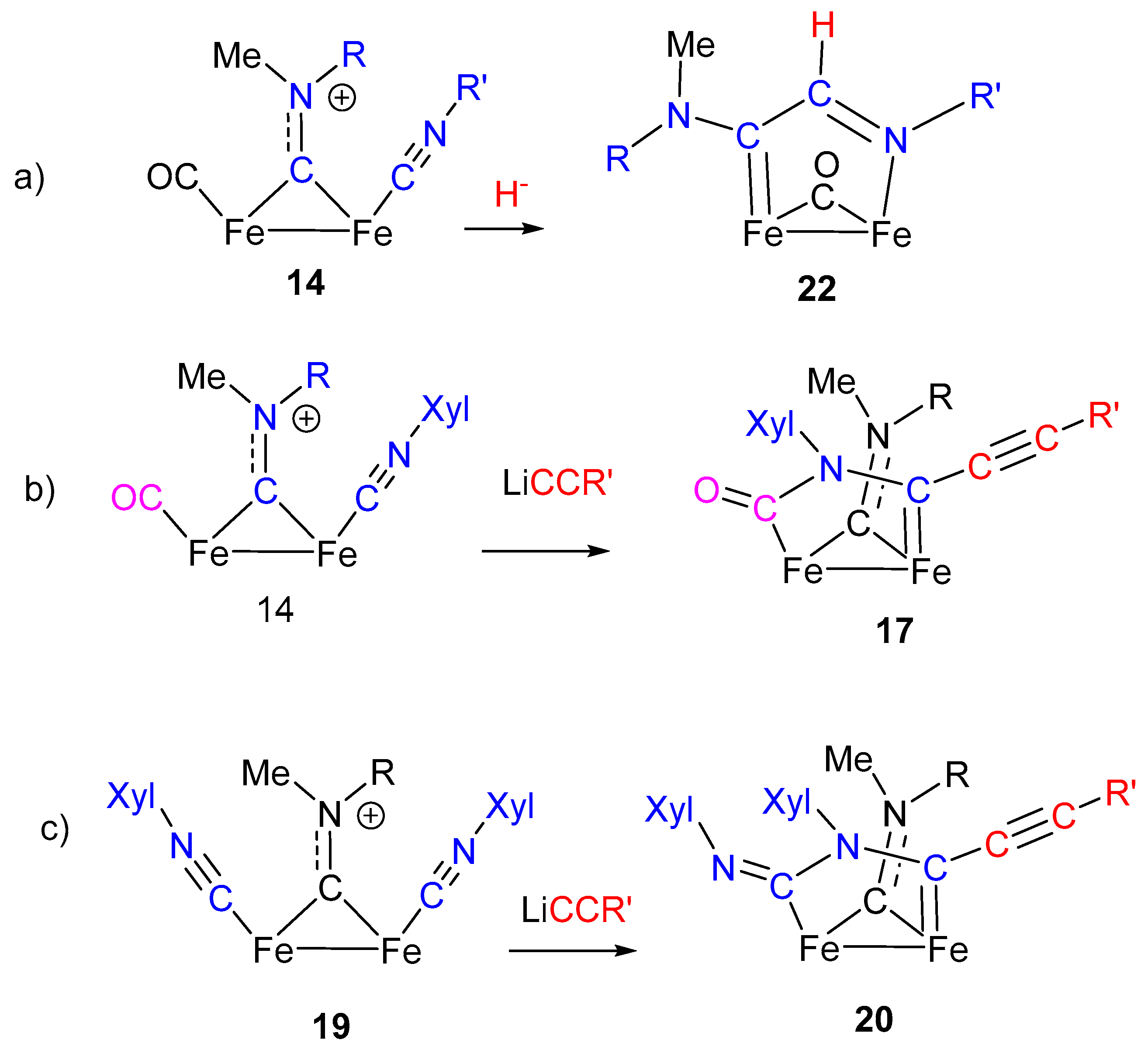
4. Insertion Reactions Involving Isocyanides in Diiron Complexes
5. Addition of Isocyanides to Coordinated Ligands
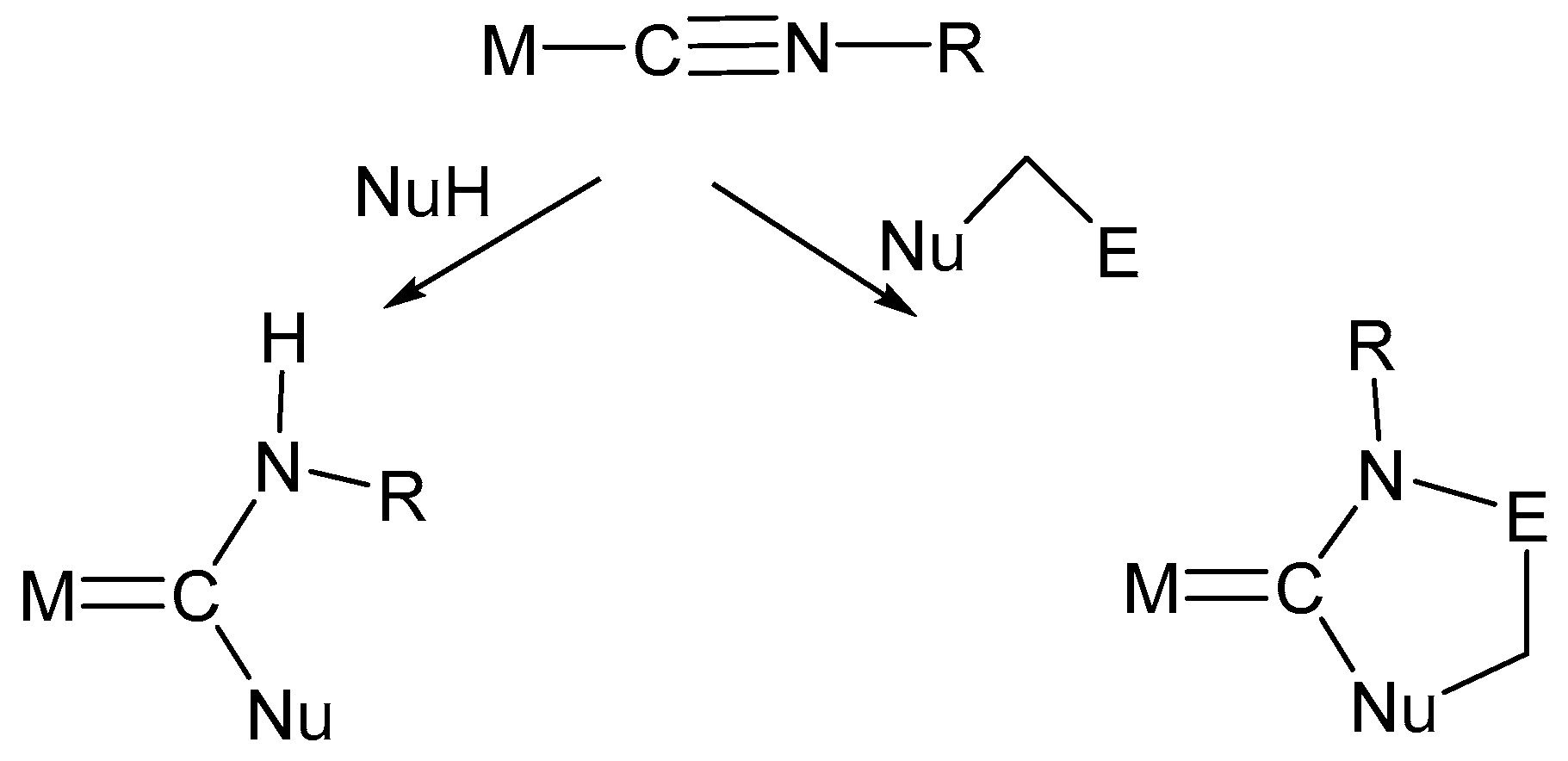
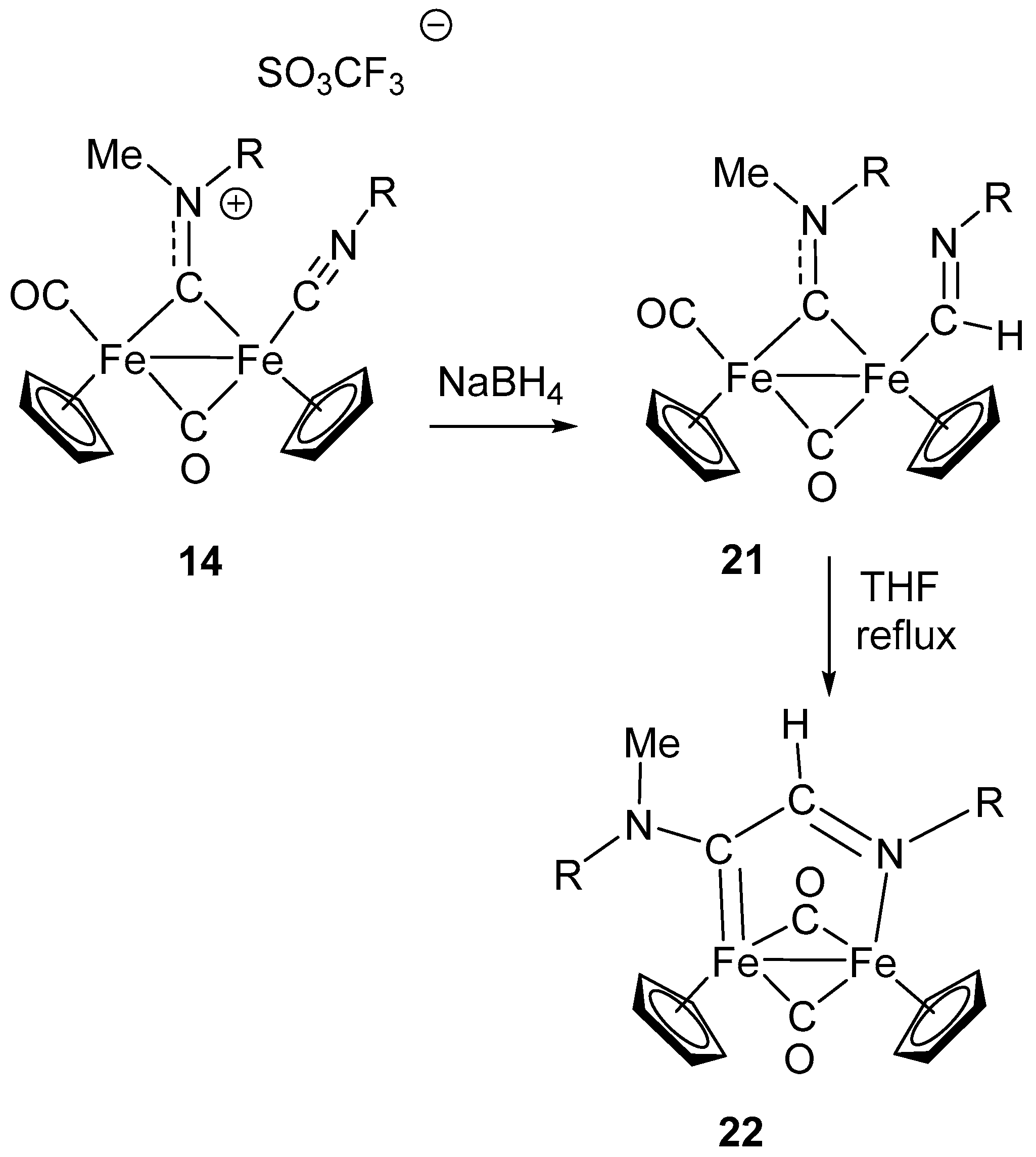
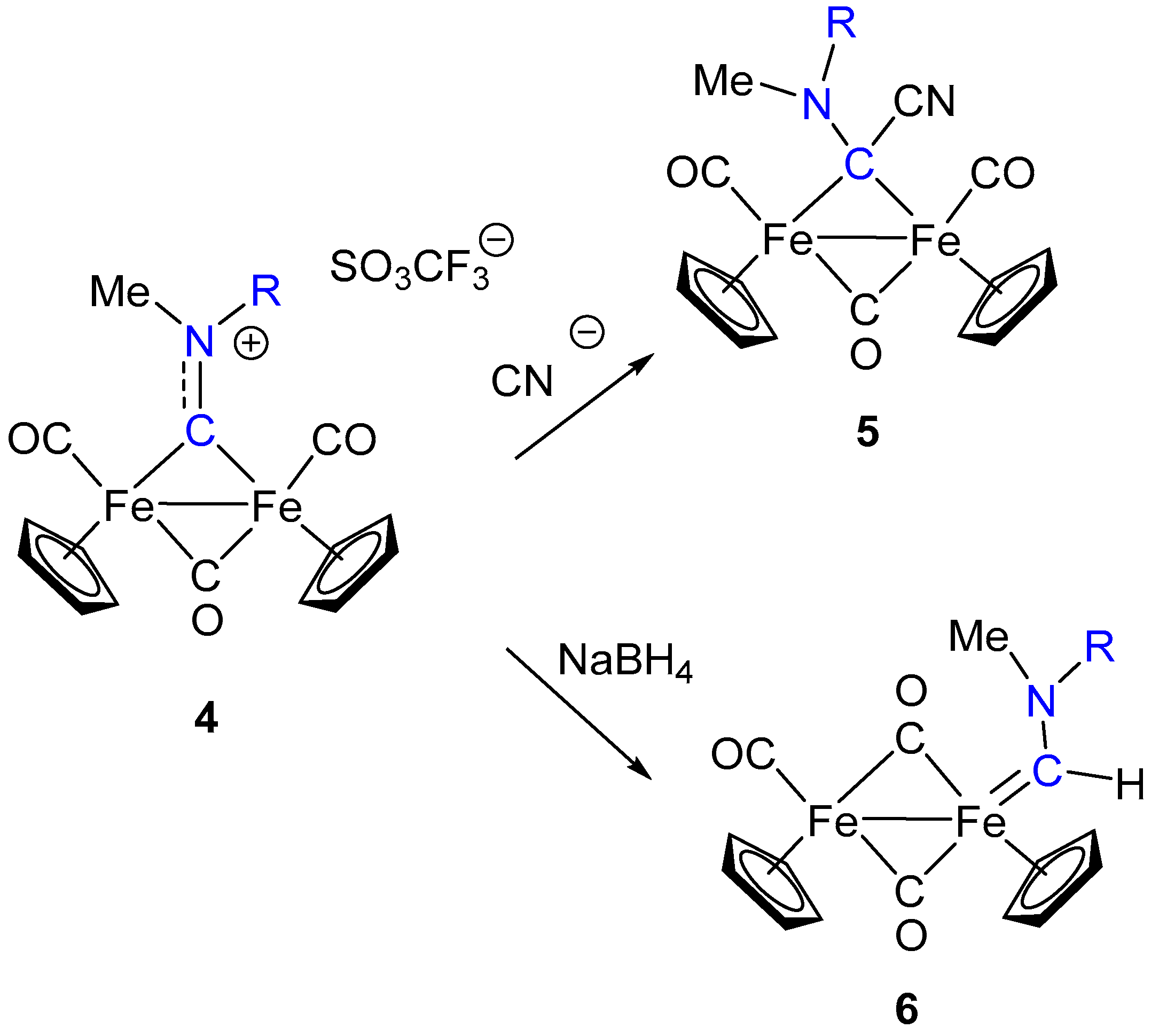
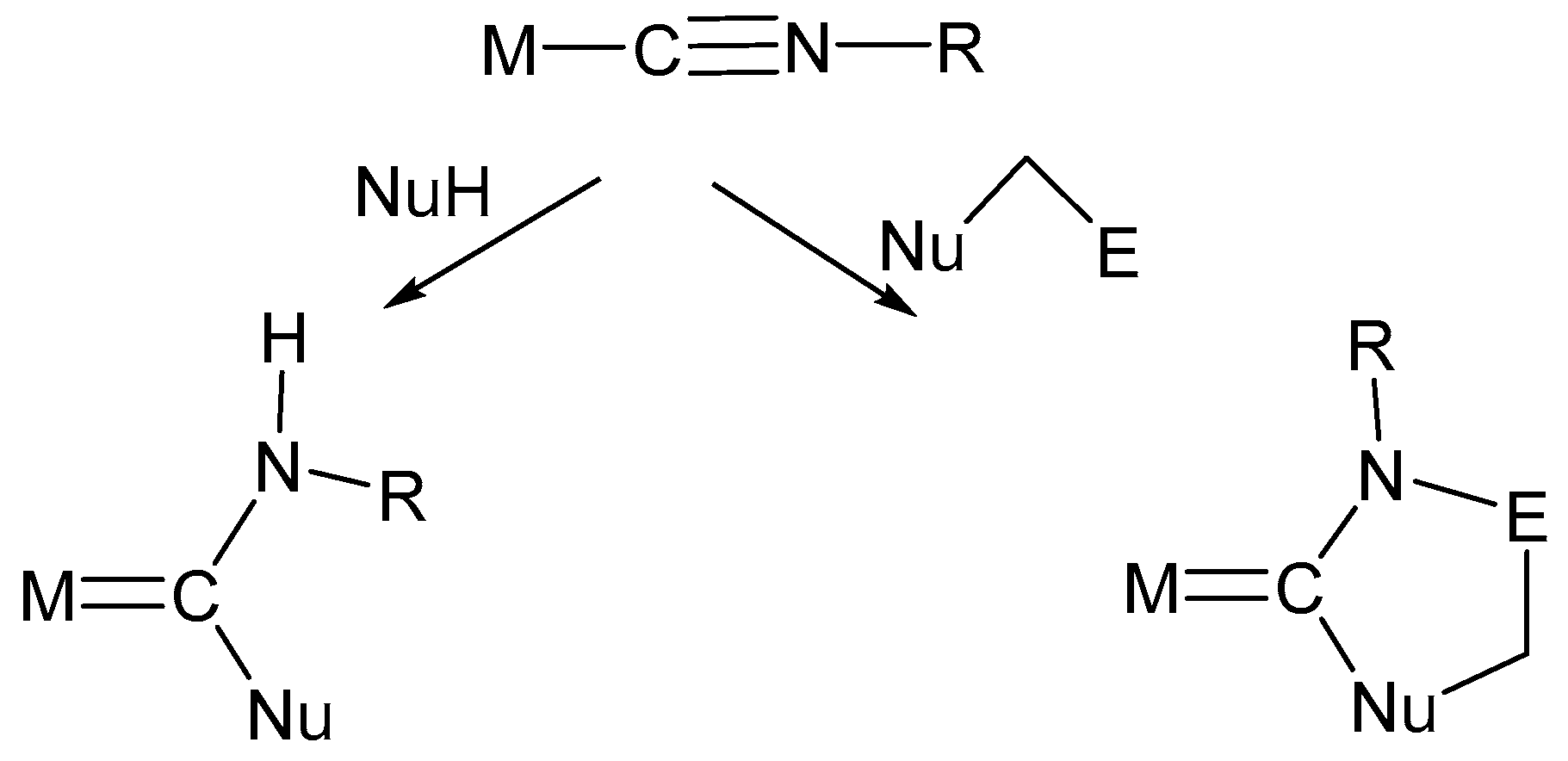
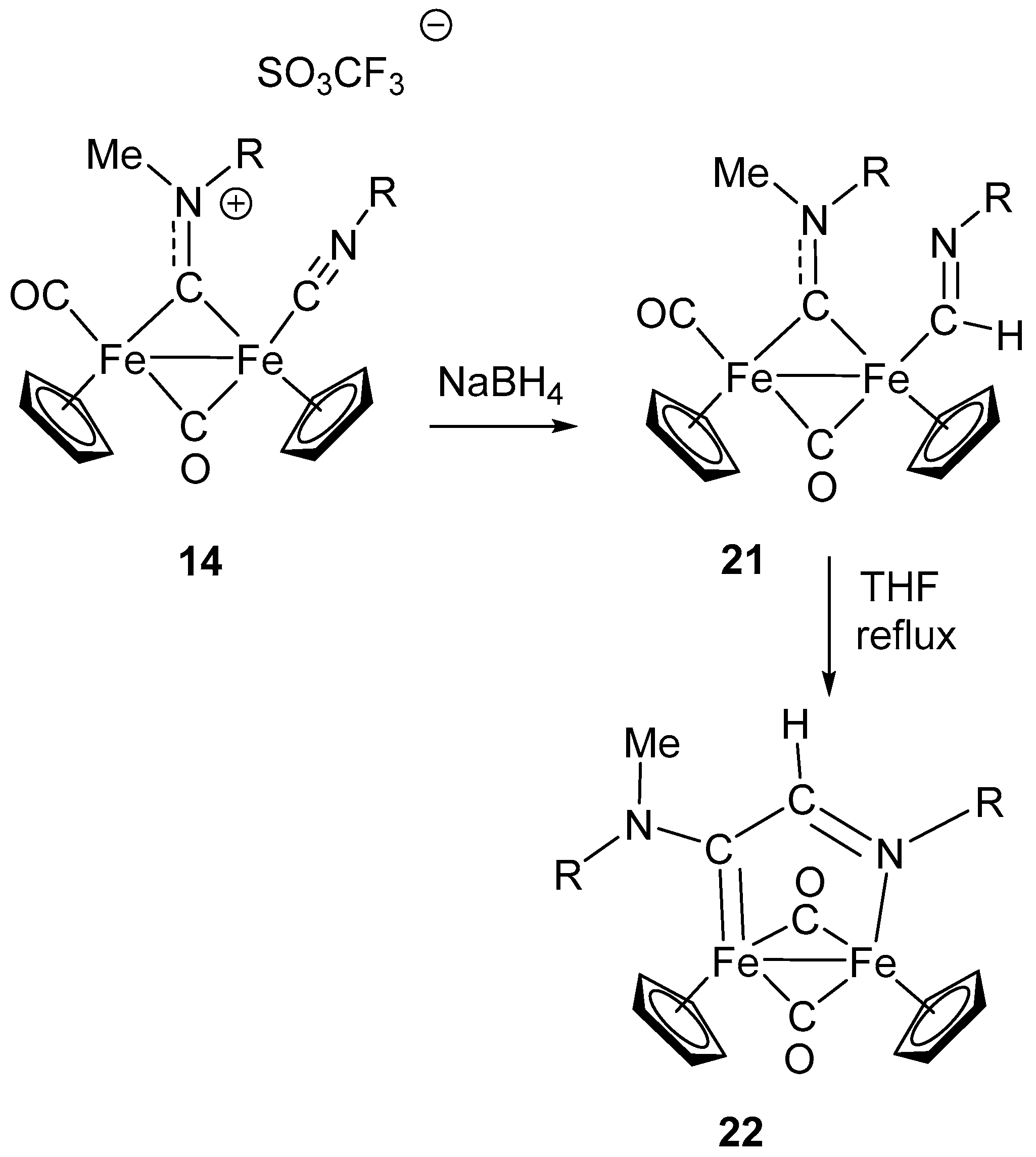
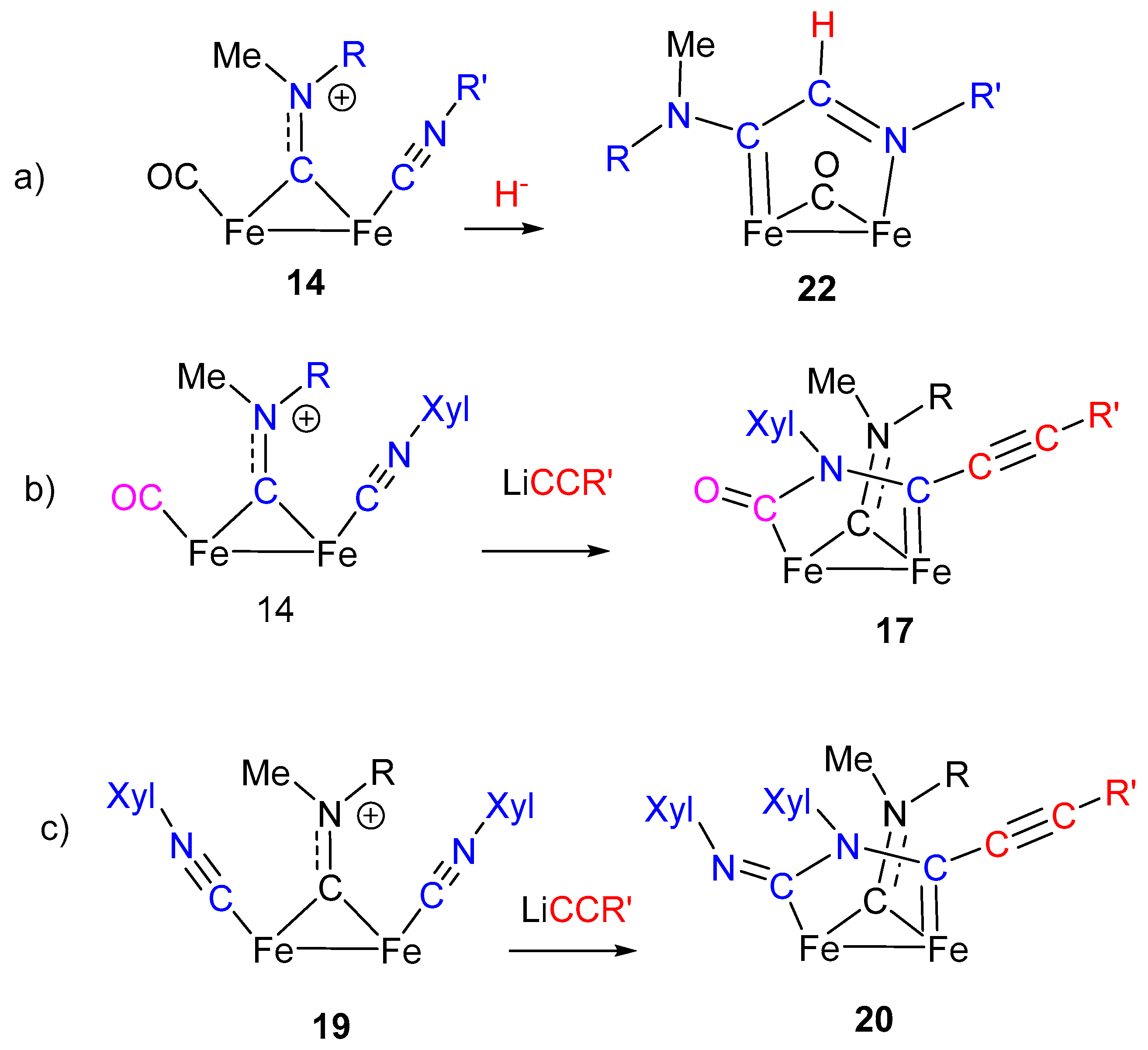
6. Nucleophilic Addition at Isocyanide Ligands
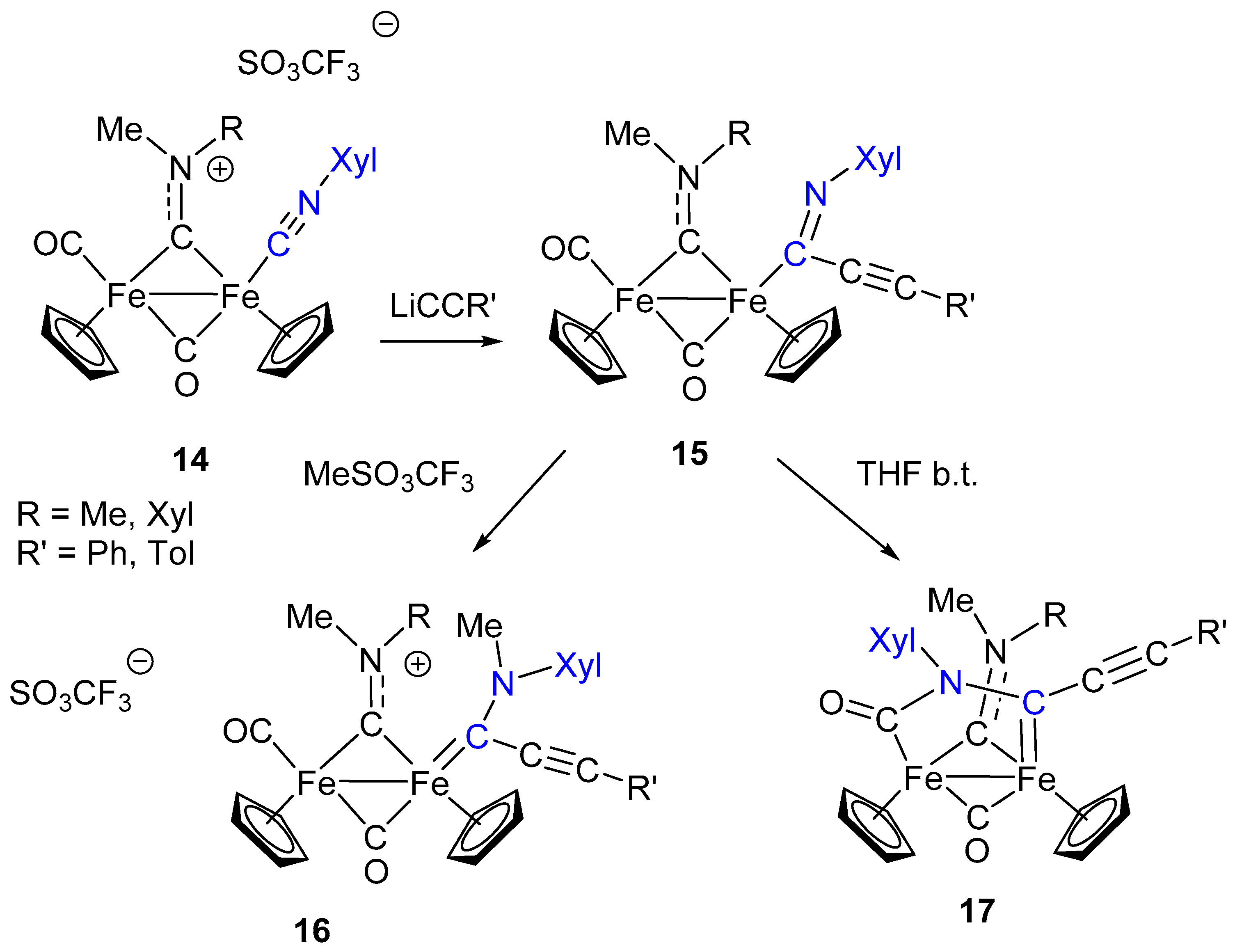
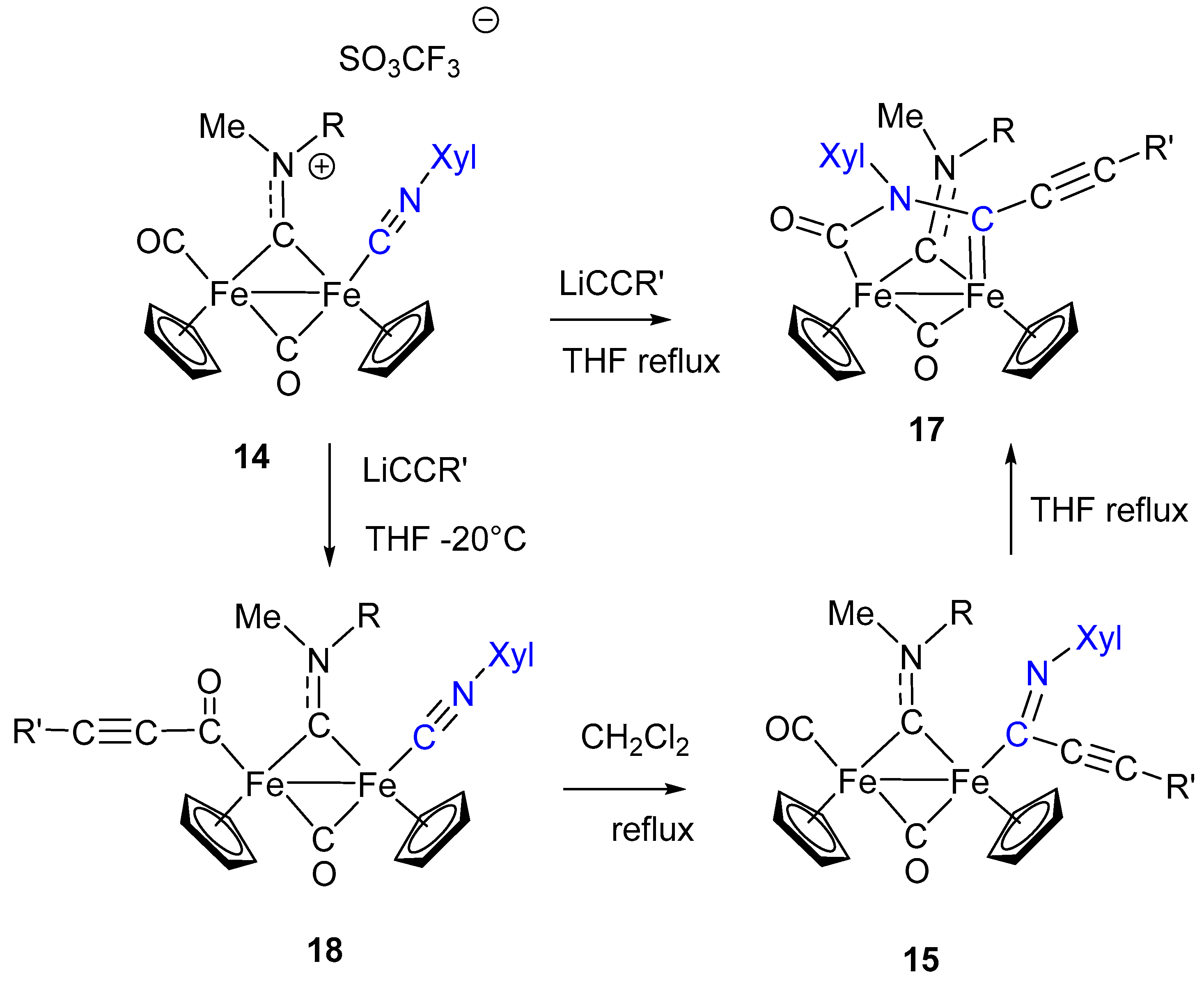
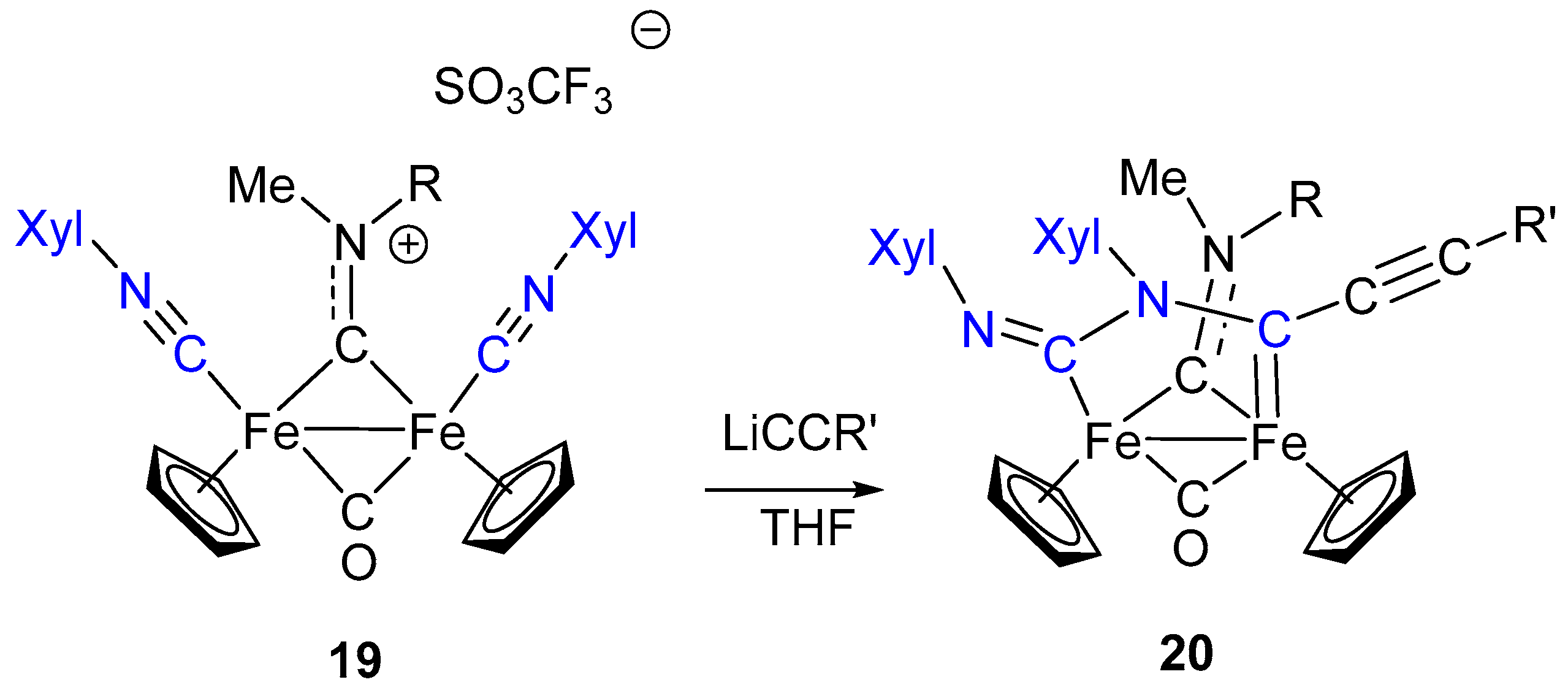
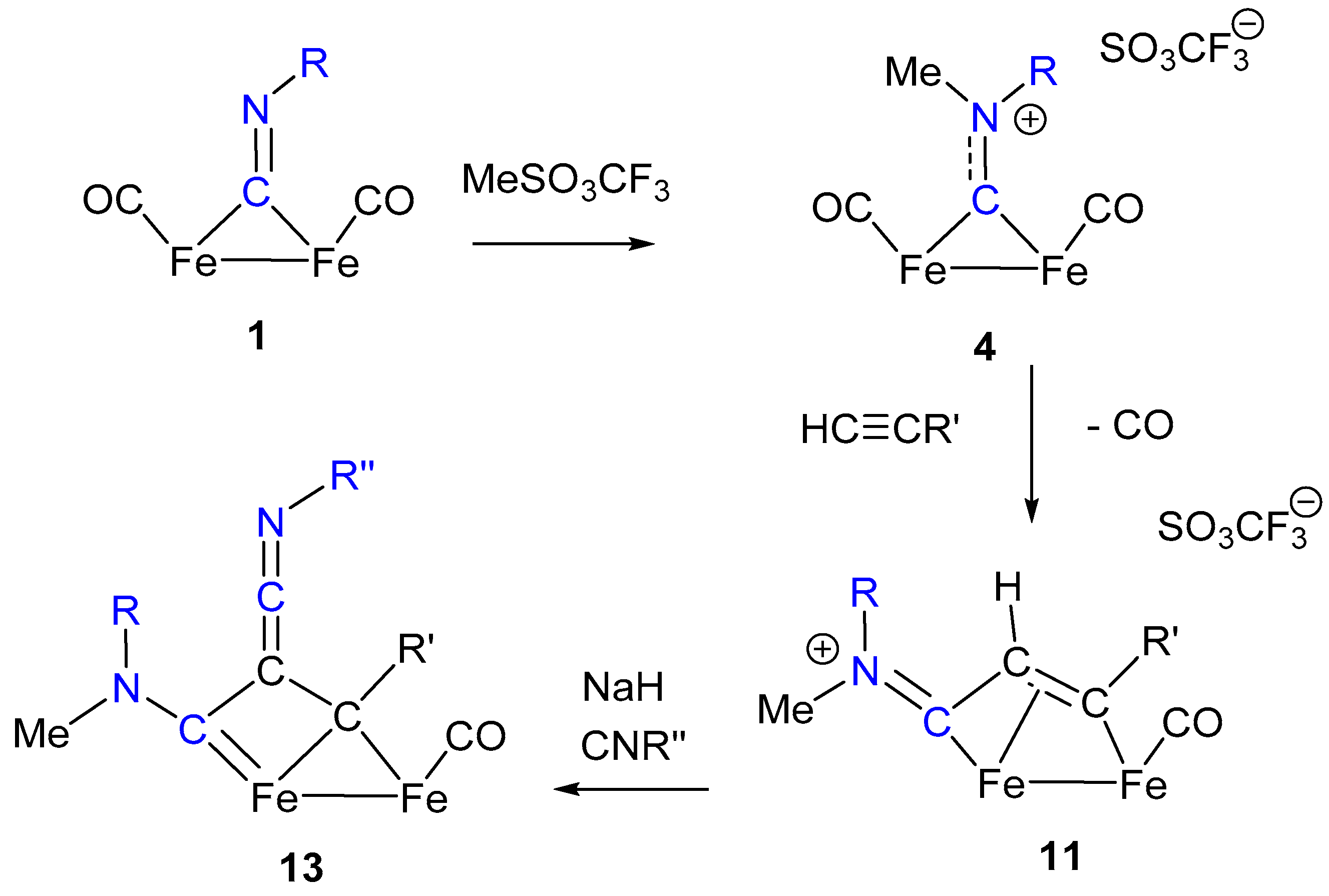
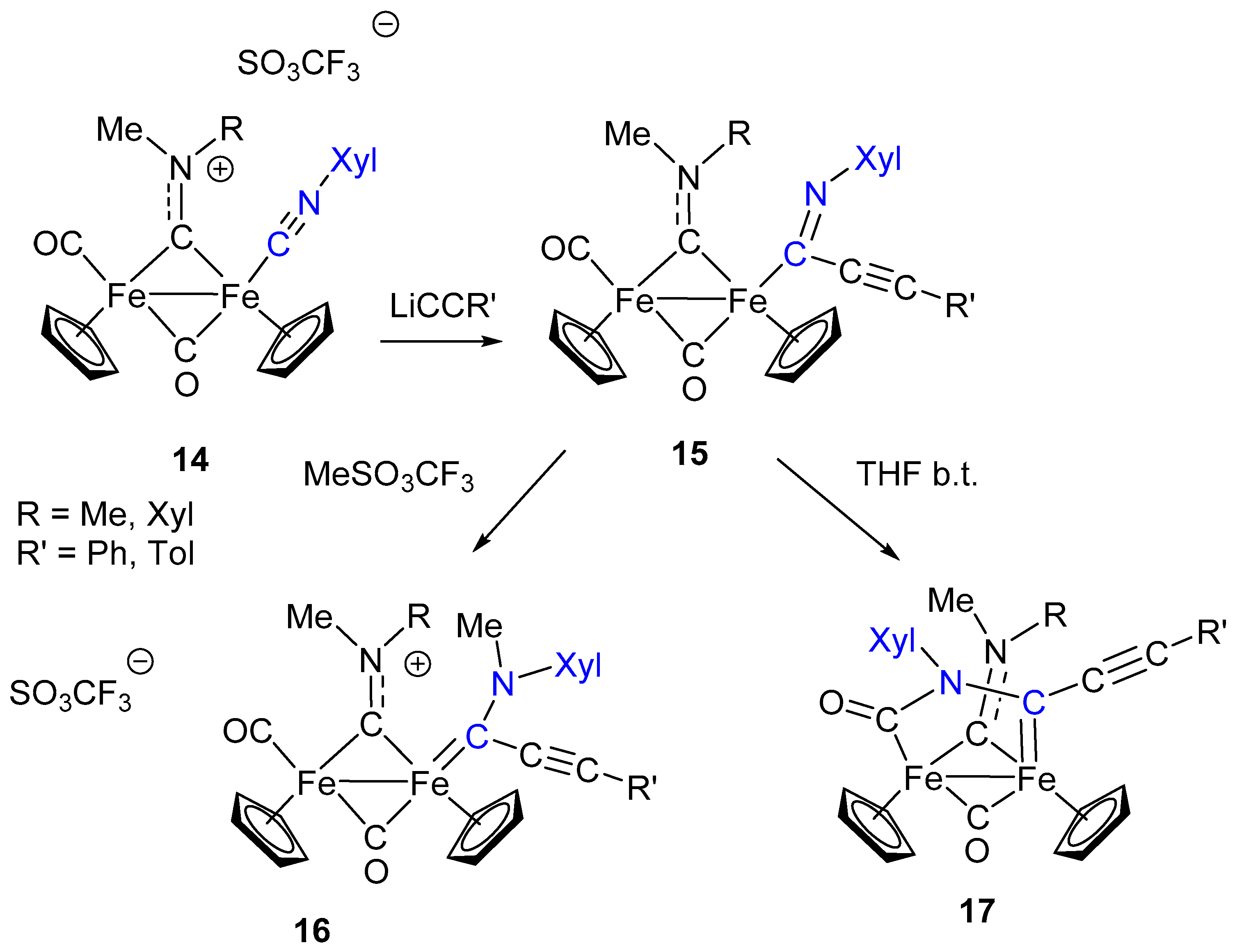
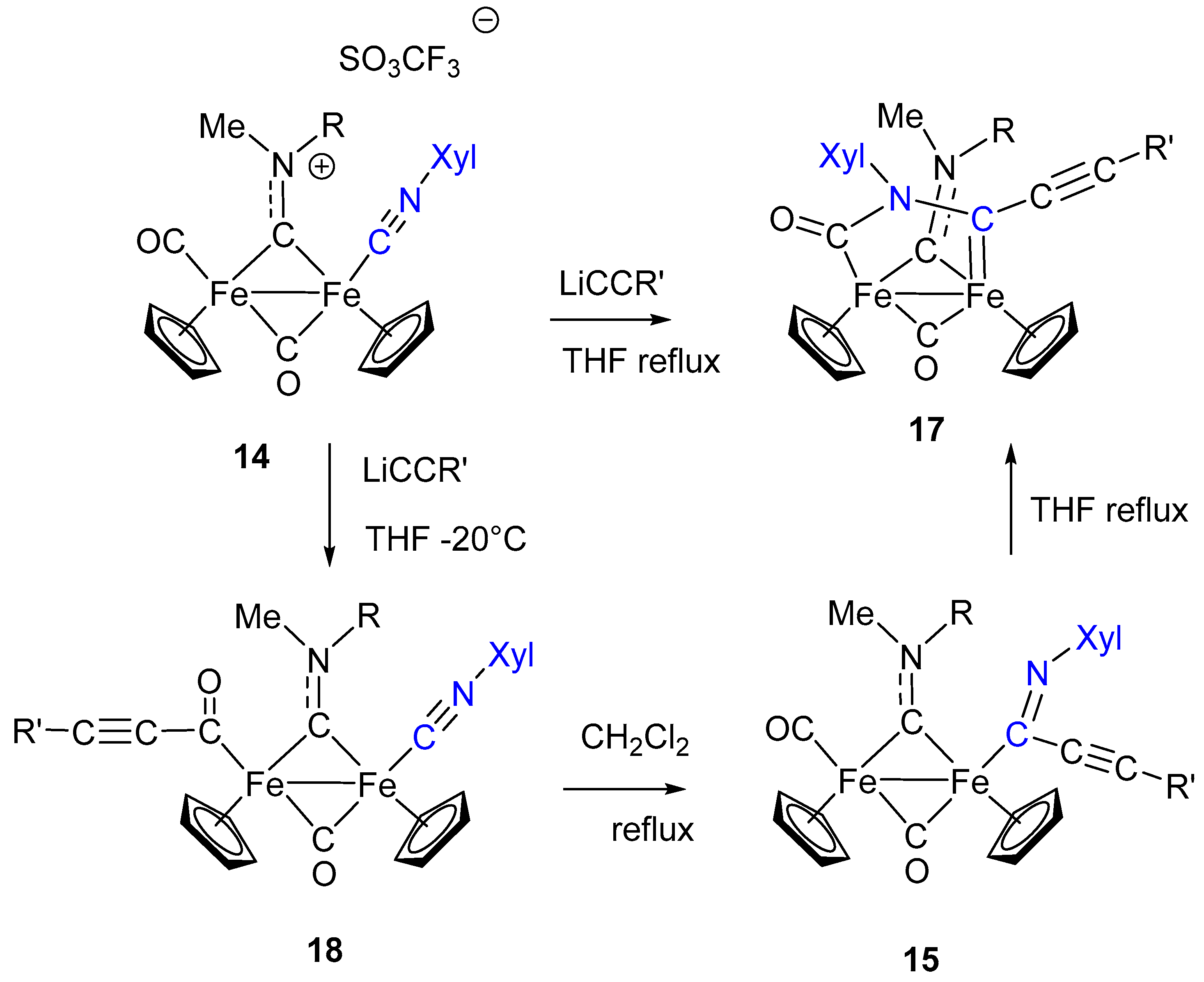
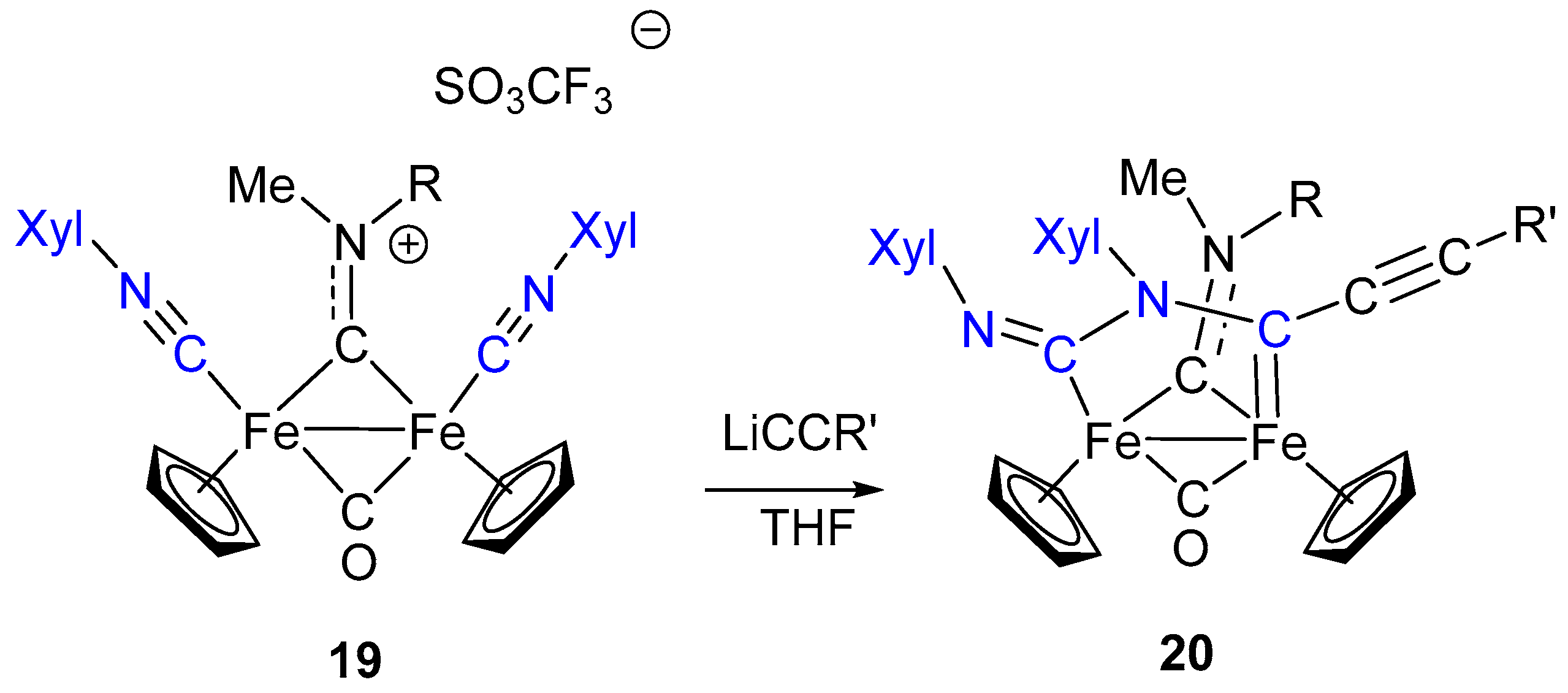
7. Cascade and Coupling Reactions Triggered by Nucleophilic Addition at Isocyanide Ligands
8. Conclusions
Funding
Conflicts of Interest
References
- Van Berkel, S.S.; Bögels, B.G.M.; Wijdeven, M.A.; Westermann, B.; Rutjes, F.P.J.T. Recent advances in asymmetric isocyanide-based multicomponent reactions. Eur. J. Org. Chem. 2012, 3543–3559. [Google Scholar] [CrossRef]
- Lygin, A.V.; de Meijere, A. Isocyanides in the synthesis of nitrogen heterocycles. Angew. Chem. Int. Ed. 2010, 49, 9094–9124. [Google Scholar] [CrossRef] [PubMed]
- Kaim, L.E.; Grimaud, L. Beyond the Ugi reaction: Less conventional interactions between isocyanides and iminium species. Tetrahedron 2009, 65, 2153–2171. [Google Scholar] [CrossRef]
- Dömling, A. Recent developments in isocyanide based multicomponent reactions in applied chemistry. Chem. Rev. 2006, 106, 17–89. [Google Scholar] [CrossRef] [PubMed]
- Cotton, F.A. Progress in Inorganic Chemistry; Interscience: New York, NY, USA, 1959; Volume 1, pp. 284–379. [Google Scholar]
- Malatesta, L.; Bonati, F. Isocyanide Complexes of Transition Metals; Wiley: New York, NY, USA, 1969. [Google Scholar]
- Yamamoto, Y. Zerovalent transition metal complexes of organic isocyanides. Coord. Chem. Rev. 1980, 32, 193–233. [Google Scholar] [CrossRef]
- Michelin, R.A.; Pombeiro, A.J.L.; da Silva, M.F.C.G. Aminocarbene complexes derived from nucleophilic addition to isocyanide ligands. Coord. Chem. Rev. 2001, 218, 75–112. [Google Scholar] [CrossRef]
- Basato, M.; Michelin, R.A.; Mozzon, M.; Sgarbossa, P.; Tassan, A. N-heterocyclic carbenes from transition metal coordinated functional isocyanides of the type o-(CH2Y)C6H4N≡C (Y = OSiMe3, OH; N3; AsR+3 ). J. Organomet. Chem. 2005, 690, 5414–5420. [Google Scholar] [CrossRef]
- Pombeiro, A.J.L.; Guedes da Silva, M.F.C. Aminocarbyne complexes derived from isocyanides activated towards electrophilic addition. Coord. Chem. Rev. 2001, 218, 43–74. [Google Scholar] [CrossRef]
- Boyarskiy, V.P.; Bokach, N.A.; Luzyanin, K.V.; Kukushkin, V.Y. Metal-mediated and metal-catalyzed reactions of isocyanides. Chem. Rev. 2015, 115, 2698–2779. [Google Scholar] [CrossRef] [PubMed]
- Mahmudov, K.T.; Kukushkin, V.Y.; Gurbanov, A.V.; Kinzhalov, M.A.; Boyarskiy, V.P.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Isocyanide metal complexes in catalysis. Coord. Chem. Rev. 2019, 384, 65–89. [Google Scholar] [CrossRef]
- Vlaar, T.; Ruijter, E.; Maes, B.U.W.; Orru, R.V.A. Palladium-catalyzed migratory insertion of isocyanides: An emerging platform in cross-coupling chemistry. Angew. Chem. Int. Ed. 2013, 52, 7084–7097. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.; Ding, Q.; Wu, J. Recent advances in isocyanide insertion chemistry. Chem. Soc. Rev. 2013, 42, 5257–5269. [Google Scholar] [CrossRef] [PubMed]
- Lang, S. Unravelling the labyrinth of palladium-catalysed reactions involving isocyanides. Chem. Soc. Rev. 2013, 42, 4867–4880. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Xu, B. Metal-catalyzed C–H functionalization involving isocyanides. Chem. Soc. Rev. 2017, 46, 1103–1123. [Google Scholar] [CrossRef] [PubMed]
- Abellán-López, A.; Chicote, M.T.; Bautista, D.; Vicente, J. Cyclopalladated complexes derived from phenylacetone oxime. Insertion reactions of carbon monoxide, isocyanides, and alkynes. Novel amidines of the isoquinoline series. Organometallics 2013, 32, 7612–7624. [Google Scholar] [CrossRef]
- Frutos-Pedreño, R.; González-Herrero, P.; Vicente, J.; Jones, P.G. Sequential insertion of alkynes and CO or isocyanides into the Pd–C bond of cyclopalladated phenylacetamides. Synthesis of eight-membered palladacycles, benzo[d]azocine-2,4(1H,3H)-diones, and highly functionalized acrylonitrile and acrylamide derivatives. Organometallics 2012, 31, 3361–3372. [Google Scholar] [CrossRef]
- Vicente, J.; Saura-Lamas, I.; García-López, J.A.; Bautista, D. Eight-membered palladacycles derived from the insertion of olefins into the Pd–C Bond of ortho-palladated pharmaceuticals phenethylamine and phentermine. Synthesis of stable Heck-type intermediates containing accessible β-hydrogens and its use in the synthesis of 2-styrylphenethylamines, tetrahydroisoquinolines, and eight-membered cyclic amidines. Organometallics 2010, 29, 4320–4338. [Google Scholar]
- Vicente, J.; Abad, J.A.; Förtsch, W.; López-Sáez, M.J.; Jones, P.G. Reactivity of Ortho-Palladated Phenol Derivatives with Unsaturated Molecules. 1. Insertion of CO, isocyanides, alkenes, and alkynes. CO/alkene, alkyne/isocyanide, and isocyanide/alkene sequential insertion reactions. Organometallics 2004, 23, 4414–4429. [Google Scholar] [CrossRef]
- Vahabi, A.H.; Alizadeh, A.; Khavasi, H.R.; Bazgir, A. Palladium-catalyzed migratory insertion of isocyanides into C(thiophene)–SMe bonds: Access to atom-transfer reactions. Eur. J. Org. Chem. 2017, 2017, 5347–5356. [Google Scholar] [CrossRef]
- Tereniak, S.J.; Stahl, S.S. Mechanistic basis for efficient, site-selective, aerobic catalytic turnover in Pd-catalyzed C–H imidoylation of heterocycle-containing molecules. J. Am. Chem. Soc. 2017, 139, 14533–14541. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, I.; San Segundo, M.; Correa, A. Iron-catalyzed C(sp3)–H functionalization of N,N-dimethylanilines with isocyanides. Chem. Commun. 2018, 54, 1627–1630. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Chen, Y.; Li, W.; Xie, Q.; Shao, L. Microwave-assisted synthesis of phenanthridines by radical insertion/cyclization of biphenyl isocyanides. J. Org. Chem. 2016, 81, 8426–8435. [Google Scholar] [CrossRef] [PubMed]
- Jones, W.D.; Foster, G.P.; Putinas, J.M. The catalytic activation and functionalization of C–H bonds. Aldimine formation by the insertion of isonitriles into aromatic C–H Bonds. J. Am. Chem. Soc. 1987, 109, 5047–5048. [Google Scholar] [CrossRef]
- Li, Y.; Rauchfuss, T.B. Synthesis of Diiron(I) dithiolato carbonyl complexes. Chem. Rev. 2016, 116, 7043–7077. [Google Scholar] [CrossRef] [PubMed]
- Capon, J.F.; Gloaguen, F.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J. Electron and proton transfers at diiron dithiolate sites relevant to the catalysis of proton reduction by the [FeFe]-hydrogenases. Coord. Chem. Rev. 2009, 253, 1476–1494. [Google Scholar] [CrossRef]
- Mejia-Rodriguez, R.; Chong, D.; Reibenspies, J.H.; Soriaga, M.P.; Darensbourg, M.Y. The hydrophilic phosphatriazaadamantane ligand in the development of H2 production electrocatalysts: Iron hydrogenase Model Complexes. J. Am. Chem. Soc. 2004, 126, 12004–12014. [Google Scholar] [CrossRef] [PubMed]
- Cheah, M.H.; Tard, C.; Borg, S.J.; Liu, X.; Ibrahim, S.K.; Pickett, C.J.; Best, S.P. Modeling [Fe–Fe] Hydrogenase: Evidence for Bridging Carbonyl and Distal Iron Coordination Vacancy in an Electrocatalytically Competent Proton Reduction by an Iron Thiolate Assembly That Operates through Fe(0)–Fe(II) Levels. J. Am. Chem. Soc. 2007, 129, 11085–11092. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, R.; Salmi, M.; Zanotti, V. C–C bond formation in diiron complexes. Chem. Eur. J. 2012, 18, 10174–10194. [Google Scholar] [CrossRef] [PubMed]
- Busetto, L.; Maitlis, P.M.; Zanotti, V. Bridging vinylalkylidene transition metal complexes. Coord. Chem. Rev. 2010, 254, 470–486. [Google Scholar] [CrossRef]
- Busetto, L.; Zanotti, V. Carbene ligands in diiron complexes. J. Organomet. Chem. 2005, 690, 5430–5440. [Google Scholar] [CrossRef]
- Basset, J.M.; Green, M.; Howard, J.A.K.; Stone, F.G.A. Formation of nona(ethyl isocyanide)di-iron from penta(ethyl isocyanide)iron and reaction of penta(t-butyl isocyanide)iron with diphenylacetylene; X-ray crystal structures of nona(ethyl isocyanide)di-iron and tris(t-butyl isocyanide){1,4-bis-(t-butylimino)-2,3-diphenylbuta-1,3-diene}iron. J. Chem. Soc. Chem. Commun. 1978, 22, 1000–1001. [Google Scholar]
- Basset, J.M.; Barker, G.K.; Green, M.; Howard, J.A.K.; Stone, F.G.A.; Wolsey, W.C. Chemistry of low-valent metal isocyanide complexes. Part 3. The synthesis, structure, and dynamic behaviour of nonakis(ethyl and isopropyl isocyanide)di-iron and -ruthenium complexes. Crystal structure of [Fe2(µ-CNEt)3(CNEt)6]. J. Chem. Soc. Dalton Trans. 1981, 219–227. [Google Scholar] [CrossRef]
- Ruiz, J.; Riera, V.; Vivanco, M.; García-Granda, S.; Pertierra, P. A homoleptic (aryl isocyanlde)iron(0) dimer. X-ray structure determination of nonakis(phenyl isocyanide)diiron. Organometallics 1992, 11, 2734–2736. [Google Scholar] [CrossRef]
- Bellerby, J.; Boylan, M.J.; Ennis, M.; Manning, A.R. An infrared spectroscopic study of the tautomeric equilibria in solutions of tricarbonylbis(η-dienyl)isocyanidedi-iron complexes. J. Chem. Soc. Dalton Trans. 1978, 9, 1185–1189. [Google Scholar] [CrossRef]
- Joshi, K.K.; Mills, O.S.; Pauson, P.L.; Shaw, B.W.; Stubbs, W.H. An iron complex with a bridging isonitrile group. Chem. Commun. 1965, 10, 181–182. [Google Scholar] [CrossRef]
- Treiche, P.M.; Stenson, J.P.; Benedict, J.J. Reactions of several cationic organometallic isocyanide complexes with sodium borohydride. Inorg. Chem. 1971, 10, 1183–1187. [Google Scholar] [CrossRef]
- Cotton, F.A.; Frenz, B.A. Low-valent metal isocyanide complexes. IV.1 Crystal and molecular structures of cis-anti-Bis(penthaptocyclopentadienyl)dicarbonylbis(μ-methyl isocyanide)-diiron(Fe–Fe), (η5-C5H5)2Fe2(CO)2(μ-CNCH3)2. Inorg. Chem. 1974, 13, 253–256. [Google Scholar] [CrossRef]
- Hunt, I.D.; Mills, O.S. Carbon compounds of the transition metals. XXlX. Crystal structure of trans-anti-Bis(pentahapto-cyclopentadienyl)dicarbonyl-bis(μ-phenylisonitrile)-diiron(Fe–Fe), (h5-C5H5)2Fe2(CO)2(μ-C NPh)2. Acta Cryst. 1977, B33, 2432–2435. [Google Scholar] [CrossRef]
- Fehlhammer, W.P.; Mayr, A.; Kehr, W. Isocyanid-und heteroallen-verbrückte metallkomplexe iv. die krjstall- und molekolstruktur von trans-anti-bis[η-cyclopentadienyl(μ-phenylisocyanid)-(phenylisocyanid)eisen](Fe–Fe),[Fe(η-C5H5)(μ-CNC6H5)CNC6H5]2. direkter stereochemischer vergleich. J. Organomet. Chem. 1980, 197, 327–334. [Google Scholar] [CrossRef]
- Coville, N.J.; Albers, M.O.; Singleton, E. An investigation of the reaction between [{Fe(η5-C5H5)(CO)2}2] and aryl isonitriles. J. Chem. Soc. Dalton Trans. 1982, 1389–1391. [Google Scholar] [CrossRef]
- Campbell, I.L.C.; Stephens, F.S. Crystal and molecular structure of cis-(isobutyl isocyanide)di-µ-carbonylcarbonylbis(π-cyclopentadienyl)di-iron. J. Chem. Soc. Dalton Trans. 1975, 982–985. [Google Scholar] [CrossRef]
- Boss, K.; Dowling, C.; Manning, A.R. Preparation, spectra and structure of [Fe2(η-C5H5)2(L)(CN)(μ-CO){ μ-CN(R’)R}],[Fe2(η-C5H5)2(CO)(CN){μ-CNMe2}2]+ and [Fe2(η-C5H5)2(CN)2(μ-CNMe2}2] zwitterions (L = CO or organoisocyanide) and their reactions with alkyl and protic electrophiles. J. Organomet. Chem. 1996, 509, 197–207. [Google Scholar] [CrossRef]
- Busetto, L.; Carlucci, L.; Zanotti, V.; Albano, V.G.; Braga, D. Synthesis, reactions, and X-ray structures of the functionalized isocyanide complexes [Fe2{µ-CNC(O)SR}(µ-CO)(CO)2(cp)2](cp = η-C5H5, R = Me or Et) and of their carbyne and carbene derivatives. J. Chem. Soc. Dalton Trans. 1990, 243–250. [Google Scholar] [CrossRef]
- Nehring, J.L.; Heinekey, D.M. Dinuclear iron isonitrile complexes: Models for the iron hydrogenase active site. Inorg. Chem. 2003, 42, 4288–4292. [Google Scholar] [CrossRef] [PubMed]
- Boyke, C.A.; Rauchfuss, T.B.; Wilson, S.R.; Rohmer, M.-M.; Bénard, M. [Fe2(SR)2(µ-CO)(CNMe)6]2+ and Analogues: A new class of diiron dithiolates as structural models for the HoxAir state of the Fe-only hydrogenase. J. Am. Chem. Soc. 2004, 126, 15151–15160. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Barton, B.E.; Chambers, G.M.; Rauchfuss, T.B.; Arrigoni, F.; Zampella, G. Preparation and Protonation of Fe2(pdt)(CNR)6, Electron-rich analogues of Fe2(pdt)(CO)6. Inorg. Chem. 2016, 55, 3401–3412. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.; Manning, A.R.; Stephens, F.S. Reactions of [Fe2(η-dienyl)2(CO)4–n(CNR)n] with strong acids. The structure of cis-µ-carbonyl-µ-methyliminiomethylene-bis[carbonyl(η-cyclopentadienyl)iron] tetrafluoroborate. J. Chem. Soc., Dalton Trans. 1979, 23–27. [Google Scholar] [CrossRef]
- Willis, S.; Manning, A.R.; Stephens, F.S. Reactions of [Fe2(η-dienyl)2(CO)4–n(CNR)n] complexes (dienyl = C5H5, C5H4Me, or C9H7; R = alkyl or benzyl; n= 1 or 2) with alkyl halides and other alkylating agents. The crystal structure of cis-[Fe2(η-C5H4Me)2(CO)3{C(NMe2)µ}]I. J. Chem. Soc. Dalton Trans. 1980, 186–191. [Google Scholar] [CrossRef]
- Cox, G.; Dowling, C.; Manning, A.R.; McArdle, P.; Cunningham, D. A reinvestigation of the reaction of [Fe,(η-C5H5)2(CO)4−n(CNR)n](n = 1 or 2) with strong alkylating agents. J. Organomet. Chem. 1992, 438, 143–158. [Google Scholar] [CrossRef]
- Agonigi, G.; Bortoluzzi, M.; Marchetti, F.; Pampaloni, G.; Zacchini, S.; Zanotti, V. Regioselective Nucleophilic Additions to Diiron Carbonyl Complexes Containing a Bridging Aminocarbyne Ligand: A Synthetic, Crystallographic and DFT Study. Eur. J. Inorg. Chem. 2018, 960–971. [Google Scholar] [CrossRef]
- Marchetti, F. Constructing organometallic architectures from aminoalkylidyne diiron complexes. Eur. J. Inorg. Chem. 2018, 2018, 3987–4003. [Google Scholar] [CrossRef]
- Zanotti, V.; Bordoni, S.; Busetto, L.; Carlucci, L.; Palazzi, A.; Serra, R.; Albano, V.G.; Monari, M.; Prestopino, F.; Laschi, F.; et al. Diiron aminoalkylidene complexes. Organometallics 1995, 14, 5232–5241. [Google Scholar] [CrossRef]
- Albano, V.G.; Busetto, L.; Camiletti, C.; Castellari, C.; Monari, M.; Zanotti, V. Selective C–C bond formation at diiron µ-aminocarbyne complexes. J. Chem. Soc. Dalton Trans. 1997, 4671–4676. [Google Scholar] [CrossRef]
- Albano, V.G.; Bordoni, S.; Busetto, L.; Marchetti, F.; Monari, M.; Zanotti, V. Carbon monoxide–isocyanide coupling promoted by acetylide addition to a diiron complex. J. Organomet. Chem. 2003, 684, 37–43. [Google Scholar] [CrossRef]
- Bellachioma, G.; Cardaci, G.; Macchioni, A.; Reichenbach, G. Mechanism of isocyanide insertion into the methyl–iron bond of [Fe(CO)2L2(CNR)CH3]BPh4. Inorg. Chem. 1992, 31, 63–66. [Google Scholar] [CrossRef]
- Bellachioma, G.; Cardaci, G.; Macchioni, A.; Zuccaccia, C. Effect of anions on the isocyanide insertion reaction in cationic alkyl complexes of iron(II): Kinetic, thermodynamic and solution interionic structural studies. J. Organomet. Chem. 2000, 593, 119–126. [Google Scholar] [CrossRef]
- Klose, A.; Solari, E.; Ferguson, R.; Floriani, C. Insertion reactions of isocyanides and nitriles into unsupported iron–aryl bonds: The synthesis of a dimeric iron(II) homoleptic iminoacyl complex. Organometallics 1993, 12, 2414–2416. [Google Scholar] [CrossRef]
- Klose, A.; Solari, E.; Floriani, C.; Chiesi-Villa, A.; Rizzoli, C.; Re, N. Magnetic properties diagnostic for the existence of Iron(II)–Iron(II) bonds in dinuclear complexes which derive from stepwise insertion reactions on unsupported iron–aryl bonds. J. Am. Chem. Soc. 1994, 116, 9123–9135. [Google Scholar] [CrossRef]
- Hogarth, G.; Lavender, M.H.; Shukri, K. Diiron–hydride complexes: synthesis, structure, and reactivity of trans-[Fe2(CO)4(µ-H)(µ-CO)(µ-PCy2)(µ-Ph2PCH2PPh2)]. Organometallics 1995, 14, 2325–2341. [Google Scholar] [CrossRef]
- Alvarez, M.A.; García, M.E.; García-Vivó, D.; Ruiz, M.A.; Vega, M.F. Insertion, rearrangement, and coupling processes in the reactions of the unsaturated hydride complex [W2(η5-C5H5)2(H)(μ-PCy2)(CO)2] with isocyanides. Organometallics 2013, 32, 4543–4555. [Google Scholar] [CrossRef]
- Alvarez, M.A.; García, M.E.; Ramos, A.; Ruiz, M.A. Reactivity of the unsaturated hydride [Mo2(η5-C5H5)2(μ-H)(μ-PCy2)(CO)2] toward P-donor bidentate ligands and unsaturated N-containing organic molecules. Organometallics 2007, 26, 1461–1472. [Google Scholar] [CrossRef]
- Beringhelli, T.; D’Alfonso, G.; Minoja, A.; Ciani, G.; Moret, M.; Sironi, A. Insertion of isocyanides into metal-hydrogen bonds in triangular hydrido-carbonyl clusters of rhenium. X-ray crystal structures of [Re3(μ-H)3(μ-η-CHNR)(CO)10]− (R = p-Tolyl) and of [Re3(μ-H)3(μ3-η2-CHNR)(CO)9]− (R = cyclohexyl). Organometallics 1991, 10, 3131–3138. [Google Scholar] [CrossRef]
- Fujita, K.; Nakaguma, H.; Hanasaka, F.; Yamaguchi, R. Synthesis of a DMPM and hydrido-bridged diiridium complex, [(Cp*Ir)2(μ-dmpm)(μ-H)2][OTf]2, and its reactivity toward alkynes and isocyanides. Organometallics 2002, 21, 3749–3757. [Google Scholar] [CrossRef]
- Albano, V.G.; Busetto, L.; Marchetti, F.; Monari, M.; Zacchini, S.; Zanotti, V. Alkyne-Isocyanide Coupling in [Fe2(CNMe)(CO)3(Cp)2]: A new route to diiron μ-vinyliminium complexes. Organometallics 2007, 26, 3448–3455. [Google Scholar] [CrossRef]
- Dyke, A.F.; Knox, S.A.R.; Naish, P.J.; Taylor, G.E. Organic chemistry of dinuclear metal centres. Part 1. Combination of alkynes with carbon monoxide at di-iron and diruthenium centres: Crystal structure of [Ru2(CO)(µ-CO){µ-σ:η3-C(O)C2Ph2}(η-C5H5)2]. J. Chem. Soc. Dalton Trans. 1982, 1297–1307. [Google Scholar] [CrossRef]
- Boni, A.; Funaioli, T.; Marchetti, F.; Pampaloni, G.; Pinzino, C.; Zacchini, S. Electrochemical, EPR and computational results on [Fe2Cp2(CO)2]-based complexes with a bridging hydrocarbyl ligand. J. Organomet. Chem. 2011, 696, 3551–3556. [Google Scholar] [CrossRef]
- Marchetti, F.; Zacchini, S.; Zanotti, V. Photochemical alkyne insertions into the iron–thiocarbonyl bond of [Fe2(CS)(CO)3(Cp)2]. Organometallics 2016, 35, 2630–2637. [Google Scholar] [CrossRef]
- Albano, V.G.; Busetto, L.; Marchetti, F.; Monari, M.; Zacchini, S.; Zanotti, V. Diiron µ-vinyliminium complexes from acetylene insertion into a metal–aminocarbyne bond. Organometallics 2003, 22, 1326–1331. [Google Scholar] [CrossRef]
- Albano, V.G.; Busetto, L.; Marchetti, F.; Monari, M.; Zacchini, S.; Zanotti, V. Stereochemistry of the insertion of disubstituted alkynes into the metal aminocarbyne bond in diiron complexes. J. Organomet. Chem. 2004, 689, 528–538. [Google Scholar] [CrossRef]
- Ciancaleoni, G.; Zacchini, S.; Zanotti, V.; Marchetti, F. DFT mechanistic insights into the alkyne insertion reaction affording diiron μ-vinyliminium complexes and new functionalization pathways. Organometallics 2018, 37, 3718–3731. [Google Scholar] [CrossRef]
- Zanotti, V. Reactions of bridging C3 ligands in diiron complexes: Unconventional routes to new functionalized organic frames. Pure Appl. Chem. 2010, 82, 1555–1568. [Google Scholar] [CrossRef]
- Albano, V.G.; Busetto, L.; Marchetti, F.; Monari, M.; Zacchini, S.; Zanotti, V. C–C bond formation by cyanide addition to dinuclear vinyliminium complexes. J. Organomet. Chem. 2006, 691, 4234–4243. [Google Scholar] [CrossRef]
- Busetto, L.; Marchetti, F.; Zacchini, S.; Zanotti, V. Reactions of diazo compounds at μ-vinyliminium ligands: synthesis of novel dinuclear azine–bis(alkylidene) complexes. Organometallics 2007, 26, 3577–3584. [Google Scholar] [CrossRef]
- Aumann, R. Ketenimine complexes from carbene complexes and isocyanides: Versatile building blocks for carbocycles and N-heterocycles [New Synthetic Methods (74)]. Angew. Chem. Int. ed. Engl. 1988, 27, 1456–1467. [Google Scholar] [CrossRef]
- Fernández, I.; Cossío, F.P.; Sierra, M.A. Mechanism of the Generation of Ketenimine–M(CO)n complexes (M = Cr, W, Fe) from Fischer carbenes and isocyanides. Organometallics 2007, 26, 3010–3017. [Google Scholar] [CrossRef]
- Cadierno, V.; García-Álvarez, J.; Gimeno, J.; Rubio-García, J. Reaction of isocyanides with iminophosphorane-based carbene ligands: Synthesis of unprecedented ketenimine–ruthenium complexes. J. Organomet. Chem. 2005, 690, 5856–5862. [Google Scholar] [CrossRef]
- Mitsudo, T.; Watanabe, H.; Komiya, Y.; Watanabe, Y.; Takaegami, Y.; Nakatsu, K.; Kinoshita, K.; Miyagawa, Y. Synthesis, crystal structure and reactivities of the first η4-vinlylketen metal complexes. J. Organomet. Chem. 1980, 190, C39–C42. [Google Scholar] [CrossRef]
- Busetto, L.; Marchetti, F.; Zacchini, S.; Zanotti, V. Addition of Isocyanides at Diiron μ-Vinyliminium Complexes: Synthesis of novel ketenimine-bis(alkylidene) complexes. Organometallics 2008, 27, 5058–5066. [Google Scholar] [CrossRef]
- Ruiz, J.; García, L.; Mejuto, C.; Vivanco, M.; Díaz, M.R.; García-Granda, S. Strong electron-donating metalla-N-heterocyclic carbenes. Chem. Commun. 2014, 50, 2129–2132. [Google Scholar] [CrossRef] [PubMed]
- Yu, I.; Wallis, C.J.; Patrick, B.O.; Diaconescu, P.L.; Mehrkhodavandi, P. Phosphine-tethered carbene ligands: Template synthesis and reactivity of cyclic and acyclic functionalized carbenes. Organometallics 2010, 29, 6065–6076. [Google Scholar] [CrossRef]
- Marchetti, F.; Zacchini, S.; Zanotti, V. Growing the molecular architecture at alkynyl(amino)carbene ligands in diiron µ-aminocarbyne complexes. Eur. J. Inorg. Chem. 2016, 2016, 4820–4828. [Google Scholar] [CrossRef]
- Wang, D.-L.; Hwang, W.-S.; Lee, L.; Chiang, M.Y. Reaction of N-(3-methyl-2-thienylmethylidene)aniline with diiron nonacarbonyl: Cyclometalation induced methyl migration, imidoyl complex formation, and hydrogenation. J. Organomet. Chem. 1999, 579, 211–216. [Google Scholar] [CrossRef]
- Lappert, M.F. Contributions to the chemistry of carbenemetal chemistry. J. Organomet. Chem. 2005, 690, 5467–5473. [Google Scholar] [CrossRef]
- Marchetti, F.; Zacchini, S.; Zanotti, V. Carbon monoxide–isocyanide coupling promoted by acetylide addition to a diiron complex. Chem. Commun. 2015, 51, 8101–8104. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, F.; Zacchini, S.; Zanotti, V. C–N coupling of isocyanide ligands promoted by acetylide addition to diiron aminocarbyne complexes. Organometallics 2015, 34, 3658–3664. [Google Scholar] [CrossRef]
- Carnahan, E.M.; Protasiewicz, J.D.; Lippard, S.J. The 15 years of reductive coupling: What have we learned? Acc. Chem. Res. 1993, 26, 90–97. [Google Scholar] [CrossRef]
- Bashall, A.; Collier, P.E.; Gade, L.H.; McPartlin, M.; Mountford, P.; Pugh, S.M.; Radojevic, S.; Schubart, M.; Scowen, I.J.; Trösch, D.J.M. C–C and C–N coupling reactions of an imidotitanium complex with isocyanides. Organometallics 2000, 19, 4784–4794. [Google Scholar] [CrossRef]
- Rehder, D.; Bottcher, C.; Collazo, C.; Hedelt, R.; Schmidt, H. Isocyanide–Group 5 complexes and metal-centred C–C coupling. J. Organomet. Chem. 1999, 585, 294–307. [Google Scholar] [CrossRef]
- Collazo, C.; Rodewald, D.; Schmidt, H.; Rehder, D. Niobium-Centered C–C Coupling of Isonitriles. Organometallics 1996, 15, 4884–4887. [Google Scholar] [CrossRef]
- Acho, J.A.; Lippard, S.J. Synthesis and Structural Characterization of [Cr(t-C4H9HNC≡CNH-t-C4H9)(CN-t-C4H9)4I]I. Reductive coupling of isocyanide ligands in a first row transition metal complex. Organometallics 1994, 13, 1294–1299. [Google Scholar] [CrossRef]
- Shen, J.; Yap, G.P.A.; Theopold, K.H. Chromium mediated reductive coupling of isonitrile forms unusual heterocycles. J. Am. Chem. Soc. 2014, 136, 3382–3384. [Google Scholar] [CrossRef] [PubMed]
- Ojo, W.S.; Petillon, F.Y.; Schollhammer, P.; Talarmin, J. C–C, C–S, and C–N coupling versus dealkylation processes in the cationic tris(thiolato)dimolybdenum(III) complexes [Mo2Cp2(μ-SMe)3L2]+ (L = xylNC, t-BuNC, CO, MeCN). Organometallics 2008, 27, 4207–4222. [Google Scholar] [CrossRef]
- Cabon, N.; Paugam, E.; Petillon, F.Y.; Schollhammer, F.; Talarmin, J. Unexpected Coupling of Cp and two RNC ligands at a {Mo2(μ-SMe)3} nucleus. Organometallics 2003, 22, 4178–4180. [Google Scholar] [CrossRef]
- Okazaki, M.; Suto, K.; Kudo, N.; Takano, M.; Ozawa, F. Synthesis and structure of cubane-type tetrairon clusters possessing μ3-isonitrile ligands. Reductive coupling of two isonitriles on redox-responsive tetrairon reaction sites. Organometallics 2012, 31, 4110–4113. [Google Scholar] [CrossRef]
- Marchetti, F.; Zacchini, S.; Zanotti, V. Coupling of isocyanide and μ-aminocarbyne ligands in diiron complexes promoted by hydride addition. Organometallics 2014, 33, 3990–3997. [Google Scholar] [CrossRef]
- Cabon, N.; Petillon, F.Y.; Orain, P.Y.; Schollhammer, F.; Talarmin, J.; Muir, K.W. Controlled nucleophilic activation of different sites in [Mo2Cp2L2(μ-SMe)2(μ-L’)]+ cations (L = ButNC, xylNC, CO; L’ = SMe or PPh2). J. Organomet. Chem. 2005, 690, 4583–4601. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzoni, R.; Marchetti, F.; Cingolani, A.; Zanotti, V. Bond Forming Reactions Involving Isocyanides at Diiron Complexes. Inorganics 2019, 7, 25. https://doi.org/10.3390/inorganics7030025
Mazzoni R, Marchetti F, Cingolani A, Zanotti V. Bond Forming Reactions Involving Isocyanides at Diiron Complexes. Inorganics. 2019; 7(3):25. https://doi.org/10.3390/inorganics7030025
Chicago/Turabian StyleMazzoni, Rita, Fabio Marchetti, Andrea Cingolani, and Valerio Zanotti. 2019. "Bond Forming Reactions Involving Isocyanides at Diiron Complexes" Inorganics 7, no. 3: 25. https://doi.org/10.3390/inorganics7030025
APA StyleMazzoni, R., Marchetti, F., Cingolani, A., & Zanotti, V. (2019). Bond Forming Reactions Involving Isocyanides at Diiron Complexes. Inorganics, 7(3), 25. https://doi.org/10.3390/inorganics7030025