Structural Dynamics of Spin Crossover in Iron(II) Complexes with Extended-Tripod Ligands


Abstract
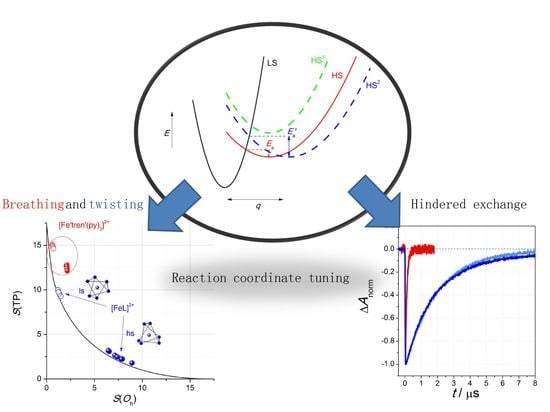
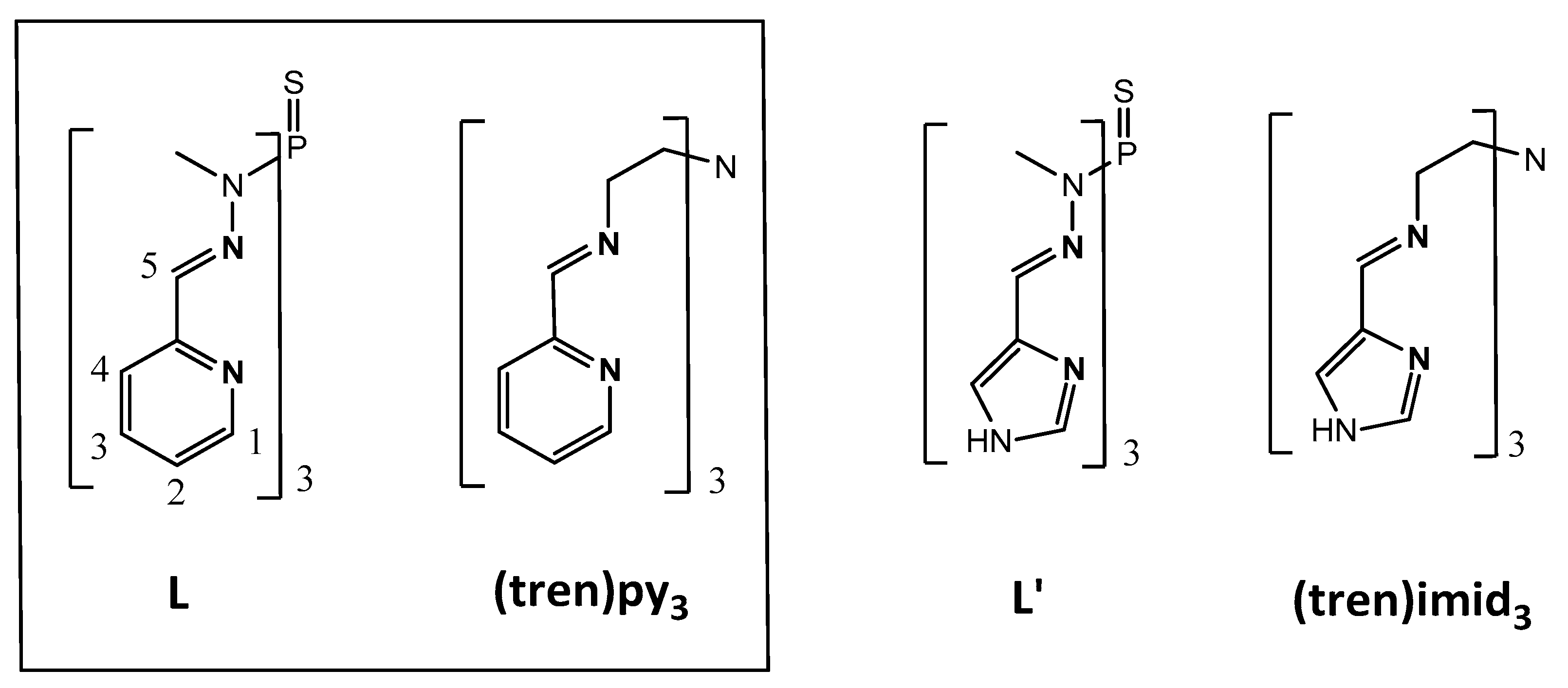
1. Introduction
2. Results
2.1. Structural Characteristics of [FeL]2+ and [ZnL]2+
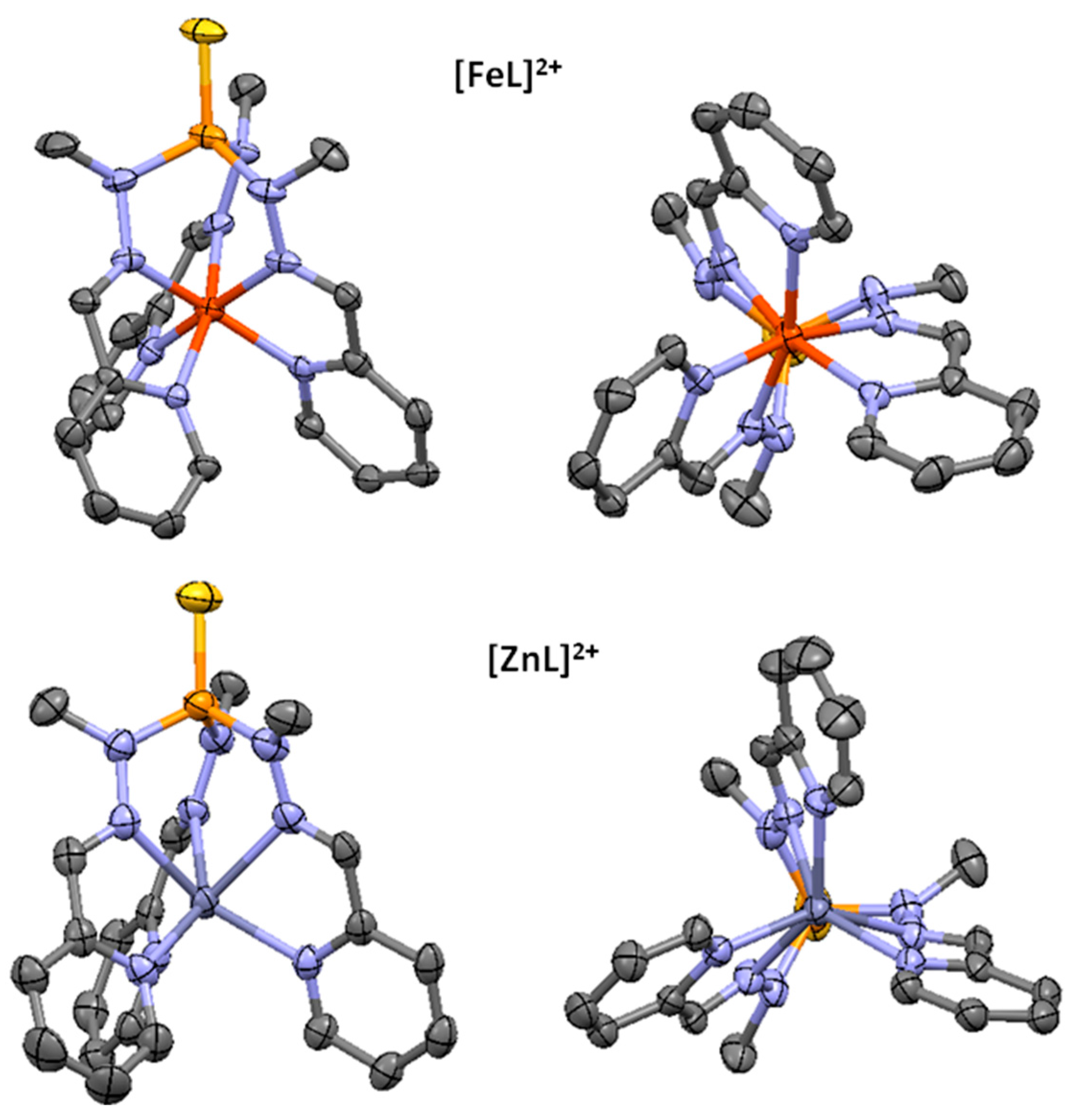
2.1.1. Complex Synthesis and Solid State Structures
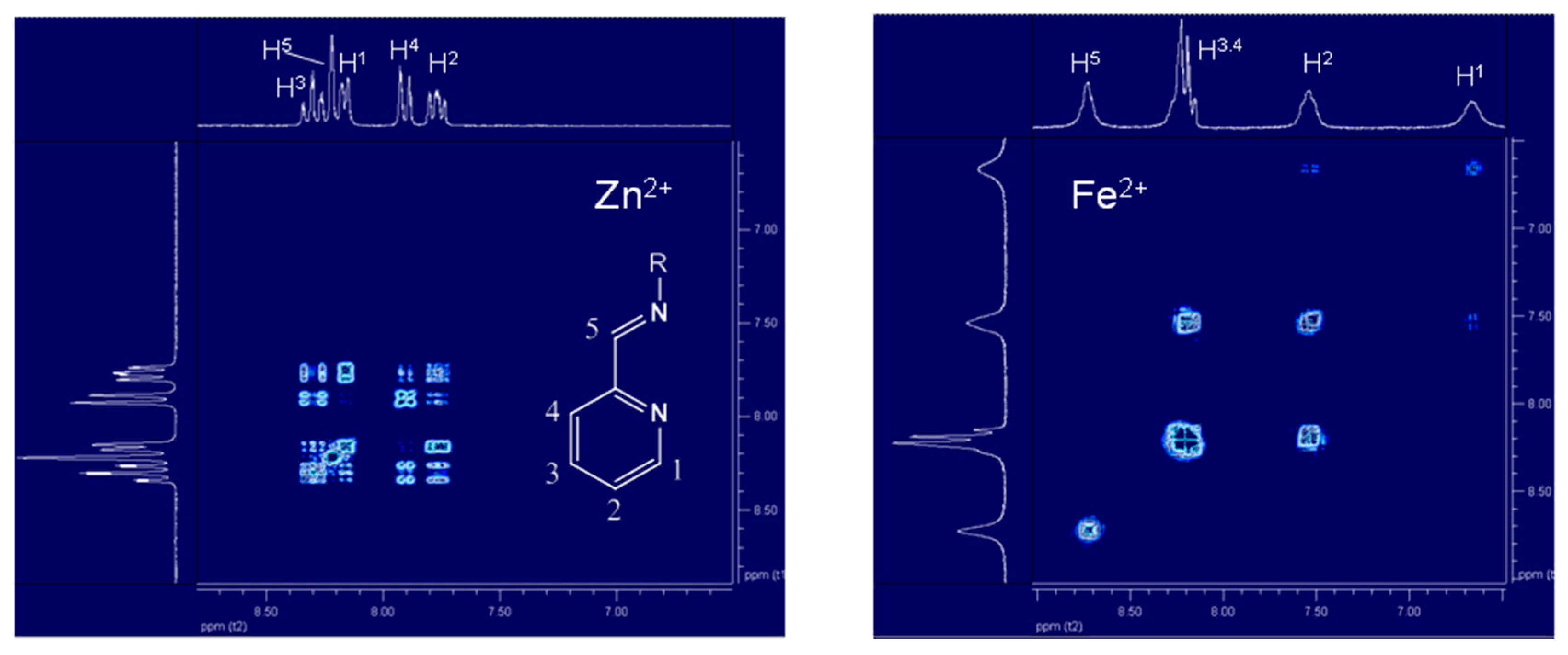
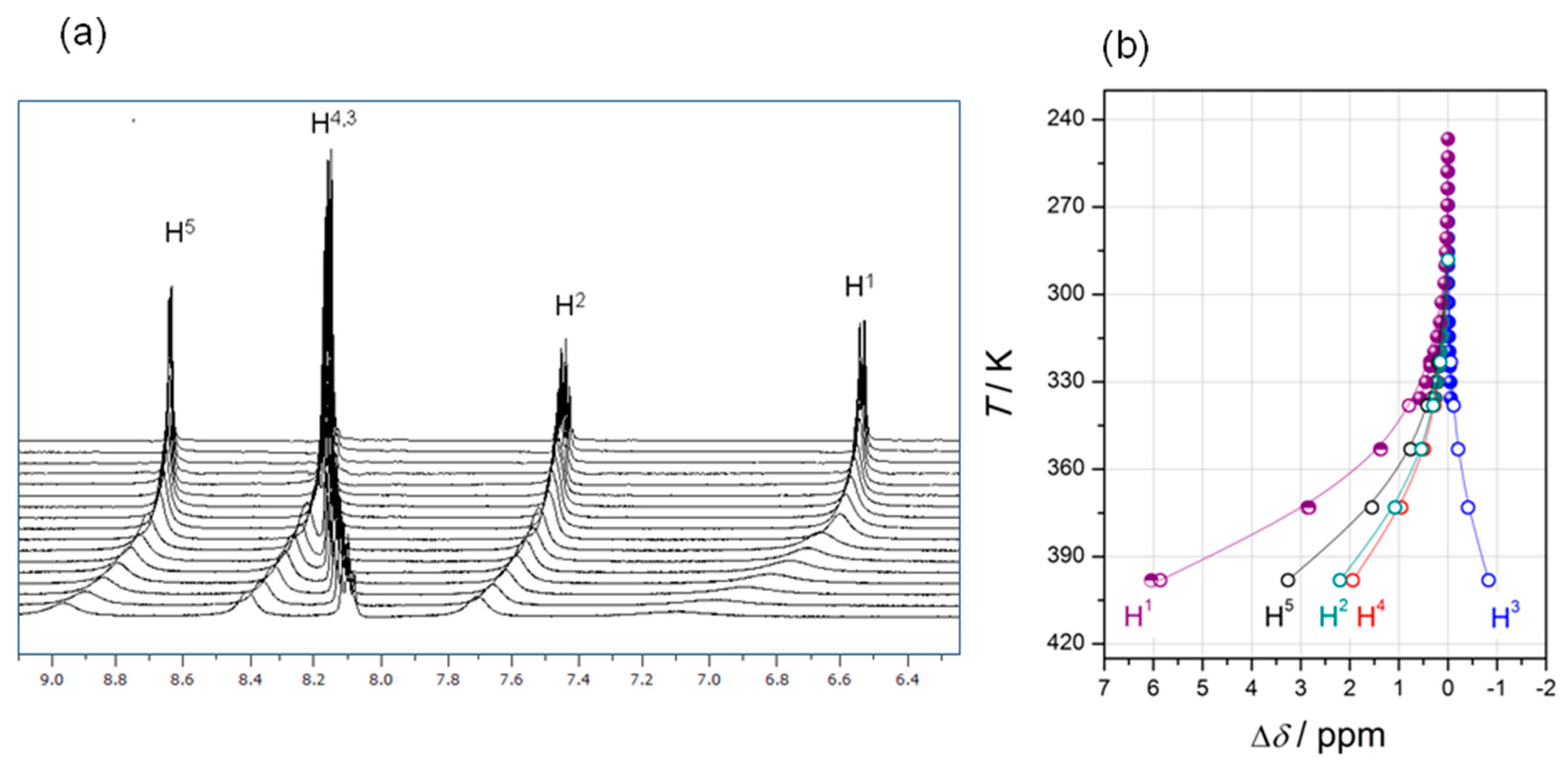
2.1.2. Complex Structures in Solution via 1H-NMR Spectroscopy
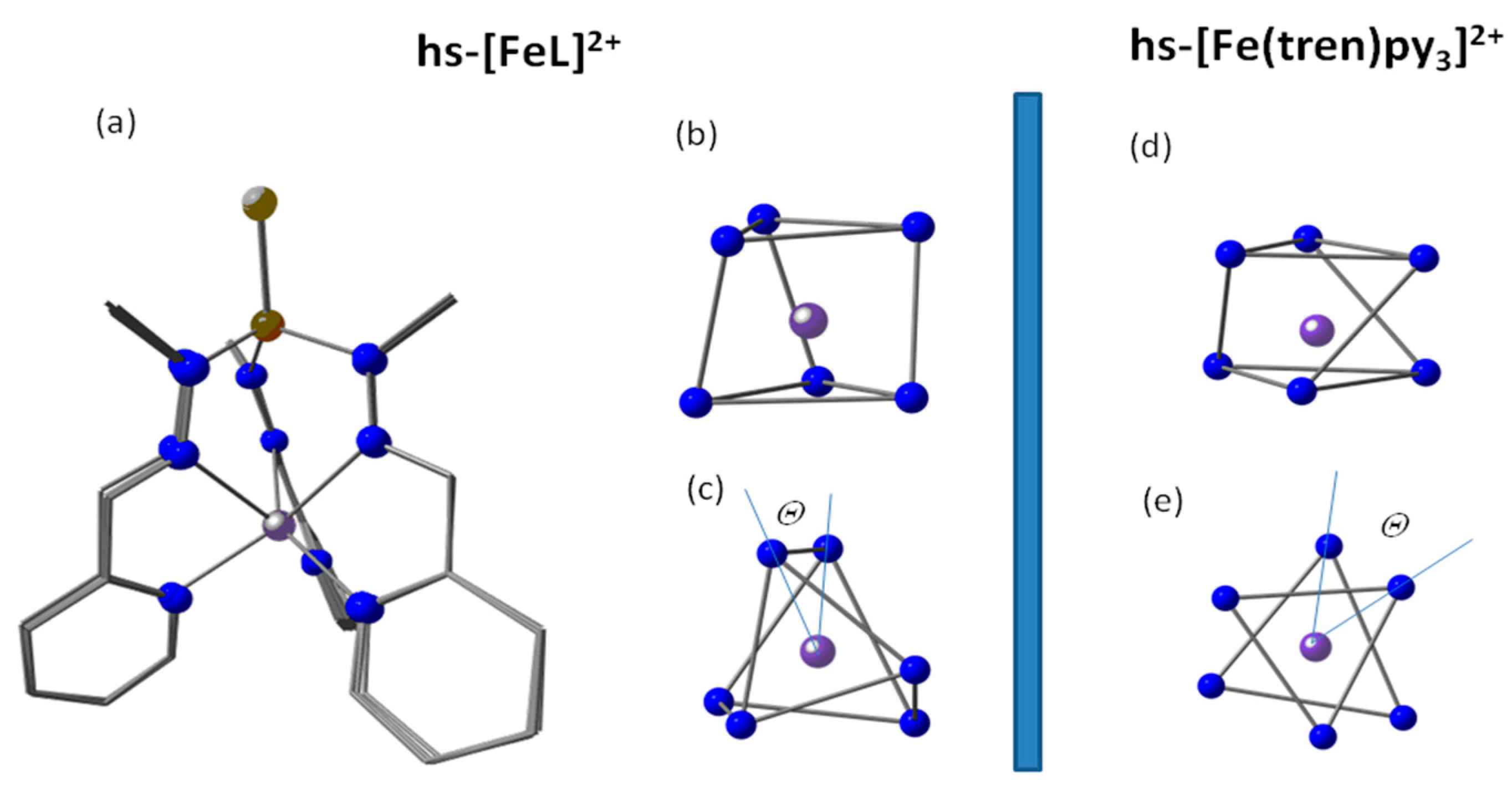
2.1.3. DFT Structure Elucidation of Elusive hs-[FeL]2+
2.2. DFT-Derived SCO Energies of [FeL]2+ and [Fe(tren)py3]2+
2.3. Dynamics of SCO in [FeL]2+ and [Fe(tren)py3]2+
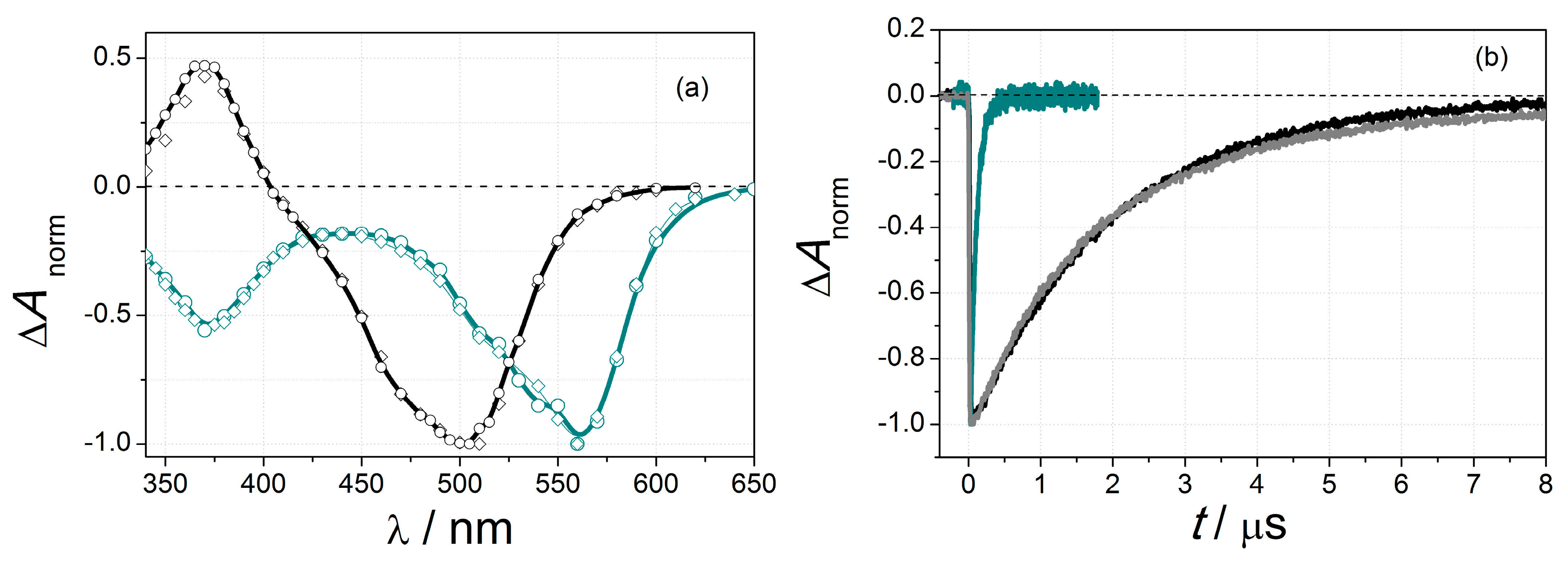
2.3.1. Optical Spectroscopy
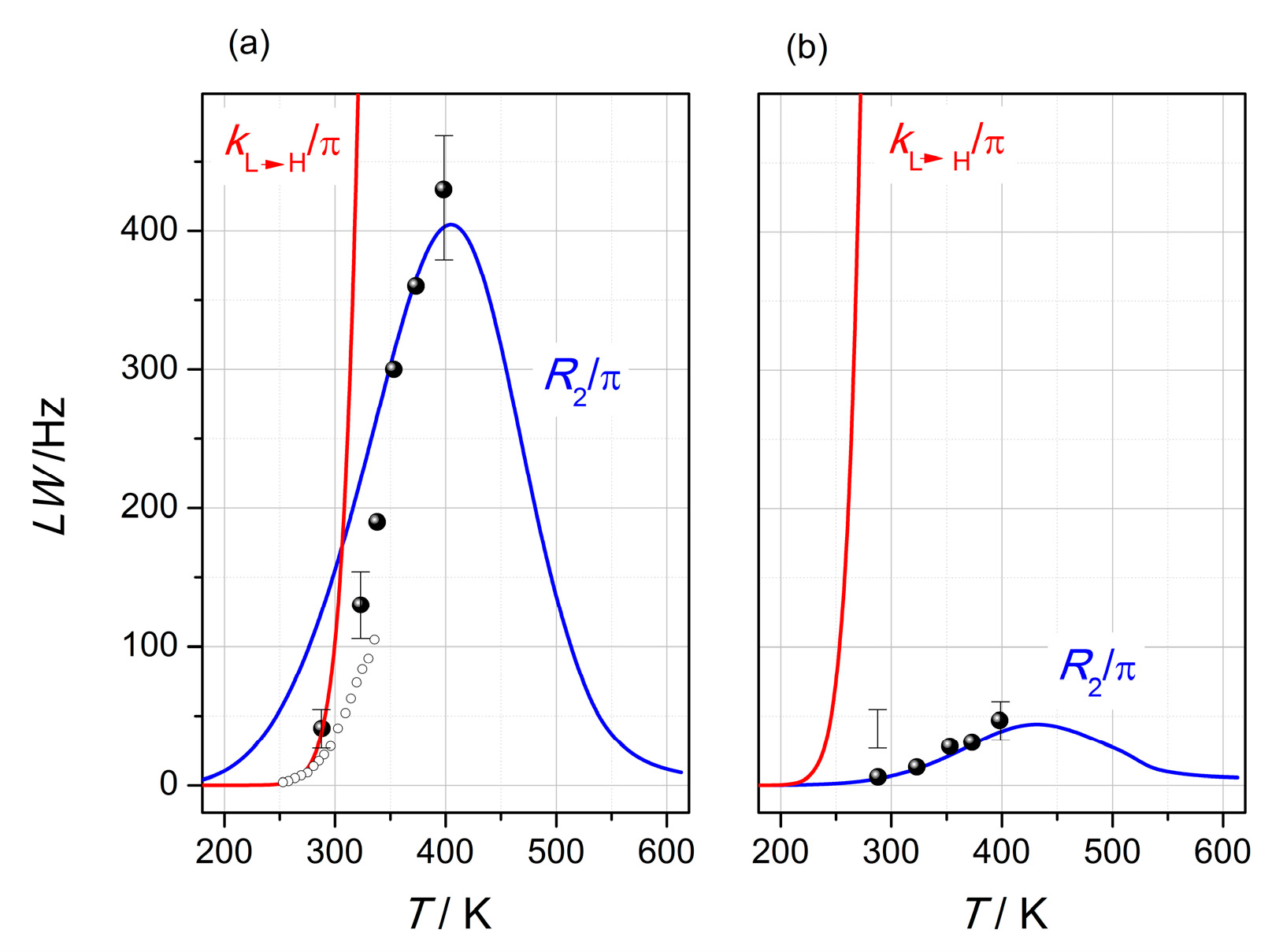
2.3.2. VT-NMR Spectroscopy
3. Discussion
4. Materials and Methods
4.1. Computational Methods
4.2. Materials and General Techniques
4.3. Instrumentation
4.4. X-ray Crystal Structure Determination
4.5. [FeL](BF4)2: [{(S)P(NMeNPy)3}Fe](BF4)2
4.6. [ZnL](ClO4)2: [{(S)P(NMeNPy)3}Zn](ClO4)2
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Halcrow, M.A. (Ed.) Spin Crossover Materials; Wiley: Chichester, UK, 2013. [Google Scholar]
- Kahn, O.; Martinez, C.J. Spin-Transition Polymers: From Molecular Materials toward Memory Devices. Science 1998, 279, 44–48. [Google Scholar] [CrossRef]
- Létard, J.-F.; Guionneau, P.; Gouz-Capes, L. Towards Spin Crossover Applications. Top. Curr. Chem. 2004, 235, 221–249. [Google Scholar] [CrossRef]
- Gütlich, P.; Gaspar, A.B.; Garcia, Y. Spin state switching in iron coordination compounds. Beilstein J. Org. Chem. 2013, 9, 342–391. [Google Scholar] [CrossRef] [PubMed]
- Bousseksou, A.; Molnar, G.; Salmon, L.; Nicolazzi, W. Molecular spin crossover phenomenon: Recent achievements and prospects. Chem. Soc. Rev. 2011, 40, 3313–3335. [Google Scholar] [CrossRef] [PubMed]
- Sato, O.; Tao, J.; Zhang, Y.-Z. Controlling Magnetic Properties through External Stimuli. Angew. Chem. Int. Ed. 2006, 46, 2152. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.S.; Ruben, M. Emerging trends in spin crossover (SCO) based functional materials and devices. Coord. Chem. Rev. 2017, 346, 176–205. [Google Scholar] [CrossRef]
- Decurtins, S.; Gütlich, P.; Köhler, C.P.; Spiering, H.; Hauser, A. Light-induced excited spin state trapping in a transition-metal complex: The hexa-1-propyltetrazole-iron (II) tetrafluoroborate spin-crossover system. Chem. Phys. Lett. 1984, 105, 1–4. [Google Scholar] [CrossRef]
- Letard, J.-F.; Asthana, S.; Shepherd, H.J.; Guionneau, P.; Goeta, A.E.; Suemura, N.; Ishikawa, R.; Kaizaki, S. Photomagnetism of a sym-cis-Dithiocyanato Iron(II) Complex with a Tetradentate N,N′-Bis(2-pyridylmethyl)1,2-ethanediamine Ligand. Chem. Eur. J. 2012, 19, 5924–5934. [Google Scholar] [CrossRef] [PubMed]
- Letard, J.-F.; Capes, L.; Chastanet, G.; Molinar, N.; Letard, S.; Real, J.-A.; Kahn, O. Critical temperature of the LIESST effect in iron(II) spin crossover compounds. Chem. Phys. Lett. 1999, 313, 115–120. [Google Scholar] [CrossRef]
- Hauser, A.; Enachescu, C.; Daku, M.L.; Vargas, A.; Amstutz, N. Low-temperature lifetimes of metastable high-spin states in spin-crossover and in low-spin iron(II) compounds: The rule and exceptions to the rule. Coord. Chem. Rev. 2006, 250, 1642–1652. [Google Scholar] [CrossRef]
- Klingele, J.; Kaase, D.; Schmucker, M.; Lan, Y.H.; Chastanet, G.; Letard, J.-F. Thermal Spin Crossover and LIESST Effect Observed in Complexes [Fe(LCh)2(NCX)2] [LCh = 2,5-Di(2-Pyridyl)-1,3,4-Chalcadiazole; Ch = O, S, Se; X = S, Se, BH3]. Inorg. Chem. 2013, 52, 6000–6010. [Google Scholar] [CrossRef] [PubMed]
- Dose, E.V.; Hoselton, M.A.; Sutin, N.; Tweedle, M.F.; Wilson, L.J. Dynamics of intersystem crossing processes in solution for six-coordinate d5, d6, and d7 spin-equilibrium metal complexes of iron(III), iron(II), and cobalt(II). J. Am. Chem. Soc. 1978, 100, 1141–1147. [Google Scholar] [CrossRef]
- Garvey, J.J.; Lawthers, I. Photochemically-induced perturbation of the 1A ⇌ 5T equilibrium in FeII complexes by pulsed laser irradiation in the metal-to-ligand charge-transfer absorption band. J. Chem. Soc. Chem. Commun. 1982, 906–907. [Google Scholar] [CrossRef]
- Beattie, J.K. Dynamics of Spin Equilibria in Metal Complexes. Adv. Inorg. Chem. 1988, 32, 1–53. [Google Scholar] [CrossRef]
- Toftlund, H. Spin equilibria in iron(II) complexes. Coord. Chem. Rev. 1989, 94, 67–108. [Google Scholar] [CrossRef]
- Xie, C.-L.; Hendrickson, D.N. Mechanism of spin-state interconversion in ferrous spin-crossover complexes: Direct evidence for quantum mechanical tunneling. J. Am. Chem. Soc. 1987, 109, 6981–6988. [Google Scholar] [CrossRef]
- Al-Obaidi, A.H.R.; Jensen, K.B.; McGarvey, J.J.; Toftlund, H.; Jensen, B.; Bell, S.E.J.; Carroll, J.G. Structural and Kinetic Studies of Spin Crossover in an Iron(II) Complex with a Novel Tripodal Ligand. Inorg. Chem. 1996, 35, 5055–5060. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-R.; McCusker, J.K.; Toftlund, H.; Wilson, S.R.; Trautwein, A.X.; Winkler, H.; Hendrickson, D.N. [Tetrakis(2-pyridylmethyl)ethylenediamine]iron(II) perchlorate, the first rapidly interconverting ferrous spin-crossover complex. J. Am. Chem. Soc. 1990, 112, 6814–6827. [Google Scholar] [CrossRef]
- McCusker, J.K.; Toftlund, H.; Rheingold, A.L.; Hendrickson, D.N. Ligand conformational changes affecting 5T2 → 1A1 intersystem crossing in a ferrous complex. J. Am. Chem. Soc. 1993, 115, 1797–1804. [Google Scholar] [CrossRef]
- Brady, C.; McGarvey, J.J.; McCusker, J.K.; Toftlund, H.; Hendrickson, D.N. Time-Resolved Relaxation Studies of Spin Crossover Systems in Solution. Top. Curr. Chem. 2004, 235, 1–22. [Google Scholar] [CrossRef]
- McCusker, J.K.; Rheingold, A.L.; Hendrickson, D.N. Variable-Temperature Studies of Laser-Initiated 5T2 → 1A1 Intersystem Crossing in Spin-Crossover Complexes: Empirical Correlations between Activation Parameters and Ligand Structure in a Series of Polypyridyl Ferrous Complexes. Inorg. Chem. 1996, 35, 2100–2112. [Google Scholar] [CrossRef]
- Hain, S.K.; Heinemann, F.W.; Gieb, K.; Müller, P.; Hörner, G.; Grohmann, A. On the Spin Behaviour of Iron(II)–Dipyridyltriazine Complexes and Their Performance as Thermal and Photonic Spin Switches. Eur. J. Inorg. Chem. 2010, 221–232. [Google Scholar] [CrossRef]
- Fatur, S.M.; Shepard, S.G.; Higgins, R.F.; Shores, M.P.; Damrauer, N.H. A Synthetically Tunable System To Control MLCT Excited-State Lifetimes and Spin States in Iron(II) Polypyridines. J. Am. Chem. Soc. 2017, 139, 4493–4505. [Google Scholar] [CrossRef] [PubMed]
- Stock, P.; Pędziński, T.; Spintig, N.; Grohmann, A.; Hörner, G. High Intrinsic Barriers against Spin-State Relaxation in Iron(II)-Complex Solutions. Chem. Eur. J. 2013, 19, 839–842. [Google Scholar] [CrossRef] [PubMed]
- Stock, P.; Spintig, N.; Scholz, J.; Epping, J.D.; Oelsner, C.; Wiedemann, D.; Grohmann, A.; Hörner, G. Spin-State Dynamics of a Photochromic Iron(II) Complex and its Immobilization on Oxide Surfaces Via Phenol Anchors. J. Coord. Chem. 2015, 68, 3099–3115. [Google Scholar] [CrossRef]
- Stock, P.; Deck, E.; Hohnstein, S.; Korzekwa, J.; Meyer, K.; Heinemann, F.W.; Breher, F.; Hörner, G. Molecular Spin Crossover in Slow Motion: Light-Induced Spin-State Transitions in Trigonal Prismatic Iron(II) Complexes. Inorg. Chem. 2016, 55, 5254–5265. [Google Scholar] [CrossRef] [PubMed]
- Bailar, J.C., Jr. Some problems in the stereochemistry of coordination compounds: Introductory lecture. J. Inorg. Nucl. Chem. 1958, 8, 165–175. [Google Scholar] [CrossRef]
- Trapp, I.; Löble, M.W.; Meyer, J.; Breher, F. Copper complexes of tripodal κ6N-donor ligands: A structural, EPR spectroscopic and electrochemical study. Inorg. Chim. Acta 2011, 374, 373–384. [Google Scholar] [CrossRef]
- Löble, M.W.; Casimiro, M.; Thielemann, D.T.; Oña-Burgos, P.; Fernandez, I.; Roesky, P.W.; Breher, F. 1H,89Y HMQC and Further NMR Spectroscopic and X-ray Diffraction Investigations on Yttrium-Containing Complexes Exhibiting Various Nuclearities. Chem. Eur. J. 2012, 18, 5325–5334. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekhar, V.; Azhakar, R.; Pandian, B.M.; Boomishankar, R.; Steiner, A. A phosphorus-supported multisite coordination ligand containing three imidazolyl arms and its metalation behaviour. An unprecedented co-existence of mononuclear and macrocyclic dinuclear Zn(II) complexes in the same unit cell of a crystalline lattice. Dalton Trans. 2008, 37, 5962–5969. [Google Scholar] [CrossRef] [PubMed]
- Marchivie, M.; Guionneau, P.; Letard, J.-F.; Chasseau, D. Photo-induced spin-transition: The role of the iron(II) environment distortion. Acta Crystallogr. Sect. B Struct. Sci. 2005, 61, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, E.B.; Gebala, A.E.; Swift, D.R.; Tasker, P.A. Trigonal prismatic-octahedral coordination. Complexes of intermediate geometry. Inorg. Chem. 1972, 11, 2775–2784. [Google Scholar] [CrossRef]
- Wentworth, R.A.D. Trigonal prismatic vs. octahedral stereochemistry in complexes derived from innocent ligands. Coord. Chem. Rev. 1972, 9, 171–187. [Google Scholar] [CrossRef]
- Purcell, K.F. Pseudorotational intersystem crossing in d6 complexes. J. Am. Chem. Soc. 1979, 101, 5147–5152. [Google Scholar] [CrossRef]
- Vanquickenborne, L.G.; Pierloot, K. Role of spin change in the stereomobile reactions of strong-field d6 transition-metal complexes. Inorg. Chem. 1981, 20, 3673–3677. [Google Scholar] [CrossRef]
- Comba, P. Coordination geometries of hexaamine cage complexes. Inorg. Chem. 1989, 28, 426–431. [Google Scholar] [CrossRef]
- Larsen, E.; La Mar, G.N.; Wagner, B.E.; Parks, J.E.; Holm, R.H. Three-dimensional macrocyclic encapsulation reactions. III. Geometrical and electronic features of tris(diimine) complexes of trigonal-prismatic, antiprismatic, and intermediate stereochemistry. Inorg. Chem. 1972, 11, 2652–2668. [Google Scholar] [CrossRef]
- Comba, P.; Sargeson, A.M.; Engelhardt, L.M.; Harrowfield, J.M.B.; White, A.H.; Horn, E.; Snow, M.R. Analysis of trigonal-prismatic and octahedral preferences in hexaamine cage complexes. Inorg. Chem. 1985, 24, 2325–2327. [Google Scholar] [CrossRef]
- Alvarez, S.; Avnir, D.; Llunell, M.; Pinsky, M. Continuous symmetry maps and shape classification. The case of six-coordinated metal compounds. New J. Chem. 2002, 26, 996–1009. [Google Scholar] [CrossRef]
- Knight, J.C.; Alvarez, S.; Amoroso, A.J.; Edwards, P.G.; Singh, N. A novel bipyridine-based hexadentate tripodal framework with a strong preference for trigonal prismatic co-ordination geometries. Dalton Trans. 2010, 39, 3870–3883. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S. Distortion Pathways of Transition Metal Coordination Polyhedra Induced by Chelating Topology. Chem. Rev. 2015, 115, 13447–13483. [Google Scholar] [CrossRef] [PubMed]
- Conti, A.J.; Xie, C.L.; Hendrickson, D.N. Tunneling in spin-state interconversion of ferrous spin-crossover complexes. Concentration dependence of apparent activation energy determined in solution by laser-flash photolysis. J. Am. Chem. Soc. 1989, 111, 1171–1180. [Google Scholar] [CrossRef]
- Brewer, G.; Olida, M.J.; Schmiedekamp, A.M.; Viragh, C.; Zavalij, P.Y. A DFT computational study of spin crossover in iron(III) and iron(II) tripodal imidazole complexes. A comparison of experiment with calculations. Dalton Trans. 2006, 35, 5617. [Google Scholar] [CrossRef] [PubMed]
- Sunatsuki, Y.; Ikuta, Y.; Matsumoto, N.; Ohta, H.; Kojima, M.; Iijima, S.; Hayami, S.; Maeda, Y.; Kaizaki, S.; Dahan, F.; Tuchagues, J.-P. An Unprecedented Homochiral Mixed-Valence Spin-Crossover Compound. Angew. Chem. Int. Ed. 2003, 42, 1614. [Google Scholar] [CrossRef] [PubMed]
- Ohta, H.; Sunatsuki, Y.; Kojima, M.; Iijima, S.; Akashi, H.; Matsumoto, N. A Tripodal Ligand Containing Three Imidazole Groups Inducing Spin Crossover in Both Fe(II) and Fe(III) Complexes; Structures and Spin Crossover Behaviors of the Complexes. Chem. Lett. 2004, 33, 350. [Google Scholar] [CrossRef]
- Sunatsuki, Y.; Ohta, H.; Kojima, M.; Ikuta, Y.; Goto, Y.; Matsumoto, N.; Iijima, S.; Akashi, H.; Kaizaki, S.; Dahan, F.; et al. Supramolecular Spin-Crossover Iron Complexes Based on Imidazole-Imidazolate Hydrogen Bonds. Inorg. Chem. 2004, 43, 4154. [Google Scholar] [CrossRef] [PubMed]
- Brewer, C.; Brewer, G.; Luckett, C.; Marbury, G.S.; Viragh, C.; Beatty, A.M.; Scheidt, W.R. Proton Control of Oxidation and Spin State in a Series of Iron Tripodal Imidazole Complexes. Inorg. Chem. 2004, 43, 2402. [Google Scholar] [CrossRef] [PubMed]
- Halcrow, M.A. Structure: Function relationships in molecular spin-crossover complexes. Chem. Soc. Rev. 2011, 40, 4119–4142. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Bauer, W.; Obel, J. An Iron(II) Spin-Crossover Complex with a 70 K Wide Thermal Hysteresis Loop. Angew. Chem Int. Ed. 2008, 47, 10098–10101. [Google Scholar] [CrossRef] [PubMed]
- Stock, P.; Erbe, A.; Buck, M.; Wiedemann, D.; Menard, H.; Hörner, G.; Grohmann, A. Thiocyanate Anchors for Salt-like Iron(II) Complexes on Au(111): Promises and Caveats. Z. Naturforsch. B A J. Chem. Sci. 2014, 69, 1164–1180. [Google Scholar] [CrossRef]
- Hostettler, M.; Törnroos, K.W.; Chernyshow, D.; Vangdal, B.; Bürgi, H.-B. Challenges in Engineering Spin Crossover: Structures and Magnetic Properties of Six Alcohol Solvates of Iron(II) Tris(2-picolylamine) Dichloride. Angew. Chem. Int. Ed. 2004, 43, 4589–4594. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, R.M.; Mealli, C.; Bailey, M.; Howe, N.; Torre, L.P.; Wilson, L.J.; Andrews, L.C.; Rose, N.J.; Lingafelter, E.C. The Variable Coordination Chemistry of a Potentially Heptadentate Ligand with a Series of 3d Transition Metal Ions. The Chemistry and the Strcutures of [M(py3tren)]2+, where M(II) = Mn, Fe, Co, Ni, Cu and Zn and (py3tren) = N{CH2CH2N=C(H)(C5H4N)}3. Coord. Chem. Rev. 1987, 77, 89–163. [Google Scholar] [CrossRef]
- Lazar, H.Z.; Forestier, T.; Barrett, S.A.; Kilner, C.A.; Letard, J.-F.; Halcrow, M.A. Thermal and light-induced spin-crossover in salts of the heptadentate complex [tris(4-{pyrazol-3-yl}-3-aza-3-butenyl)amine]iron(II). Dalton Trans. 2007, 36, 4276–4285. [Google Scholar] [CrossRef] [PubMed]
- Reiher, M.; Salomon, O.; Hess, B.A. Reparameterization of hybrid functionals based on energy differences of states of different multiplicity. Theor. Chem. Acc. 2001, 107, 48–55. [Google Scholar] [CrossRef]
- Lawson Daku, M.L.; Vargas, A.; Hauser, A.; Fouqueau, A.; Casida, M.E. Assessment of Density Functionals for the High-Spin/Low-Spin Energy Difference in the Low-Spin Iron(II) Tris(2,2’-bipyridine) Complex. ChemPhysChem 2005, 6, 1393–1410. [Google Scholar] [CrossRef] [PubMed]
- Bowman, D.N.; Jakubikova, E. Low-Spin versus High-Spin Ground State in Pseudo-Octahedral Iron Complexes. Inorg. Chem. 2012, 51, 6011–6019. [Google Scholar] [CrossRef] [PubMed]
- Bowman, D.N.; Bondarev, A.; Mukherjee, S.; Jakubikova, E. Tuning the Electronic Structure of Fe(II) Polypyridines via Donor Atom and Ligand Scaffold Modifications: A Computational Study. Inorg. Chem. 2015, 54, 8786–8793. [Google Scholar] [CrossRef] [PubMed]
- Mengel, A.K.C.; Förster, C.; Breivogel, A.; Mack, K.; Ochsmann, J.R.; Laquai, F.; Ksenofontov, V.; Heinze, K. Heteroleptic Push-Pull Substituted Iron(II) Bis(tridentate) Complex with Low Energy Charge Transfer States. Chem. Eur. J. 2015, 21, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Jamula, L.L.; Brown, A.M.; Guo, D.; McCusker, J.K. Synthesis and characterization of a high-symmetry ferrous polypyridyl complex: Approaching the 5T2/3T1 crossing point for FeII. Inorg. Chem. 2014, 53, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Buhks, E.; Navon, G.; Bixon, M.; Jortner, J. Spin Conversion Processes in Solutions. J. Am. Chem. Soc. 1980, 102, 2918–2923. [Google Scholar] [CrossRef]
- Barrett, S.A.; Kilner, C.A.; Halcrow, M.A. Spin-crossover in [Fe(3-bpp)2][BF4]2 in different solvents—A dramatic stabilisation of the low-spin state in water. Dalton Trans. 2011, 40, 12021–12024. [Google Scholar] [CrossRef] [PubMed]
- Petzold, H.; Djomgoue, P.; Hörner, G.; Speck, J.M.; Rüffer, T.; Schaarschmidt, D. 1H-NMR spectroscopic elucidation in solution of the kinetics and thermodynamics of spin crossover for an exceptionally robust Fe2+ complex. Dalton Trans. 2016, 45, 13798–13809. [Google Scholar] [CrossRef] [PubMed]
- Petzold, H.; Djomgoue, P.; Hörner, G.; Heider, S.; Lochenie, C.; Weber, B.; Rüffer, T.; Schaarschmidt, D. Spin state variability in Fe2+ complexes of substituted (2-(pyridin-2-yl)-1,10-phenanthroline) ligands as versatile terpyridine analogues. Dalton Trans. 2017, 46, 6218–6229. [Google Scholar] [CrossRef] [PubMed]
- Petzold, H.; Djomgoue, P.; Hörner, G.; Lochenie, C.; Weber, B.; Rüffer, T. Bis-meridional Fe2+ SCO complexes of phenyl and pyridyl substituted 2-(pyridin-2-yl)-1,10-phenanthrolines. Dalton Trans. 2017. (under Review). [Google Scholar]
- Renz, F.; Oshio, H.; Ksenofontov, V.; Waldeck, M.; Spiering, H.; Gütlich, P. Strong Field Iron(II) Complex Converted by Light into a Long-Lived High-Spin State. Angew. Chem. Int. Ed. 2000, 39, 3699–3700. [Google Scholar] [CrossRef]
- Vanko, G.; Bordage, A.; Papai, M.; Haldrup, K.; Glatzel, P.; March, A.M.; Doumy, G.; Britz, A.; Galler, A.; Assefa, T.; et al. Detailed Characterization of a Nanosecond-Lived Excited State: X-ray and Theoretical Investigation of the Quintet State in Photoexcited [Fe(terpy)2]2+. J. Phys. Chem. C 2015, 119, 5888–5902. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lawson Daku, M.L.; Zhang, J.; Suarez-Alcantara, K.; Jennings, G.; Kurtz, C.A.; Canton, S.E. Dynamic Jahn–Teller Effect in the Metastable High-Spin State of Solvated [Fe(terpy)2]2+. J. Phys. Chem. C 2015, 119, 3312–3321. [Google Scholar] [CrossRef]
- Canton, S.E.; Zhang, X.; Lawson Daku, M.L.; Liu, Y.; Zhang, J.; Alvarez, S. Mapping the Ultrafast Changes of Continuous Shape Measures in Photoexcited Spin Crossover Complexes without Long-Range Order. J. Phys. Chem. C 2015, 119, 3322–3330. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Kanai, Y.; Wang, X.; Selloni, A.; Car, R. Testing the TPSS meta-generalized-gradient-approximation exchange-correlation functional in calculations of transition states and reaction barriers. J. Chem. Phys. 2006, 125, 234104. [Google Scholar] [CrossRef] [PubMed]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.; Perdew, J.P. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J. Chem. Phys. 2003, 119, 12129. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Pędziński, T.; Markiewicz, A.; Marciniak, B. Photosensitized oxidation of methionine derivatives. Laser flash photolysis studies. Res. Chem. Intermed. 2009, 35, 497–506. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; Cabral de Menezes, S.M.; Granger, P.; Hoffman, R.E.; Zilm, K.W. Further conventions for NMR shielding and chemical shifts. Pure Appl. Chem. 2008, 80, 59–84. [Google Scholar] [CrossRef]
- CrysAlisPro Software System, version 1.171.37. Intelligent Data Collection and Processing Software for Small Molecule and Protein Crystallography. Agilent Technologies: Oxford, UK, 2014.
- Clark, R.C.; Reid, J.S. The analytical calculation of absorption in multifaceted crystals. Acta Crystallogr. Sect. A Found. Crystallogr. 1995, 51, 887–897. [Google Scholar] [CrossRef]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Kratzert, D.; Holstein, J.J.; Krossing, I. DSR: Enhanced modelling and refinement of disordered structures with SHELXL. J. Appl. Crystallogr. 2015, 48, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Platon Squeeze: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 9–18. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | [FeL](BF4)2·1.5 CH3CN | [ZnL](ClO4)2·1.5 CH3CN |
|---|---|---|
| Sum formula | C24H28.5B2F8FeN10.5PS | C24H28.5Cl2N10.5O8PSZn |
| M (g·mol−1) | 756.57 | 791.37 |
| Shape and color | black plate | colorless column |
| Size (mm3) | 0.41 × 0.31 × 0.16 | 0.44 × 0.25 × 0.21 |
| crystal system | orthorhombic | monoclinic |
| space group | P212121 | P21/c |
| a (pm) | 1134.21(5) | 909.34(12) |
| b (pm) | 1532.91(7) | 3165.3(3) |
| c (pm) | 3673.55(18) | 1628.1(2) |
| α (°) | 90 | 90 |
| β (°) | 90 | 136.39(3) |
| γ (°) | 90 | 90 |
| V (106 pm3) | 6387.0(5) | 3232.2(19) |
| µ (mm−1) | 0.671 | 1.104 |
| ρcalcd. (g·cm−3) | 1.574 | 1.626 |
| Z | 8 | 4 |
| T (K) | 150(1) | 150(1) |
| 2 θmax (°) | 52.00 | 52.00 |
| reflns. Measured | 27,331 | 24,831 |
| reflns. Unique | 12,295 | 6322 |
| parameters/restraints | 932/253 | 475/114 |
| R1 (I ≥ 2σ(I)) | 0.0578 | 0.0699 |
| R1 (all data) | 0.0810 | 0.0820 |
| wR2 (I ≥ 2σ(I)) | 0.1078 | 0.1606 |
| wR2 (all data) | 0.1180 | 0.1680 |
| u, v | 0.0427, 1.3949 | 0.0472, 12.7098 |
| S | 1.018 | 1.109 |
| Flack parameter x | 0.012 (12) | n/a |
| ρmax/ρmin (e × 10−6 pm−3) | 0.50/−0. 53 | 0.99/−0.90 |
| CCDC number | 1564278 | 1564279 |
| [FeL](BF4)2 | ls-[FeL]2+ | [ZnL](ClO4)2 | [ZnL]2+ | |
|---|---|---|---|---|
| distances (Å) | ||||
| dFe–N(ald) | 1.924(7) | 1.930(1) | 2.244(16) | 2.254(2) |
| dFe–N(py) | 1.979(10) | 1.974(1) | 2.133(11) | 2.134(2) |
| dFe–N | 1.952(15) | 1.952(11) | 2.19(2) | 2.19(3) |
| bite | 2.520(6) | 2.535(2) | 2.657(4) | 2.677(1) |
| Fe–P | 3.123(3) | 3.162 | 3.482(5) | 3.509 |
| h b | 2.225 | 2.216 | 2.587 | 2.586 |
| δ c | −0.017 | −0.014 | +0.425 | +0.389 |
| angles (°) | ||||
| cis Nald–Fe–Nald | 90.4(9) | 90.6(1) | 79.8(6) | 81.2(2) |
| cis Npy–Fe–Npy | 91.1(14) | 91.2(1) | 97(3) | 95.9(1) |
| bite angle | 80.4(2) | 81.0(1) | 74.7(4) | 75.1(3) |
| trans Npy–Fe–Nald | 166.2(12) | 167.7(1) | 145(2) | 148.1(2) |
| distortion | ||||
| ∑cis/° d | 76.2 | 57.9 | 163.3 | 164.4 |
| θ° e | 43.4(8) | 45.0(1) | 19(2) | 22.3(1) |
| S (Oh) f | 1.464 | 1.168 | 8.613 | 7.147 |
| S (TP) f | 9.306 | 10.007 | 2.135 | 2.700 |
| B3LYP a0 a = | BP86 | PBE | TPSS0 | TPSSh | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0.00 | 0.05 | 0.10 | 0.15 | 0.20 | 0.25 | |||||
| Bond lengths [Å] | ||||||||||
| dFe–N(ald) | 2.197 | 2.211 | 2.226 | 2.230 | 2.238 | 2.253 | 2.199 | 2.200 | 2.205 | 2.198 |
| dFe–N(py) | 2.132 | 2.150 | 2.169 | 2.175 | 2.186 | 2.198 | 2.134 | 2.138 | 2.161 | 2.152 |
| dFe–N | 2.164 | 2.181 | 2.198 | 2.203 | 2.212 | 2.225 | 2.167 | 2.169 | 2.183 | 2.175 |
| distortion | ||||||||||
| ∑cis/° b | 150.1 | 154.7 | 160.2 | 161.2 | 163.9 | 171.2 | 150.5 | 151.2 | 159.8 | 155.6 |
| θ/° c | 24.0 | 22.2 | 20.5 | 20.3 | 20.2 | 17.8 | 24.3 | 24.3 | 20.4 | 21.5 |
| S(Oh) d | 6.54 | 7.13 | 7.78 | 7.86 | 7.91 | 8.92 | 6.51 | 6.45 | 7.82 | 7.39 |
| S(TP) d | 3.09 | 2.66 | 2.27 | 2.24 | 2.24 | 1.79 | 3.12 | 3.19 | 2.24 | 2.48 |
| ΔSCOE (kJ·mol−1) | |||||
|---|---|---|---|---|---|
| a0 b = 0.00 | 0.05 | 0.10 | 0.15 | 0.20 | |
| [FeL]2+ | 153.4 | 116.7 | 79.4 | 46.6 | 10.3 |
| [Fe(tren)py3]2+ | 154.2 | 116.3 | 80.0 | 46.2 | 14.8 |
| [FeL’]2+ | 127.7 | 89.7 | 54.1 | 21.2 | −9.0 |
| [Fe(tren)imid3]2+ | 123.3 | 87.3 | 52.8 | 20.5 | −9.0 |
| [FeL]2+ | [Fe(tren)py3]2+ | |
|---|---|---|
| Ea [kJ·mol−1] | 22(1) a | 7.8(3) b |
| kH→L [106 s−1] b | 0.46 | 15.9 |
| kL→H [106 s−1] c | <0.005 | <0.2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stock, P.; Wiedemann, D.; Petzold, H.; Hörner, G. Structural Dynamics of Spin Crossover in Iron(II) Complexes with Extended-Tripod Ligands. Inorganics 2017, 5, 60. https://doi.org/10.3390/inorganics5030060
Stock P, Wiedemann D, Petzold H, Hörner G. Structural Dynamics of Spin Crossover in Iron(II) Complexes with Extended-Tripod Ligands. Inorganics. 2017; 5(3):60. https://doi.org/10.3390/inorganics5030060
Chicago/Turabian StyleStock, Philipp, Dennis Wiedemann, Holm Petzold, and Gerald Hörner. 2017. "Structural Dynamics of Spin Crossover in Iron(II) Complexes with Extended-Tripod Ligands" Inorganics 5, no. 3: 60. https://doi.org/10.3390/inorganics5030060
APA StyleStock, P., Wiedemann, D., Petzold, H., & Hörner, G. (2017). Structural Dynamics of Spin Crossover in Iron(II) Complexes with Extended-Tripod Ligands. Inorganics, 5(3), 60. https://doi.org/10.3390/inorganics5030060