
Silylation of Dinitrogen Catalyzed by Hydridodinitrogentris(Triphenylphosphine)Cobalt(I)

Abstract
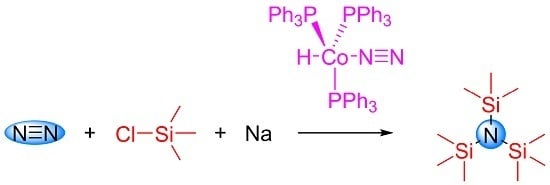
:
1. Introduction
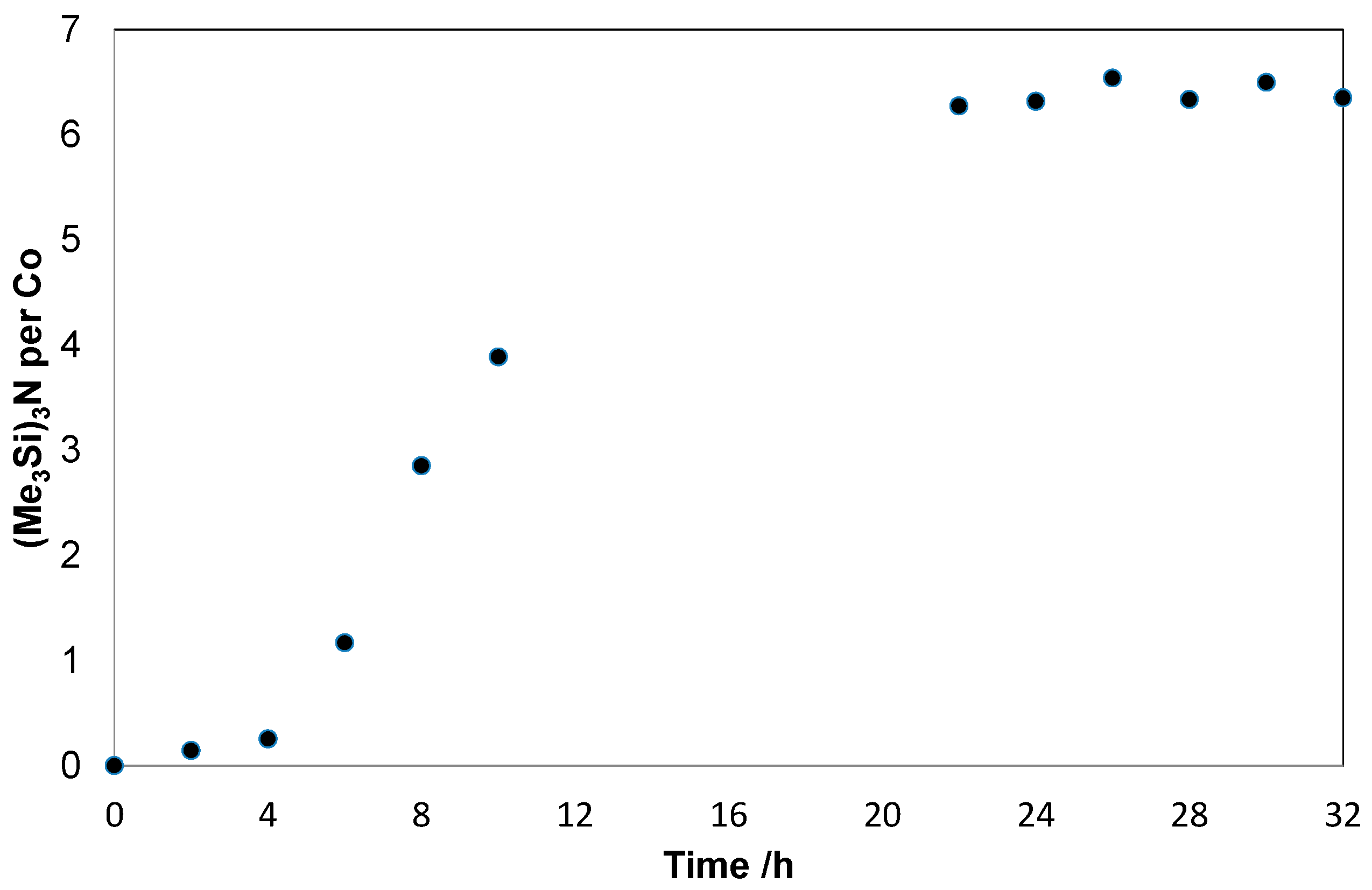
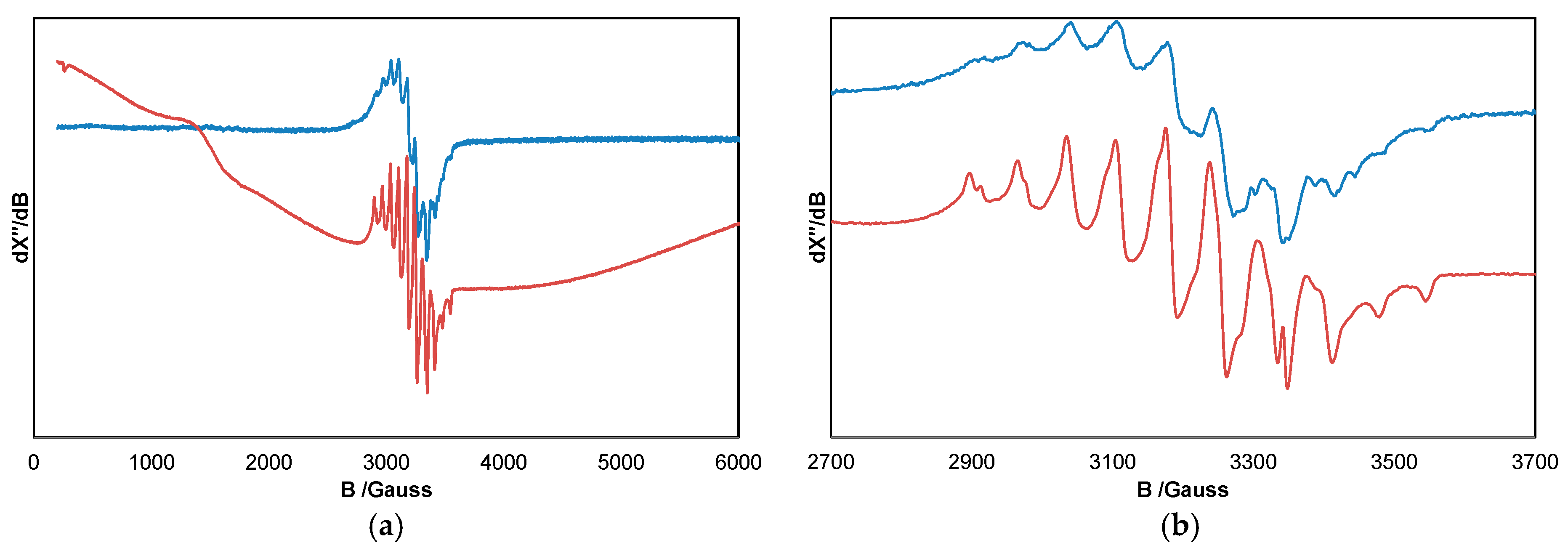
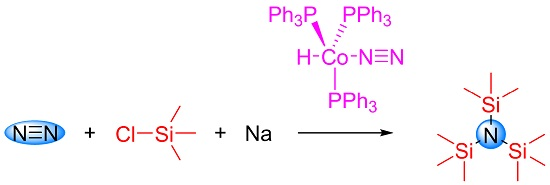
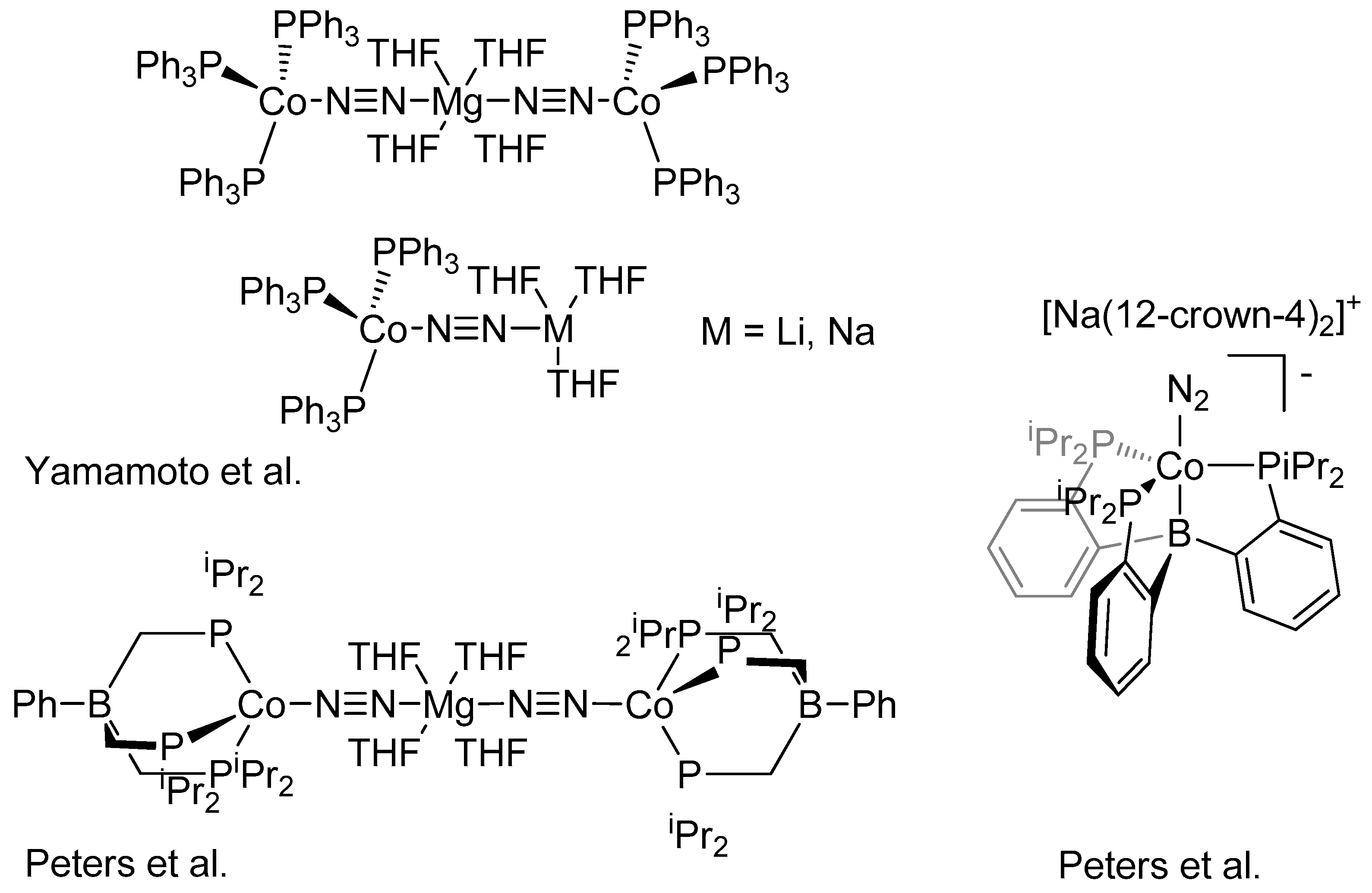
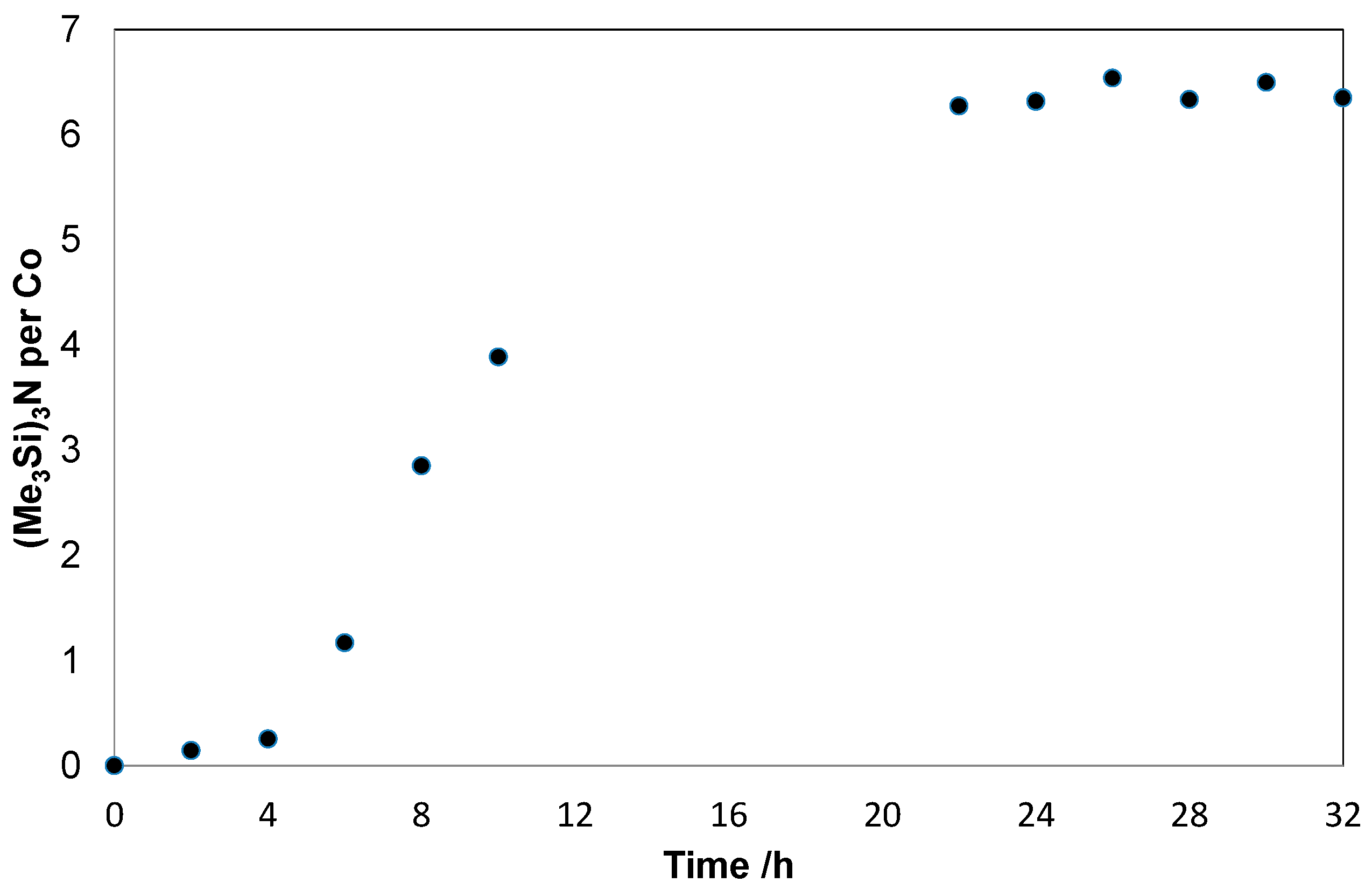
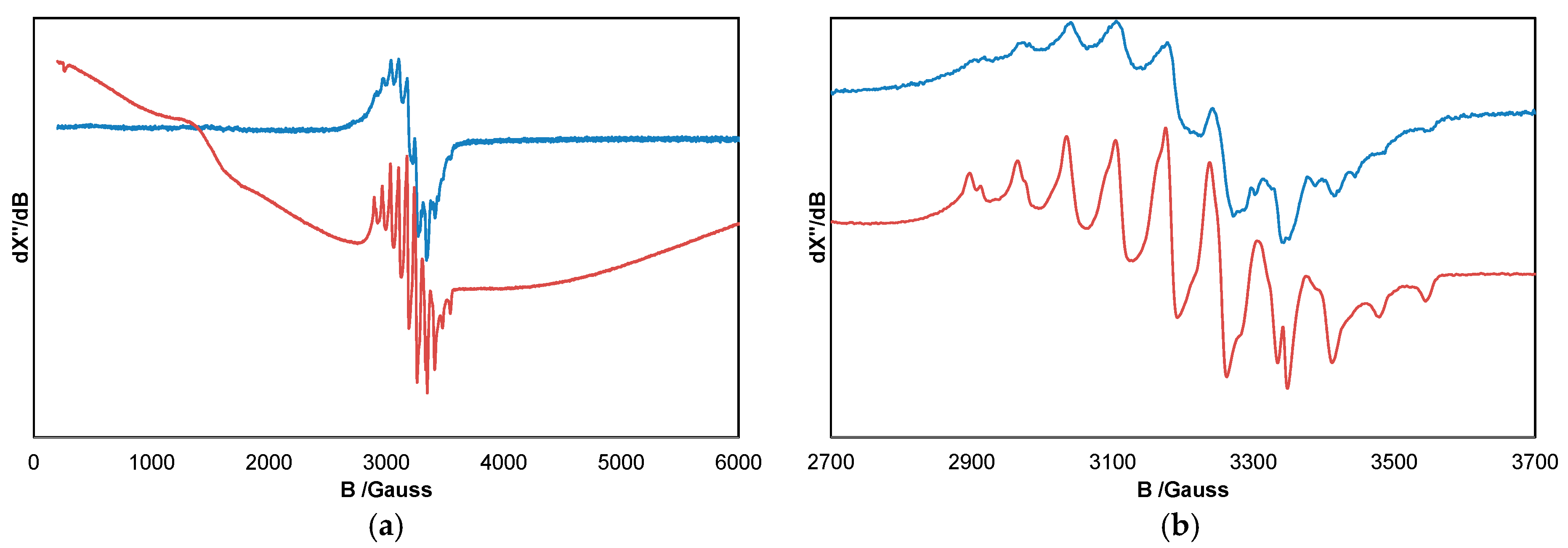
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
Abbreviations
| acac | acetylacetonato |
| DLS | dynamic light scattering |
| EPR | electron paramagnetic resonance |
| GC | gas chromatography |
| IR | infrared |
| MS | mass spectrometry |
| THF | Tetrahydrofuran |
| TON | turnover number |
References
- Bothe, H.; Schmitz, O.; Yates, M.G.; Newton, W.E. Nitrogen Fixation and Hydrogen Metabolism in Cyanobacteria. Microbiol. Mol. Biol. Rev. 2010, 74, 529–551. [Google Scholar] [CrossRef] [PubMed]
- Burgess, B.K.; Lowe, D.J. Mechanism of Molybdenum Nitrogenase. Chem. Rev. 1996, 96, 2983–3012. [Google Scholar] [CrossRef] [PubMed]
- Eady, R.R. Structure-Function Relationships of Alternative Nitrogenases. Chem. Rev. 1996, 96, 3013–3030. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, B.M.; Lukoyanov, D.; Yang, Z.-Y.; Dean, D.R.; Seefeldt, L.C. Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage. Chem. Rev. 2014, 114, 4041–4062. [Google Scholar] [CrossRef] [PubMed]
- Hidai, M.; Mizobe, Y. Recent Advances in the Chemistry of Dinitrogen Complexes. Chem. Rev. 1995, 95, 1115–1133. [Google Scholar] [CrossRef]
- Bazhenova, T.A.; Shilov, A.E. Nitrogen fixation in solution. Coord. Chem. Rev. 1995, 144, 69–145. [Google Scholar]
- Fryzuk, M.D.; Johnson, S.A. The continuing story of dinitrogen activation. Coord. Chem. Rev. 2000, 200–202, 379–409. [Google Scholar] [CrossRef]
- MacKay, B.A.; Fryzuk, M.D. Dinitrogen coordination chemistry: On the biomimetic borderlands. Chem. Rev. 2004, 104, 385–401. [Google Scholar] [CrossRef] [PubMed]
- Hidai, M.; Mizobe, Y. Research inspired by the chemistry of nitrogenase. Novel metal complexes and their reactivity toward dinitrogen, nitriles, and alkynes. Can. J. Chem. 2005, 83, 358–374. [Google Scholar] [CrossRef]
- Hinrichsen, S.; Broda, H.; Gradert, C.; Söncksen, L.; Tuczek, F. Recent developments in synthetic nitrogen fixation. Ann. Rep. Prog. Chem. Sect. A Inorg. Chem. 2012, 108, 17–47. [Google Scholar] [CrossRef]
- Van der Ham, C.J.M.; Koper, M.T.M.; Hetterscheid, D.G.H. Challenges in reduction of dinitrogen by proton and electron transfer. Chem. Soc. Rev. 2014, 43, 5183–5191. [Google Scholar] [CrossRef] [PubMed]
- Khoenkhoen, N.; de Bruin, B.; Reek, J.N.H.; Dzik, W.I. Reactivity of Dinitrogen Bound to Mid- and Late-Transition-Metal Centers. Eur. J. Inorg. Chem. 2015, 567–598. [Google Scholar] [CrossRef]
- Bezdek, M.J.; Chirik, P.J. Expanding Boundaries: N2 Cleavage and Functionalization beyond Early Transition Metals. Angew. Chem. Int. Ed. 2016, 55. [Google Scholar] [CrossRef] [PubMed]
- Hazari, N. Homogeneous iron complexes for the conversion of dinitrogen into ammonia and hydrazine. Chem. Soc. Rev. 2010, 39, 4044–4056. [Google Scholar] [CrossRef] [PubMed]
- Crossland, J.L.; Tyler, D.R. Iron-dinitrogen coordination chemistry: Dinitrogen activation and reactivity. Coord. Chem. Rev. 2010, 254, 1883–1894. [Google Scholar] [CrossRef]
- MacLeod, K.C.; Holland, P.L. Recent developments in the homogeneous reduction of dinitrogen by molybdenum and iron. Nat. Chem. 2013, 5, 559–565. [Google Scholar]
- Tanabe, Y.; Nishibayashi, Y. Developing more sustainable processes for ammonia synthesis. Coord. Chem. Rev. 2013, 257, 2551–2564. [Google Scholar] [CrossRef]
- Jia, H.-P.; Quadrelli, E.A. Mechanistic aspects of dinitrogen cleavage and hydrogenation to produce ammonia in catalysis and organometallic chemistry: Relevance of metal hydride bonds and dihydrogen. Chem. Soc. Rev. 2014, 43, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Yandulov, D.V.; Schrock, R.R. Catalytic reduction of dinitrogen to ammonia at a single molybdenum center. Science 2003, 301, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Arashiba, K.; Miyake, Y.; Nishibayashi, Y. A molybdenum complex bearing PNP-type pincer ligands leads to the catalytic reduction of dinitrogen into ammonia. Nat. Chem. 2011, 3, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Arashiba, K.; Kinoshita, E.; Kuriyama, S.; Eizawa, A.; Nakajima, K.; Tanaka, H.; Yoshizawa, K.; Nishibayashi, Y. Catalytic Reduction of Dinitrogen to Ammonia by Use of Molybdenum–Nitride Complexes Bearing a Tridentate Triphosphine as Catalysts. J. Am. Chem. Soc. 2015, 137, 5666–5669. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.S.; Rittle, J.; Peters, J.C. Catalytic conversion of nitrogen to ammonia by an iron model complex. Nature 2013, 501, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Del Castillo, T.J.; Thompson, N.B.; Peters, J.C. A Synthetic Single-Site Fe Nitrogenase: High Turnover, Freeze-Quench 57Fe Mössbauer Data, and a Hydride Resting State. J. Am. Chem. Soc. 2016, 138, 5341–5350. [Google Scholar] [CrossRef] [PubMed]
- Shiina, K. Reductive silylation of molecular nitrogen via fixation to tris(trialkylsilyl)amine. J. Am. Chem. Soc. 1972, 94, 9266–9267. [Google Scholar] [CrossRef]
- Komori, K.; Oshita, H.; Mizobe, Y.; Hidai, M. Catalytic conversion of molecular nitrogen into silylamines using molybdenum and tungsten dinitrogen complexes. J. Am. Chem. Soc. 1989, 111, 1939–1940. [Google Scholar] [CrossRef]
- Tanaka, H.; Sasada, A.; Kouno, T.; Yuki, M.; Miyake, Y.; Nakanishi, H.; Nishibayashi, Y.; Yoshizawa, K. Molybdenum-Catalyzed Transformation of Molecular Dinitrogen into Silylamine: Experimental and DFT Study on the Remarkable Role of Ferrocenyldiphosphine Ligands. J. Am. Chem. Soc. 2011, 133, 3498–3506. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; Saffon-Merceron, N.; Mézailles, N. Catalytic Dinitrogen Reduction at the Molybdenum Center Promoted by a Bulky Tetradentate Phosphine Ligand. Angew. Chem. Int. Ed. 2014, 53, 14206–14210. [Google Scholar] [CrossRef] [PubMed]
- Yuki, M.; Tanaka, H.; Sasaki, K.; Miyake, Y.; Yoshizawa, K.; Nishibayashi, Y. Iron-catalysed transformation of molecular dinitrogen into silylamine under ambient conditions. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Ung, G.; Peters, J.C. Low Temperature N2 Binding to 2-coordinate L2Fe0 Enables Reductive Trapping of L2FeN2− and NH3 Generation. Angew. Chem. Int. Ed. 2015, 54, 532–535. [Google Scholar]
- Lloyd, L. Handbook of Industrial Catalysts; Springer: New York, NY, USA, 2011. [Google Scholar]
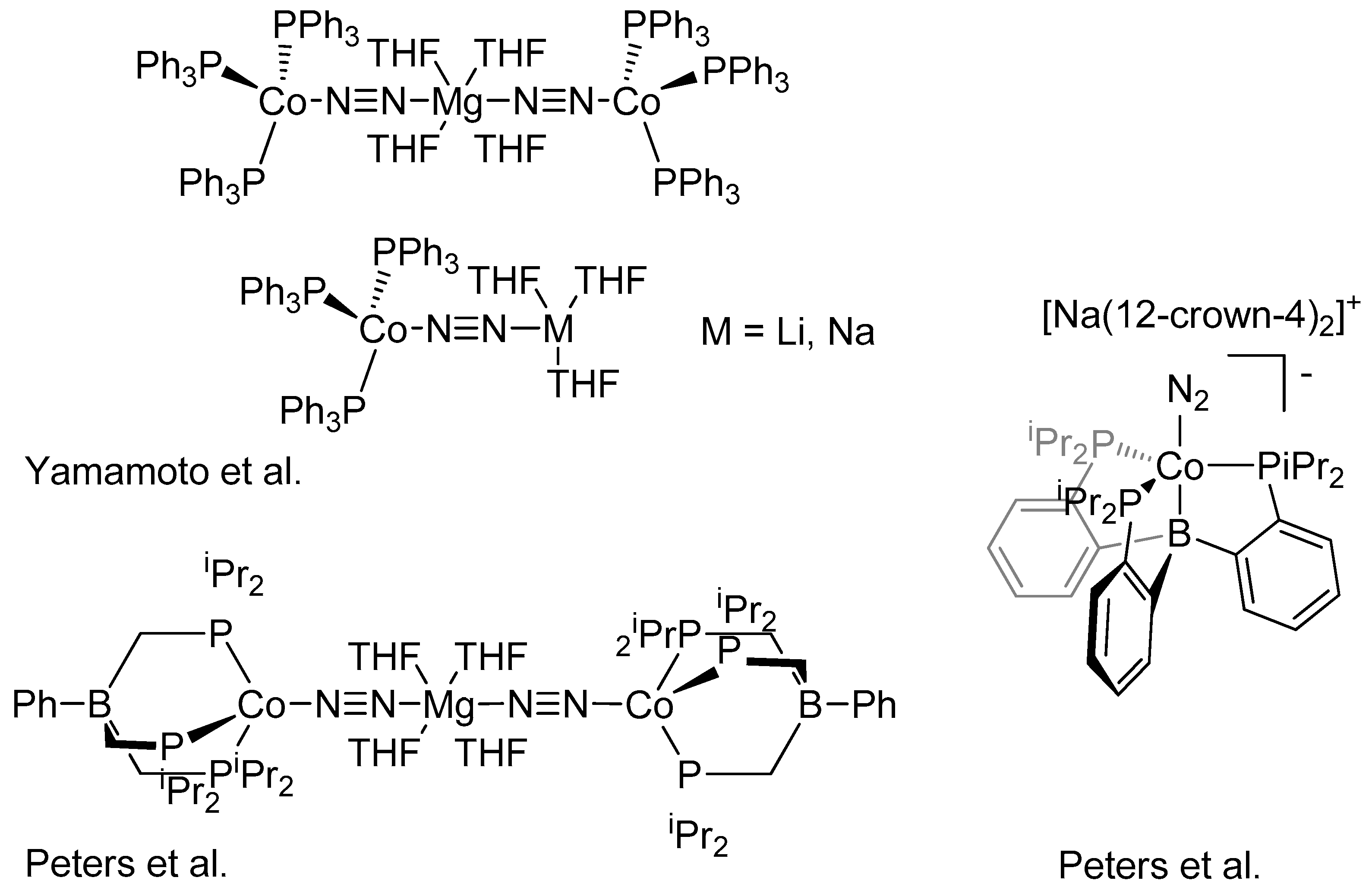
- Yamamoto, A.; Miura, Y.; Ito, T.; Chen, H.; Iri, K.; Ozawa, F. Preparation, X-ray molecular structure determination, and chemical properties of dinitrogen-coordinated cobalt complexes containing triphenylphosphine ligands and alkali metal or magnesium. Protonation of the coordinated dinitrogen to ammonia and hydrazine. Organometallics 1983, 2, 1429–1436. [Google Scholar] [CrossRef]
- Betley, T.A.; Peters, J.C. Dinitrogen Chemistry from Trigonally Coordinated Iron and Cobalt Platforms. J. Am. Chem. Soc. 2003, 125, 10782–10783. [Google Scholar] [CrossRef] [PubMed]
- Siedschlag, R.B.; Bernales, V.; Vogiatzis, K.D.; Planas, N.; Clouston, L.J.; Bill, E.; Gagliardi, L.; Lu, C.C. Catalytic Silylation of Dinitrogen with a Dicobalt Complex. J. Am. Chem. Soc. 2015, 137, 4638–4641. [Google Scholar] [CrossRef] [PubMed]
- Imayoshi, R.; Tanaka, H.; Matsuo, Y.; Yuki, M.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Cobalt-Catalyzed Transformation of Molecular Dinitrogen into Silylamine under Ambient Reaction Conditions. Chem. Eur. J. 2015, 21, 8905–8909. [Google Scholar] [CrossRef] [PubMed]
- Del Castillo, T.J.; Thompson, N.B.; Suess, D.L.M.; Ung, G.; Peters, J. Evaluating Molecular Cobalt Complexes for the Conversion of N2 to NH3. Inorg. Chem. 2015, 54, 9256–9262. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Workman, P.S. Accessing Lanthanide Diiodide Reactivity for Coupling Alkyl Chlorides to Carbonyl Compounds via the NdI3/Alkali Metal Reduction System. Organometallics 2005, 24, 1989–1991. [Google Scholar] [CrossRef]
- Jenkins, D.M.; Di Bilio, A.J.; Allen, M.J.; Betley, T.A.; Peters, J.C. Elucidation of a Low Spin Cobalt(II) System in a Distorted Tetrahedral Geometry. J. Am. Chem. Soc. 2002, 124, 15336–15350. [Google Scholar] [CrossRef] [PubMed]
- Korstanje, T.J.; van der Vlugt, J.I.; Elsevier, C.J.; de Bruin, B. Hydrogenation of carboxylic acids with a homogeneous cobalt catalyst. Science 2015, 360, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Hojilla Atienza, C.C.; Milsmann, C.; Semproni, S.P.; Turner, Z.R.; Chirik, P.J. Reversible Carbon–Carbon Bond Formation Induced by Oxidation and Reduction at a Redox-Active Cobalt Complex. Inorg. Chem. 2013, 52, 5403–5417. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Kitazume, S.; Pu, L.S.; Ikeda, S. Synthesis and properties of hydridodinitrogentris(triphenylphosphine)cobalt(I) and the related phosphine-cobalt complexes. J. Am. Chem. Soc. 1971, 93, 371–380. [Google Scholar] [CrossRef]
- Aresta, M.; Rossi, M.; Sacco, A. Tetrahedral complexes of cobalt(I). Inorg. Chim. Acta 1969, 3, 227–231. [Google Scholar] [CrossRef]
- Grutters, M.M.P.; Müller, C.; Vogt, D. Highly Selective Cobalt-Catalyzed Hydrovinylation of Styrene. J. Am. Chem. Soc. 2006, 128, 7414–7415. [Google Scholar] [CrossRef] [PubMed]
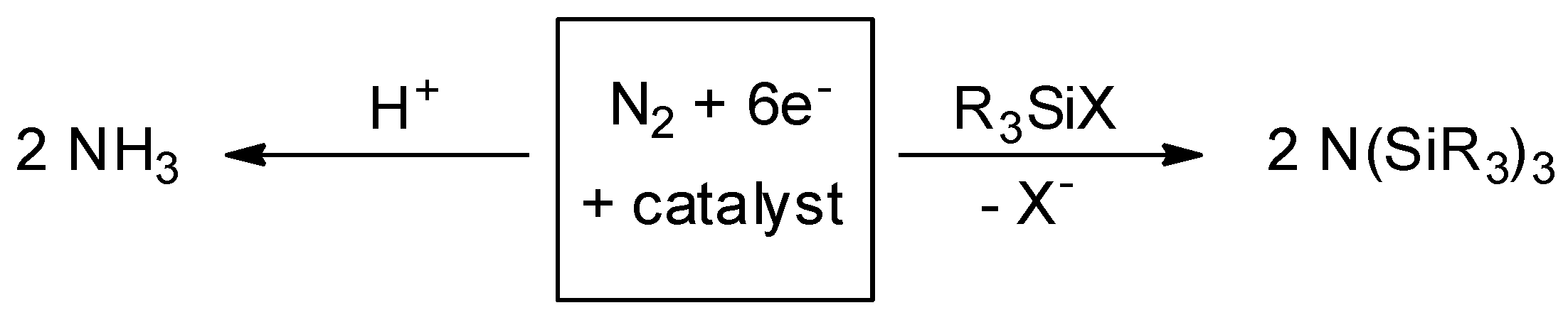
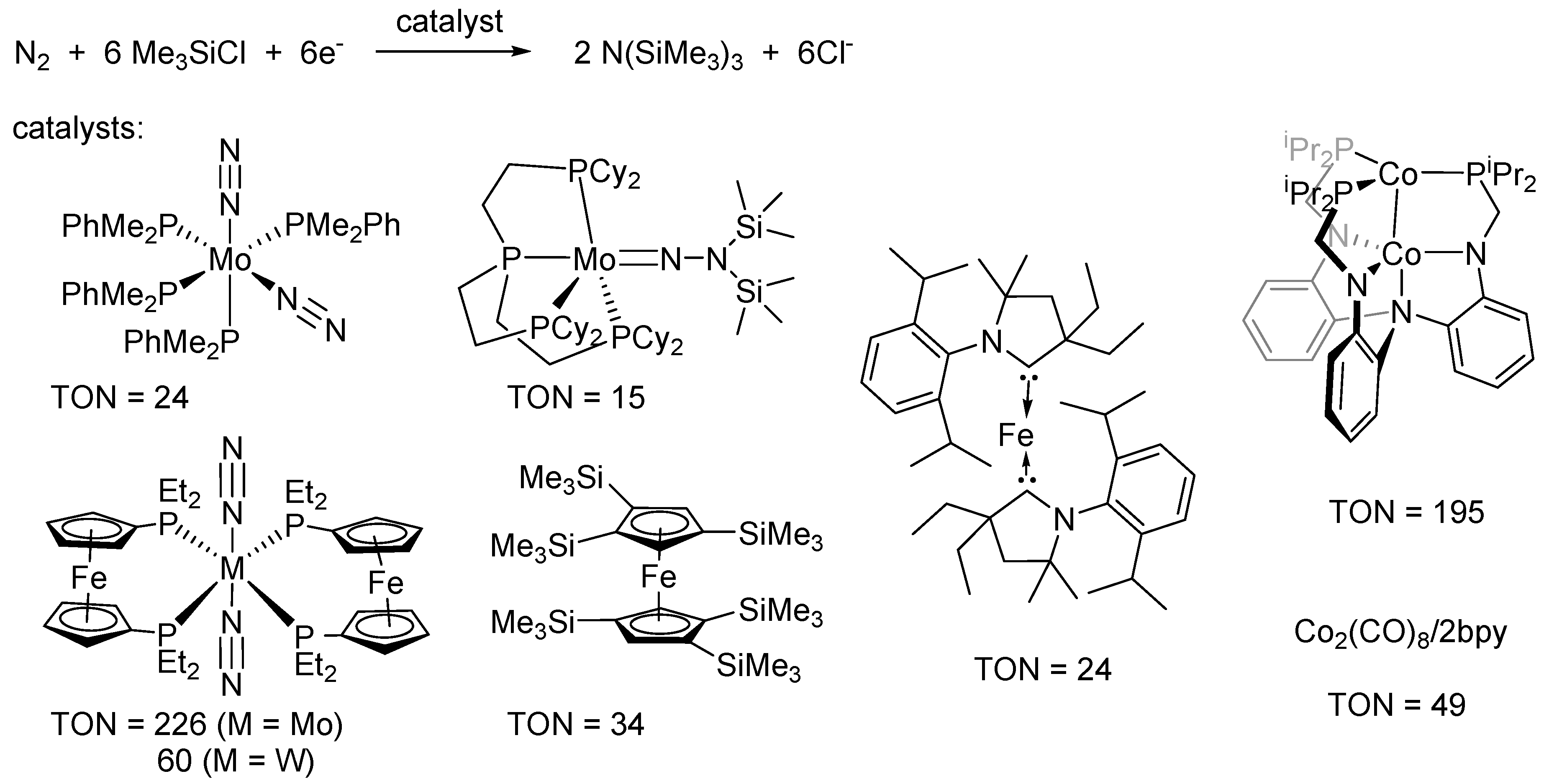

{kind=link}
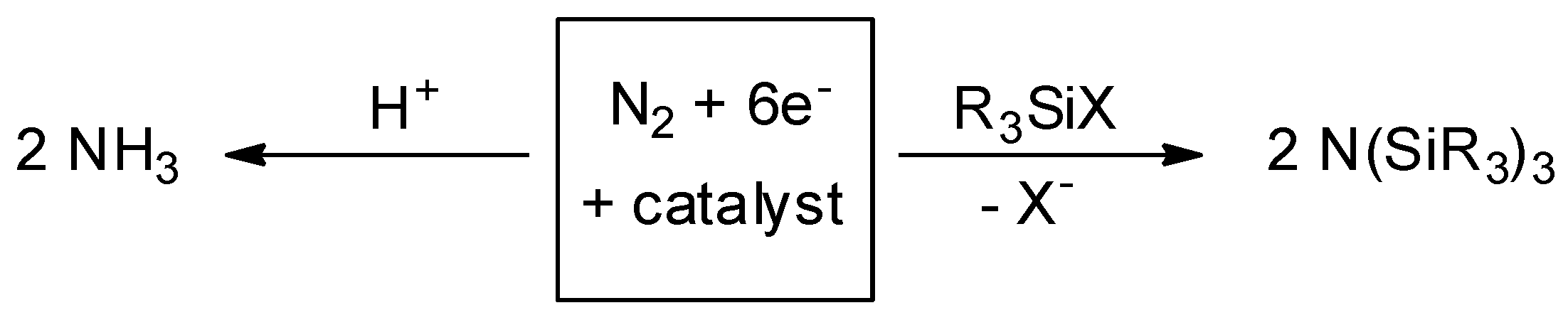{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Catalyst | Solvent | Vol./mL | Reduct. | Yield/% b | TON c |
|---|---|---|---|---|---|---|
| 1 d | CoH(PPh3)3N2 | THF | 10 | Na | 11.7 ± 1.6 | 6.7 ± 1.0 |
| 2 e | Co(acac)3 | THF | 10 | Na | 0.9 ± 0.6 | 0.5 ± 0.4 |
| 3 e | CoBr2 | THF | 10 | Na | 0 | 0 |
| 4 | Co | THF | 10 | Na | 0 | 0 |
| 5 f | Co(Cp)2 | THF | 10 | Na | 15.9 ± 3.4 | 8.3 ± 2.3 |
| 6 e | - | THF | 10 | Na | 0 | 0 |
| 7 e | CoCl2(PPh3)2 | THF | 10 | Na | 1.2 ± 0.6 | 0.7 ± 0.3 |
| 8 e | CoCl(PPh3)3 | THF | 10 | Na | 6.1 ± 1.2 | 3.7 ± 0.7 |
| 9 | CoH(PPh3)3N2 | Et2O | 10 | Na | 9.1 | 5.8 |
| 10 | CoH(PPh3)3N2 | C6H6 | 10 | Na | 0 | 0 |
| 11 | CoH(PPh3)3N2 | THF | 5 | Na | 10.8 | 6.8 |
| 12 | CoH(PPh3)3N2 | THF | 20 | Na | 11.6 | 7.3 |
| 13 | CoH(PPh3)3N2 | THF | 10 | Li | 8.7 | 5.5 |
| 14 | CoH(PPh3)3N2 | THF | 10 | K | 0 | 0 |
| 15 | CoH(PPh3)3N2 | THF | 10 | KC8 | 5.8 | 3.5 |
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dzik, W.I. Silylation of Dinitrogen Catalyzed by Hydridodinitrogentris(Triphenylphosphine)Cobalt(I). Inorganics 2016, 4, 21. https://doi.org/10.3390/inorganics4030021
Dzik WI. Silylation of Dinitrogen Catalyzed by Hydridodinitrogentris(Triphenylphosphine)Cobalt(I). Inorganics. 2016; 4(3):21. https://doi.org/10.3390/inorganics4030021
Chicago/Turabian StyleDzik, Wojciech I. 2016. "Silylation of Dinitrogen Catalyzed by Hydridodinitrogentris(Triphenylphosphine)Cobalt(I)" Inorganics 4, no. 3: 21. https://doi.org/10.3390/inorganics4030021
APA StyleDzik, W. I. (2016). Silylation of Dinitrogen Catalyzed by Hydridodinitrogentris(Triphenylphosphine)Cobalt(I). Inorganics, 4(3), 21. https://doi.org/10.3390/inorganics4030021