
Monoanionic Tin Oligomers Featuring Sn–Sn or Sn–Pb Bonds: Synthesis and Characterization of a Tris(Triheteroarylstannyl)Stannate and -Plumbate

Abstract
:1. Introduction
2. Results
3. Experimental
3.1. Syntheses
3.2. X-ray Crystallography
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Davies, A.G. Recent advances in the chemistry of the organotin hydrides. J. Chem. Res. 2006, 2006, 141–148. [Google Scholar] [CrossRef]
- Jurkschat, K.; Mehring, M. Organometallic polymers of germanium, tin and lead. In The Chemistry of Organic Germanium, Tin and Lead Compounds; Rappoport, Z., Ed.; Wiley: Chichester, UK; New York, NY, USA, 2002; Volume 2, pp. 1–130. [Google Scholar]
- Amadoruge, M.L.; Weinert, C.S. Singly bonded catenated germanes: Eighty years of progress. Chem. Rev. 2008, 108, 4253–4294. [Google Scholar] [CrossRef] [PubMed]
- Weinert, C.S. Syntheses, structures and properties of linear and branched oligogermanes. Dalton Trans. 2009, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Marschner, C.; Hlina, J. Catenated Compounds–Group 14 (Ge, Sn, Pb). In Comprehensive Inorganic Chemistry II; Reedijk, J., Poeppelmeier, K., Eds.; Elsevier B.V.: Amsterdam, the Netherlands, 2013; Volume 1, pp. 83–117. [Google Scholar]
- Gillman, H.; Cartledge, F.K. Tetrakis(triphenylstannyl)tin. J. Organomet. Chem. 1966, 5, 48–56. [Google Scholar] [CrossRef]
- Jurkschat, K.; Klaus, C.; Dargatz, M.; Tzschach, A.; Meunier-Piret, J.; Mahieu, B. Reaction of 3-dimethylamino-(1,1-dimethyl)propyl magnesium chloride with tin(IV) and tin(II) chlorides. Stabilization of a SnCl+ cation in the new tin cluster [Me2NCH2CH2C(Me2)SnCl]3·SnCl2. Z. Anorg. Allg. Chem. 1989, 577, 122–134. [Google Scholar] [CrossRef]
- Sita, L.R. Structure/property relationships of polystannanes. Adv. Organomet. Chem. 1995, 38, 189–243. [Google Scholar]
- Willemsens, L.C.; van der Kerk, G.J.M. Studies in group IV organometallic chemistry: XIII. Organometallic compounds with five metal atoms in neopentane configuration. J. Organomet. Chem. 1964, 2, 260–264. [Google Scholar] [CrossRef]
- Willemsens, L.C.; van der Kerk, G.J.M. Investigations on organolead compounds: I. A novel red organolead compound a reinvestigation of krause’s red diphenyllead. J. Organomet. Chem. 1964, 2, 271–276. [Google Scholar] [CrossRef]
- Stabenow, F.; Saak, W.; Weidenbruch, M. Tris(triphenylplumbyl)plumbate: An anion with three stretched lead–lead bonds. Chem. Commun. 2003, 18, 2342–2343. [Google Scholar] [CrossRef]
- Wang, Y.; Quillian, B.; Wei, P.; Yang, X.-J.; Robinson, G.H. New Pb–Pb bonds: Syntheses and molecular structures of hexabiphenyldiplumbane and tri(trisbiphenylplumbyl)plumbate. Chem. Commun. 2004, 19, 2224–2225. [Google Scholar] [CrossRef] [PubMed]
- Wells, W.L.; Brown, T.L. An investigation of methyltin–lithium compounds preparation and properties of tris(trimethylstannyl)stannyllithium tris(tetrahydrofuran). J. Organomet. Chem. 1968, 11, 271–280. [Google Scholar] [CrossRef]
- Kobayashi, K.; Kawanisi, M.; Hitomi, T.; Kozima, S. Mechanistic studies of decomposition of trialkylstannyllithiums. J. Organomet. Chem. 1982, 233, 299–311. [Google Scholar] [CrossRef]
- Westerhausen, M. Synthesis and crystal structure of calcium bis(trimethylstannanide)·4 THF. Angew. Chem. Int. Ed. Engl. 1994, 33, 1493–1495. [Google Scholar] [CrossRef]
- Englich, U.; Ruhlandt-Senge, K.; Uhlig, F. Novel triphenyltin substituted derivatives of heavier alkaline earth metals. J. Organomet. Chem. 2000, 613, 139–147. [Google Scholar] [CrossRef]
- Flacke, F.; Jacobs, H. [Li(NH3)4][Sn(SnPh3)3]·C6H6, crystal structure of a stannide with trigonal pyramidal tin skeleton. Eur. J. Solid State Inorg. Chem. 1997, 34, 495–501. [Google Scholar]
- Bochkarev, L.N.; Grachev, O.V.; Ziltsov, S.F.; Zakharov, L.N.; Struchkov, Y.T. Synthesis and structure of organotin complexes of ytterbium. J. Organomet. Chem. 1992, 436, 299–311. [Google Scholar] [CrossRef]
- Bochkarev, L.N.; Grachev, O.V.; Molosnova, N.E.; Ziltsov, S.F.; Zakharov, L.N.; Fukin, G.K. Novel polynuclear organotin complexes of samarium and ytterbium. J. Organomet. Chem. 1993, 443, C26–C28. [Google Scholar] [CrossRef]
- Drost, C.; Hildebrand, M.; Lönnecke, P. Synthesis and crystal structure of a novel distannylstannanediyl and a rare pentastannapropellane. Main Group Met. Chem. 2002, 25, 93–98. [Google Scholar] [CrossRef]
- Eichler, B.E.; Phillips, A.D.; Power, P.P. Reactions of phenyllithium with the stannylene Ar*SnPh (Ar* = C6H3-2,6-Trip2; Trip = C6H2-2,4,6-iPr3) and the synthesis of the distannylstannylene Sn(SnPh2Ar*)2: Contrasting behavior in methyl and phenyl derivatives. Organometallics 2003, 22, 5423–5426. [Google Scholar] [CrossRef]
- Zeckert, K.; Zahn, S.; Kirchner, B. Tin–lanthanoid donor–acceptor bonds. Chem. Commun. 2010, 46, 2638–2640. [Google Scholar] [CrossRef]
- Zeckert, K. Syntheses and structures of lanthanoid(II) complexes featuring Sn–M (M = Al, Ga, In) bonds. Dalton Trans. 2012, 41, 14101–14106. [Google Scholar] [CrossRef] [PubMed]
- Reichart, F.; Kischel, M.; Zeckert, K. Lanthanide(II) complexes of a dual functional tris(2-pyridyl)stannate derivative. Chem. Eur. J. 2009, 15, 10018–10020. [Google Scholar] [CrossRef] [PubMed]
- Leung, W.-P.; Weng, L.-H.; Kwok, W.-H.; Zhou, Z.-Y.; Zhang, Z.-Y.; Mak, T.C.W. Synthesis of a novel binuclear chlorotin(II) alkyl and a lithium trialkylstannate zwitterionic cage molecule: Crystal structures of [Sn(Cl)RN]2 and [(SnRN3)Li(μ3-Cl)(Li(tmeda)]2 [RN = CH(SiButMe2)C5H4N-2]. Organometallics 1999, 18, 1482–1485. [Google Scholar] [CrossRef]
- Drost, C.; Lönnecke, P.; Sieler, J. Stannylplumbylenes: Bonding between tetravalent tin and divalent lead. Chem. Commun. 2012, 48, 3778–3780. [Google Scholar] [CrossRef] [PubMed]
- Cardin, C.J.; Cardin, D.J.; Constantine, S.P.; Todd, A.K.; Teat, S.J.; Coles, S. The first structurally authenticated compound containing a bond between divalent tin and tetravalent tin. Organometallics 1998, 17, 2144–2146. [Google Scholar] [CrossRef]
- Phillips, A.D.; Hino, S.; Power, P.P. A reversible valence equilibrium in a heavier main group compound. J. Am. Chem. Soc. 2003, 125, 7520–7521. [Google Scholar] [CrossRef] [PubMed]
- Setaka, W.; Hirai, K.; Tomioka, H.; Sakamoto, K.; Kira, M. Formation of a stannylstannylene via intramolecular carbene addition of a transient stannaacetylene (RSn≡CR′). Chem. Commun. 2008, 28, 6558–6560. [Google Scholar] [CrossRef] [PubMed]
- Henning, J.; Eichele, K.; Fink, R.F.; Wesemann, L. Structural and spectroscopic characterization of tin–tin double bonds in cyclic distannenes. Organometallics 2014, 33, 3904–3918. [Google Scholar] [CrossRef]
- Jurkschat, K.; Abicht, H.-P.; Tzschach, A.; Mahieu, B. Zur Umsetzung von o-Brommagnesiumbenzyldiphenylphosphin mit SnCl2; Isolierung einer Spezies mit direkter SnII–SnIV-Bindung. J. Organomet. Chem. 1986, 309, C47–C50. [Google Scholar] [CrossRef]
- Kennedy, J.D.; McFarlane, W. Nuclear spin–spin coupling between directly bound tin atoms studied by a novel form of INDOR spectroscopy. Dalton Trans. 1976, 1219–1223. [Google Scholar] [CrossRef]
- Schneider, C.; Dräger, M. Über gemischte Gruppe 14-Gruppe 14-Bindungen IV. Hexa-p-tolylethan-Analoga p-ToI6Sn2, p-ToI6PbSn und p-ToI6Pb2: Darstellung, Homöotype Strukturen, NMR-Kopplungen und Valenzschwingungen. J. Organomet. Chem. 1991, 415, 349–362. [Google Scholar] [CrossRef]
- Schneider, C.; Behrends, K.; Dräger, M. Über gemischte Gruppe 14–Gruppe 14-Bindungen: VI. Hexa-o-tolylethan-Analoga o-ToI6Sn2, o-ToI6PbSn und o-ToI6Pb2: Ein Vergleich von Bindungsstärke und Polarität in der Reihung Sn–Sn, Pb–Sn, Pb–Pb. J. Organomet. Chem. 1993, 448, 29–38. [Google Scholar] [CrossRef]
- Riviere, P.; Castel, A.; Riviere-Baudet, M. Alkaline and alkaline earth metal-14 compounds: Preparation, spectroscopy, structure and reactivity. In The Chemistry of Organic Germanium, Tin and Lead Compounds; Rappoport, Z., Ed.; Wiley: Chichester, UK, New York, NY, USA, 2002; Volume 2, pp. 653–748. [Google Scholar]
- Zeckert, K.; Griebel, J.; Kirmse, R.; Weiß, M.; Denecke, R. Versatile reactivity of a lithium tris(aryl)plumbate(II) towards organolanthanoid compounds: Stable lead–lanthanoid-metal bonds or redox processes. Chem. Eur. J. 2013, 19, 7718–7722. [Google Scholar] [CrossRef] [PubMed]
- García-Rodríguez, R.; Wright, D.S. Direct synthesis of the Janus-head ligand (MePy)3Sn–Sn(MePy)3 using an unusual pyridyl-transfer reaction (MePy = 6-methyl-2-pyridyl). Dalton Trans. 2014, 43, 14529–14532. [Google Scholar] [CrossRef] [PubMed]
- Gynane, M.J.S.; Harris, D.H.; Lappert, M.F.; Power, P.P.; Rivière, P.; Rivière-Baudet, M. Subvalent group 4B metal alkyls and amides. Part 5. The synthesis and physical properties of thermally stable amides of germanium(II), tin(II), and lead(II). Dalton Trans. 1977, 2004–2009. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXS-97 and SHELXL-97, Programs for Crystal Structure Analysis; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]

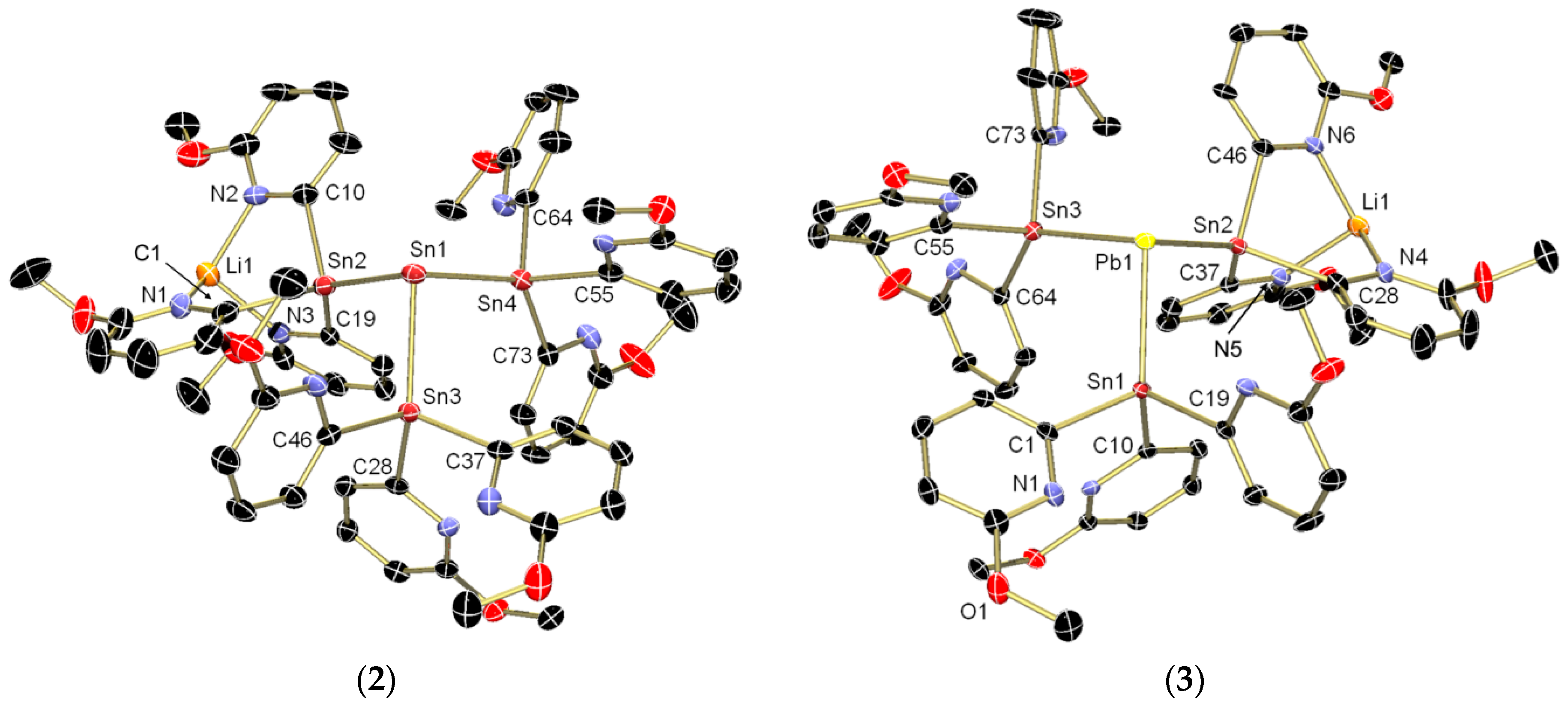

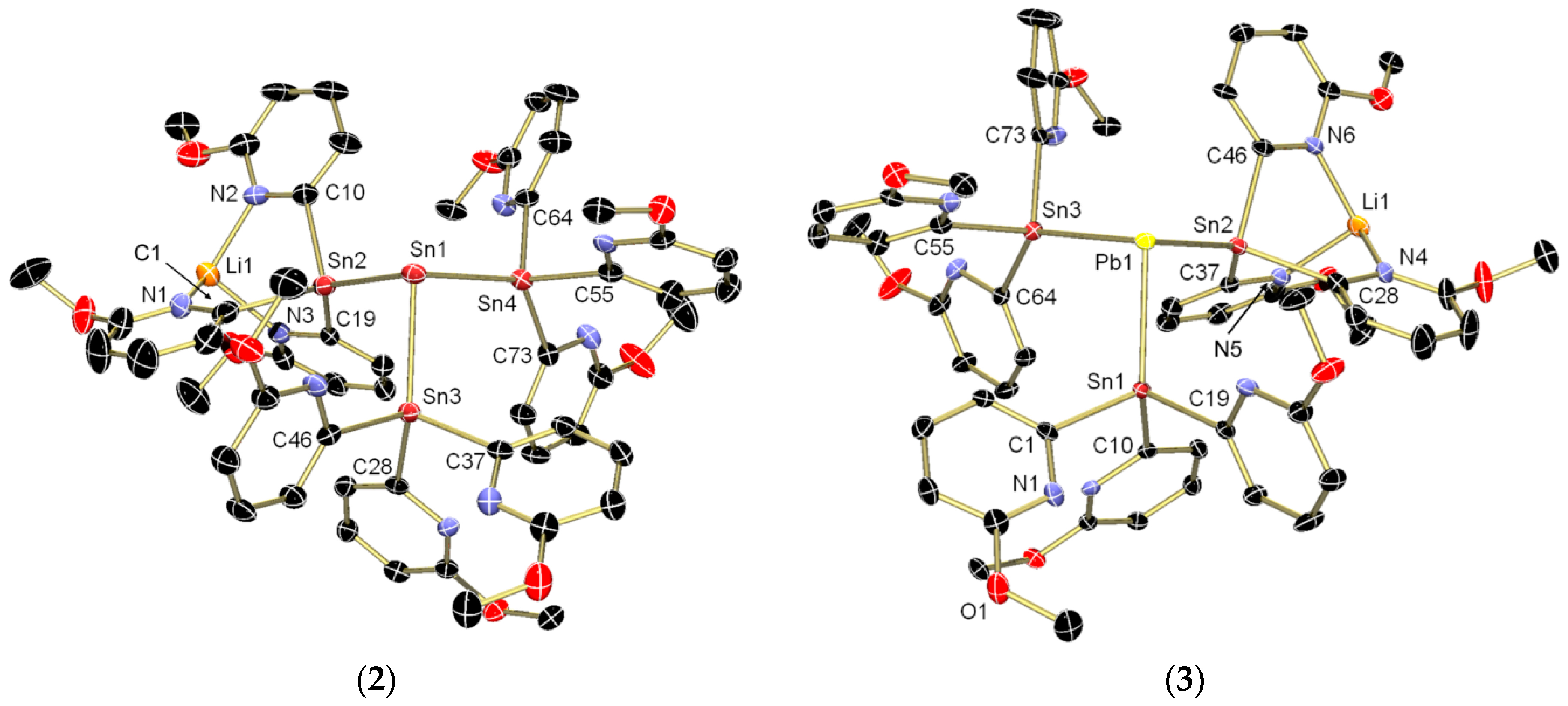{kind=link}
{kind=link}
| 2 | 3 | ||
|---|---|---|---|
| Sn(1)–Sn(2) | 2.8127(7) | Pb(1)–Sn(1) | 2.8743(4) |
| Sn(1)–Sn(3) | 2.8181(6) | Pb(1)–Sn(2) | 2.8919(4) |
| Sn(1)–Sn(4) | 2.8217(6) | Pb(1)–Sn(3) | 2.8818(4) |
| Sn(2)···Li(1) | 3.339(14) | Sn(2)···Li(1) | 3.321(10) |
| Sn(2)–Cipso | 2.168(7), 2.179(7), 2.184(8) | Sn(1)–Cipso | 2.154(5), 2.167(6), 2.182(5) |
| Sn(3)–Cipso | 2.161(6), 2.165(7), 2.183(6) | Sn(2)–Cipso | 2.172(6), 2.183(6), 2.191(6) |
| Sn(4)–Cipso | 2.163(6), 2.167(6), 2.172(7) | Sn(3)–Cipso | 2.167(6), 2.169(5), 2.177(5) |
| Li(1)–N | 2.024(13)–2.038(15) | Li(1)–N | 1.996(11)–2.030(11) |
| Sn(2)–Sn(1)–Sn(3) | 92.52(2) | Sn(1)–Pb(1)–Sn(2) | 90.90(1) |
| Sn(2)–Sn(1)–Sn(4) | 93.22(2) | Sn(1)–Pb(1)–Sn(3) | 94.49(1) |
| Sn(3)–Sn(1)–Sn(4) | 96.13(2) | Sn(2)–Pb(1)–Sn(3) | 90.85(1) |
| Cipso–Sn(2)–Cipso | 93.8(2), 98.9(3), 99.8(3) | Cipso–Sn(1)–Cipso | 101.2(2), 103.2(2), 104.8(2) |
| Cipso–Sn(3)–Cipso | 100.8(2), 102.8(2), 104.7(2) | Cipso–Sn(2)–Cipso | 92.4(2), 98.6(2), 100.0(2) |
| Cipso–Sn(4)–Cipso | 100.4(2), 101.2(3), 103.6(2) | Cipso–Sn(3)–Cipso | 101.8(2), 103.3(2), 104.3(2) |
| Cipso–Sn(2)–Sn(1) | 112.1(2), 114.0(2), 132.5(2) | Cipso–Sn(1)–Pb(1) | 109.2(1), 112.5(1), 123.8(1) |
| Cipso–Sn(3)–Sn(1) | 109.2(2),112.6(2), 124.5(1) | Cipso–Sn(2)–Pb(1) | 111.8(2), 113.7(2), 134.1(2) |
| Cipso–Sn(4)–Sn(1) | 106.0(2), 113.0(2), 129.7(2) | Cipso–Sn(3)–Pb(1) | 104.6(1), 107.9(1), 132.0(1) |
| 2 | 3 | |
|---|---|---|
| Formula | C81H108LiN9O9Sn4 | C81H108LiN9O9PbSn3 |
| Mr (g·mol−1) | 1833.46 | 1921.96 |
| Cryst system | triclinic | triclinic |
| Space group | ||
| a (Å) | 16.2901(9) | 17.2766(4) |
| b (Å) | 17.2775(8) | 19.4489(6) |
| c (Å) | 19.5167(8) | 28.6285(7) |
| α (°) | 103.329(4) | 88.874(2) |
| β (°) | 101.969(4) | 85.776(2) |
| γ (°) | 118.018(5) | 65.256(3) |
| V (Å3) | 4388.4(4) | 8711.9(4) |
| Z | 2 | 4 |
| F(000) | 1864 | 3856 |
| T (K) | −180(2) | −130(2) |
| ρcalcd (g cm3) | 1.388 | 1.465 |
| μ (mm−1) | 1.181 | 2.833 |
| Reflns collected | 31672 | 74704 |
| Reflns unique | 16052 | 41501 |
| Rint | 0.0620 | 0.0391 |
| Final R1 (I > 2σ(I)) | 0.0574 | 0.0531 |
| Final wR2 (F2) (all data) | 0.1172 | 0.1050 |
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeckert, K. Monoanionic Tin Oligomers Featuring Sn–Sn or Sn–Pb Bonds: Synthesis and Characterization of a Tris(Triheteroarylstannyl)Stannate and -Plumbate. Inorganics 2016, 4, 19. https://doi.org/10.3390/inorganics4020019
Zeckert K. Monoanionic Tin Oligomers Featuring Sn–Sn or Sn–Pb Bonds: Synthesis and Characterization of a Tris(Triheteroarylstannyl)Stannate and -Plumbate. Inorganics. 2016; 4(2):19. https://doi.org/10.3390/inorganics4020019
Chicago/Turabian StyleZeckert, Kornelia. 2016. "Monoanionic Tin Oligomers Featuring Sn–Sn or Sn–Pb Bonds: Synthesis and Characterization of a Tris(Triheteroarylstannyl)Stannate and -Plumbate" Inorganics 4, no. 2: 19. https://doi.org/10.3390/inorganics4020019
APA StyleZeckert, K. (2016). Monoanionic Tin Oligomers Featuring Sn–Sn or Sn–Pb Bonds: Synthesis and Characterization of a Tris(Triheteroarylstannyl)Stannate and -Plumbate. Inorganics, 4(2), 19. https://doi.org/10.3390/inorganics4020019