
A Structural and Spectroscopic Study of the First Uranyl Selenocyanate, [Et4N]3[UO2(NCSe)5]



Abstract
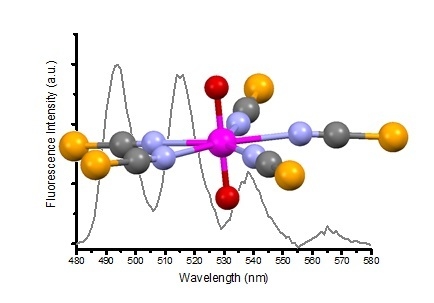
:
1. Introduction
2. Results and Discussion
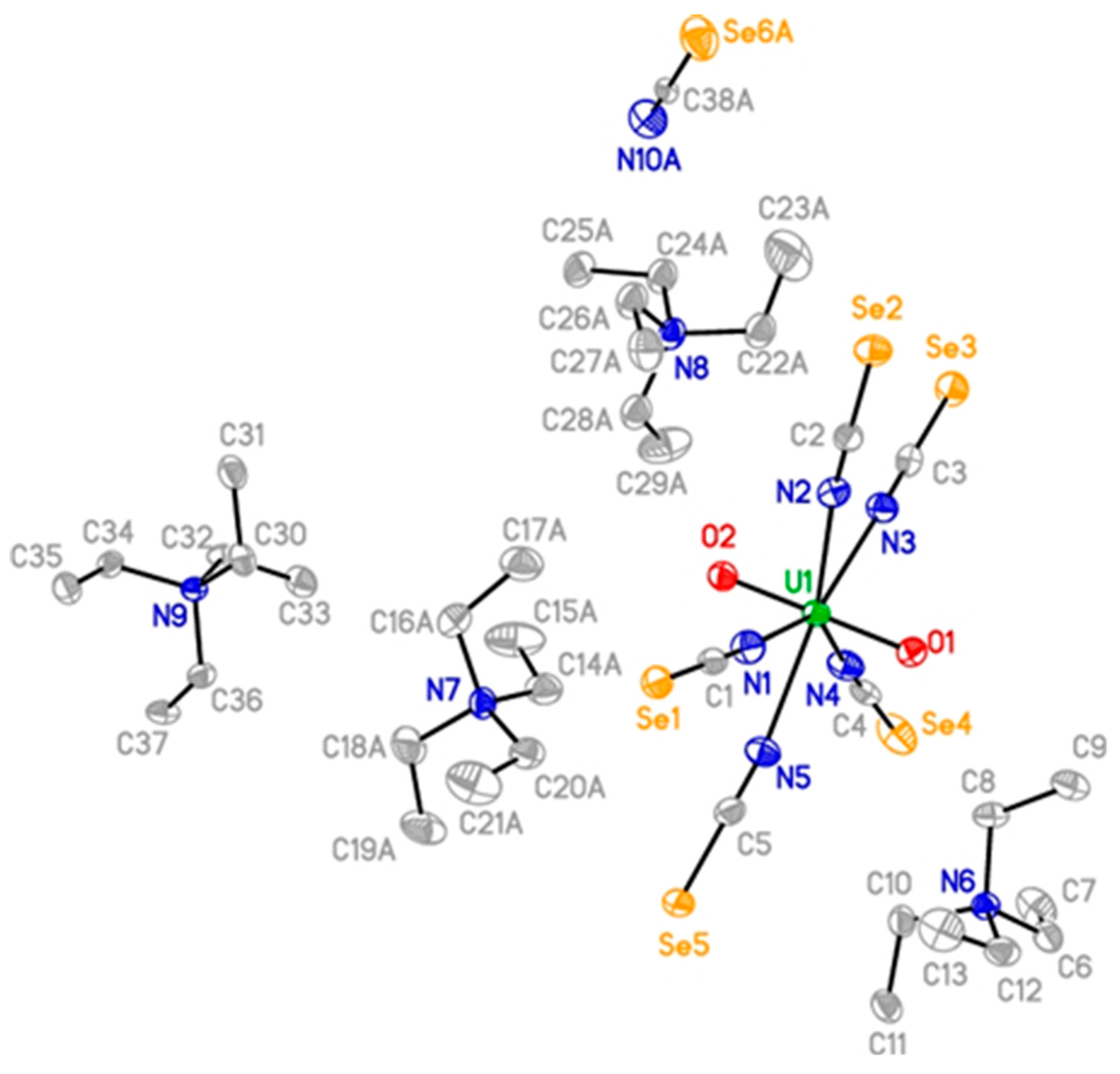
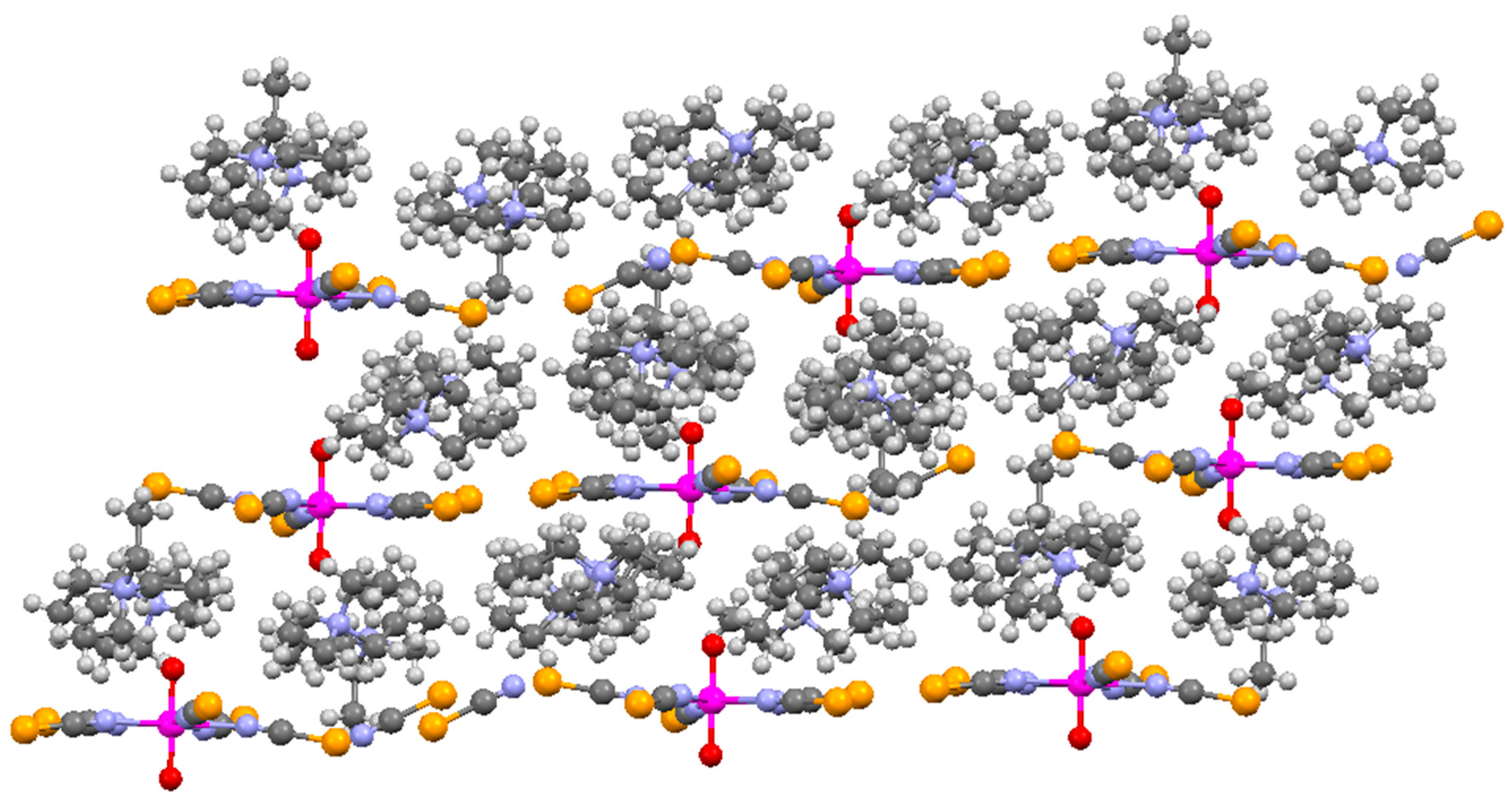
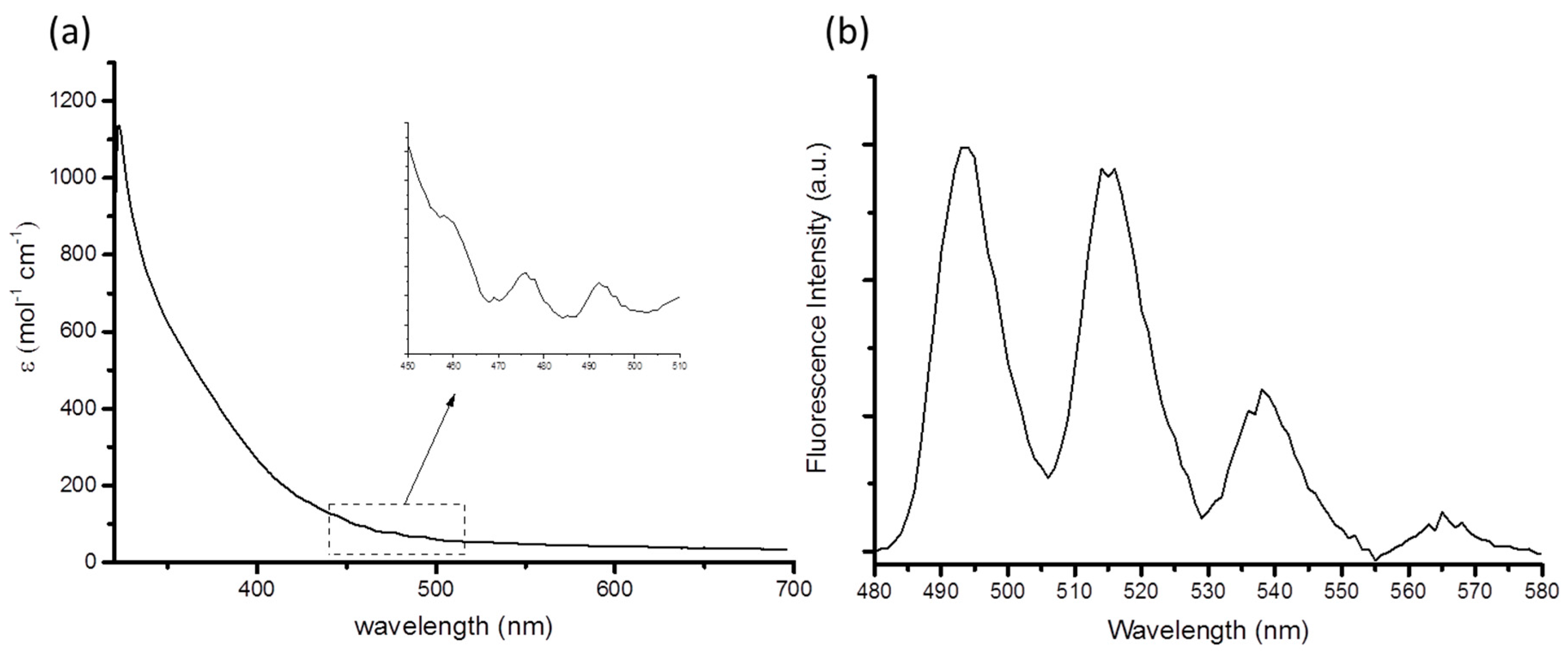

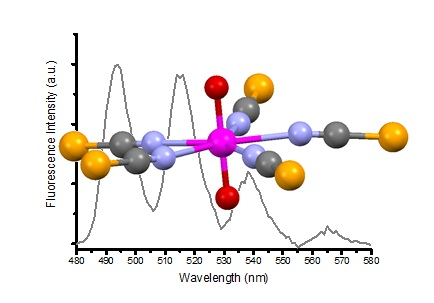{kind=link}
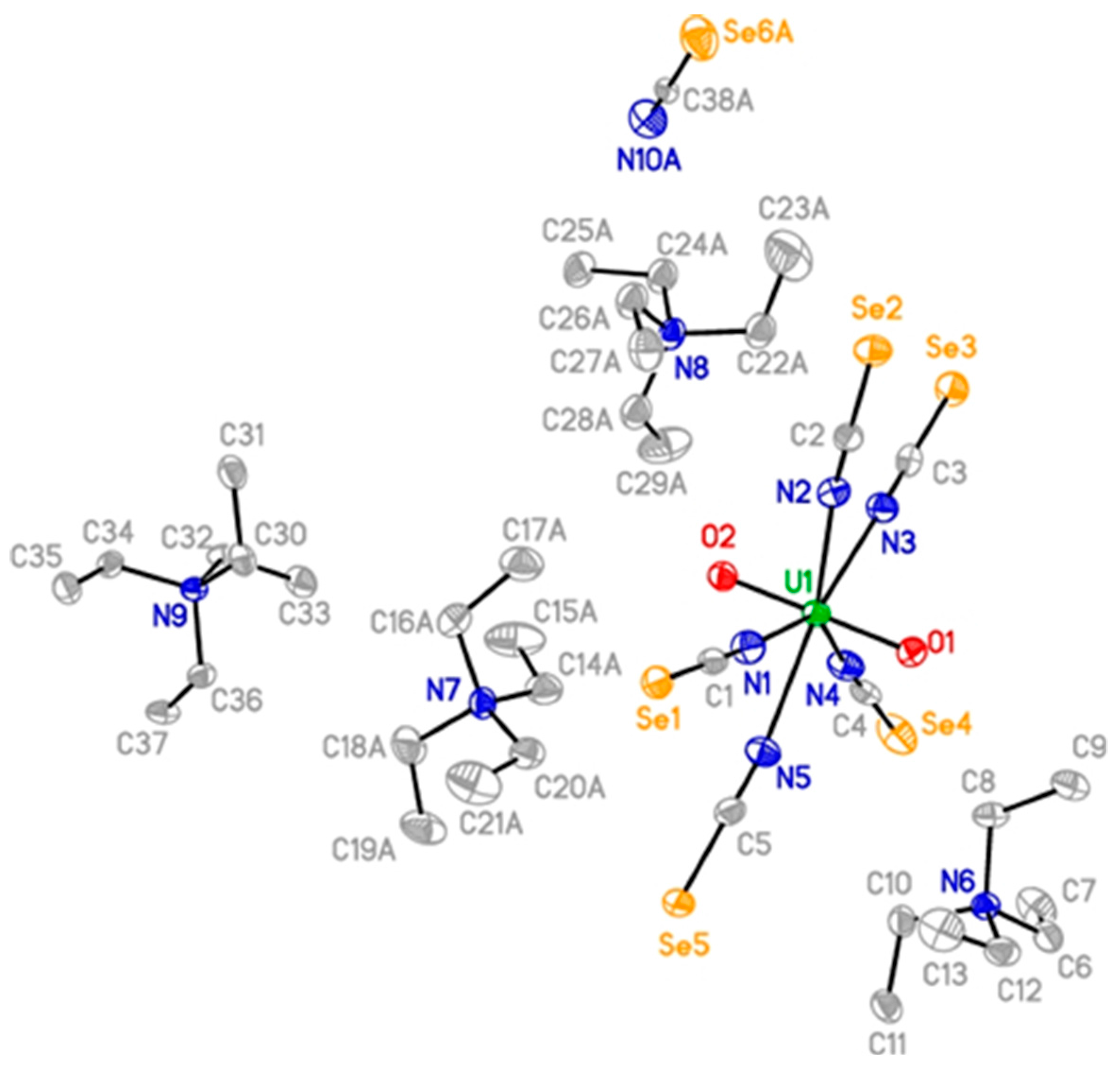{kind=link}
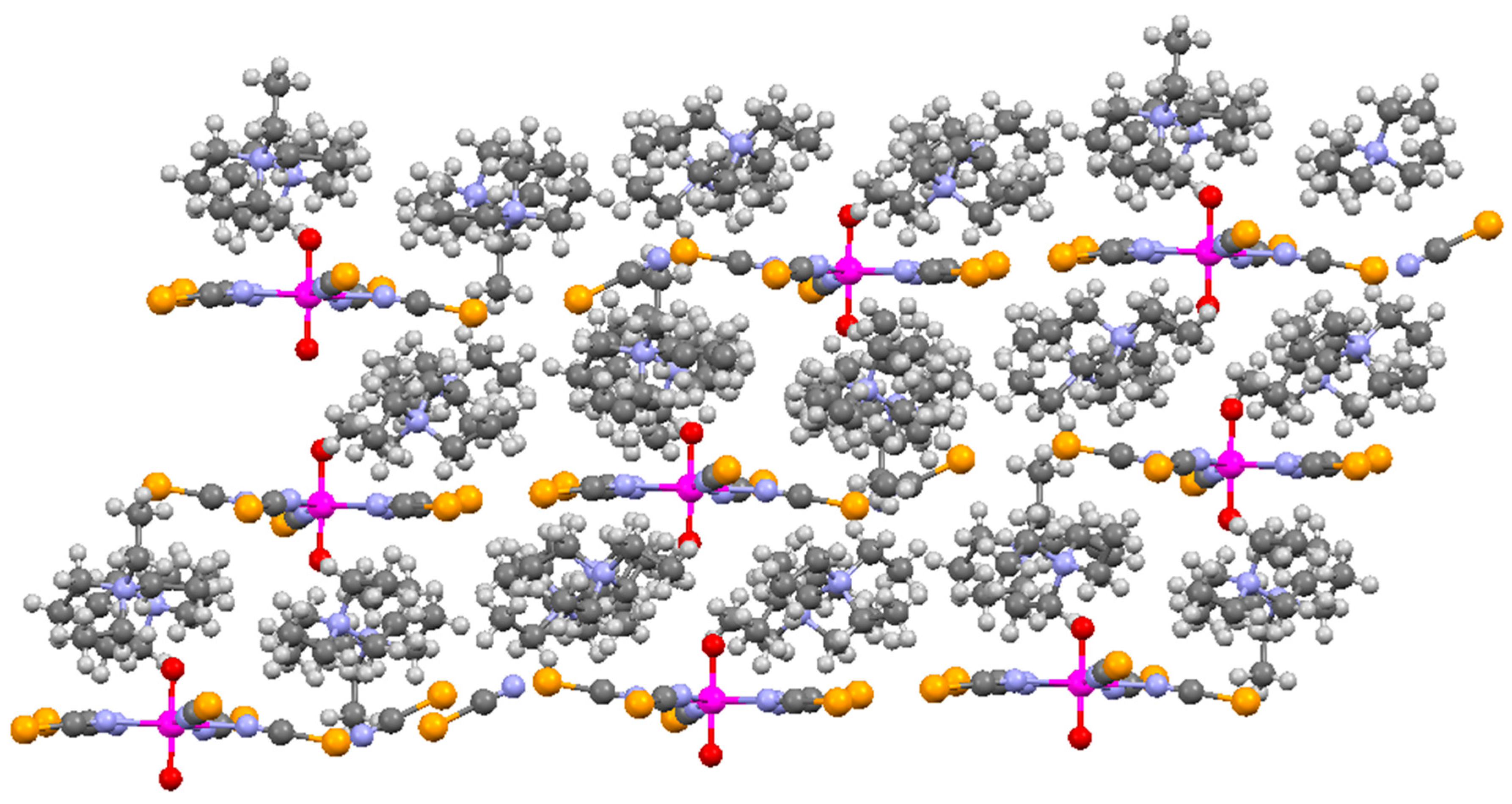{kind=link}
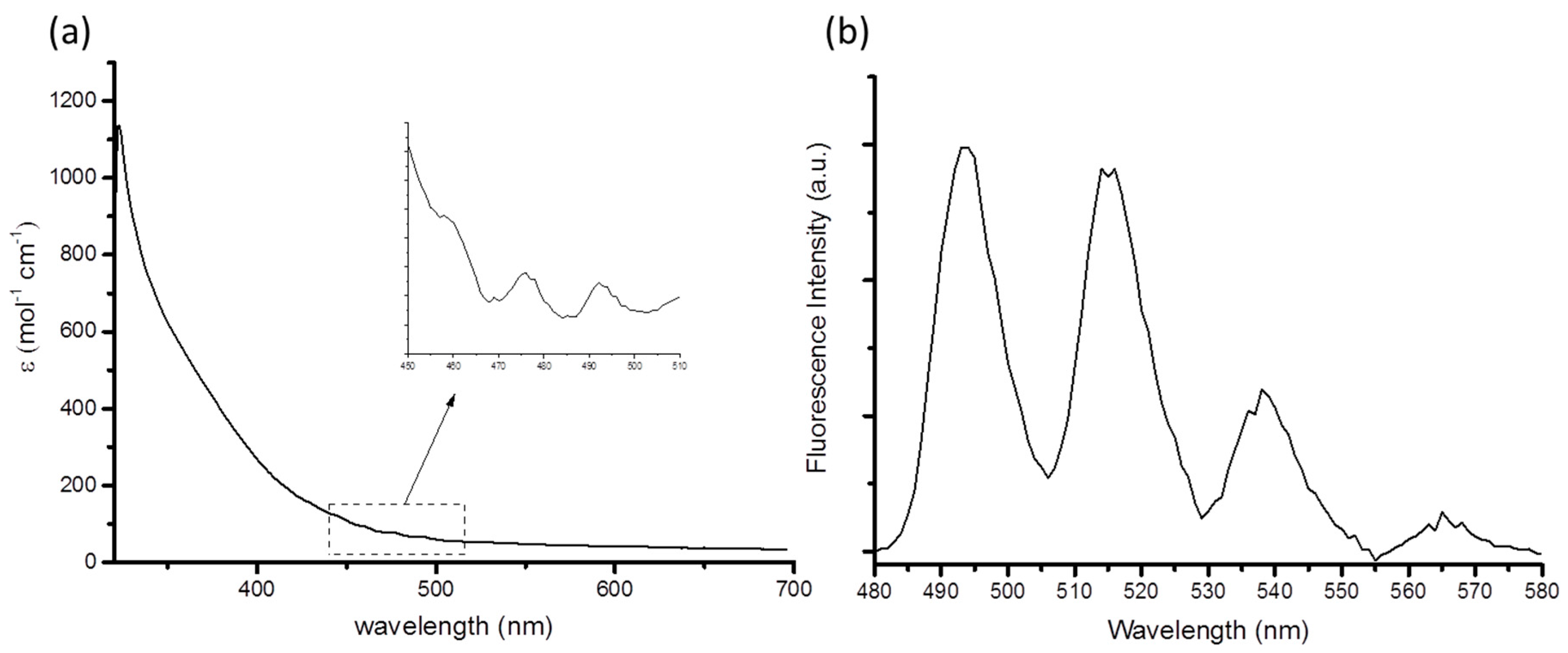{kind=link}
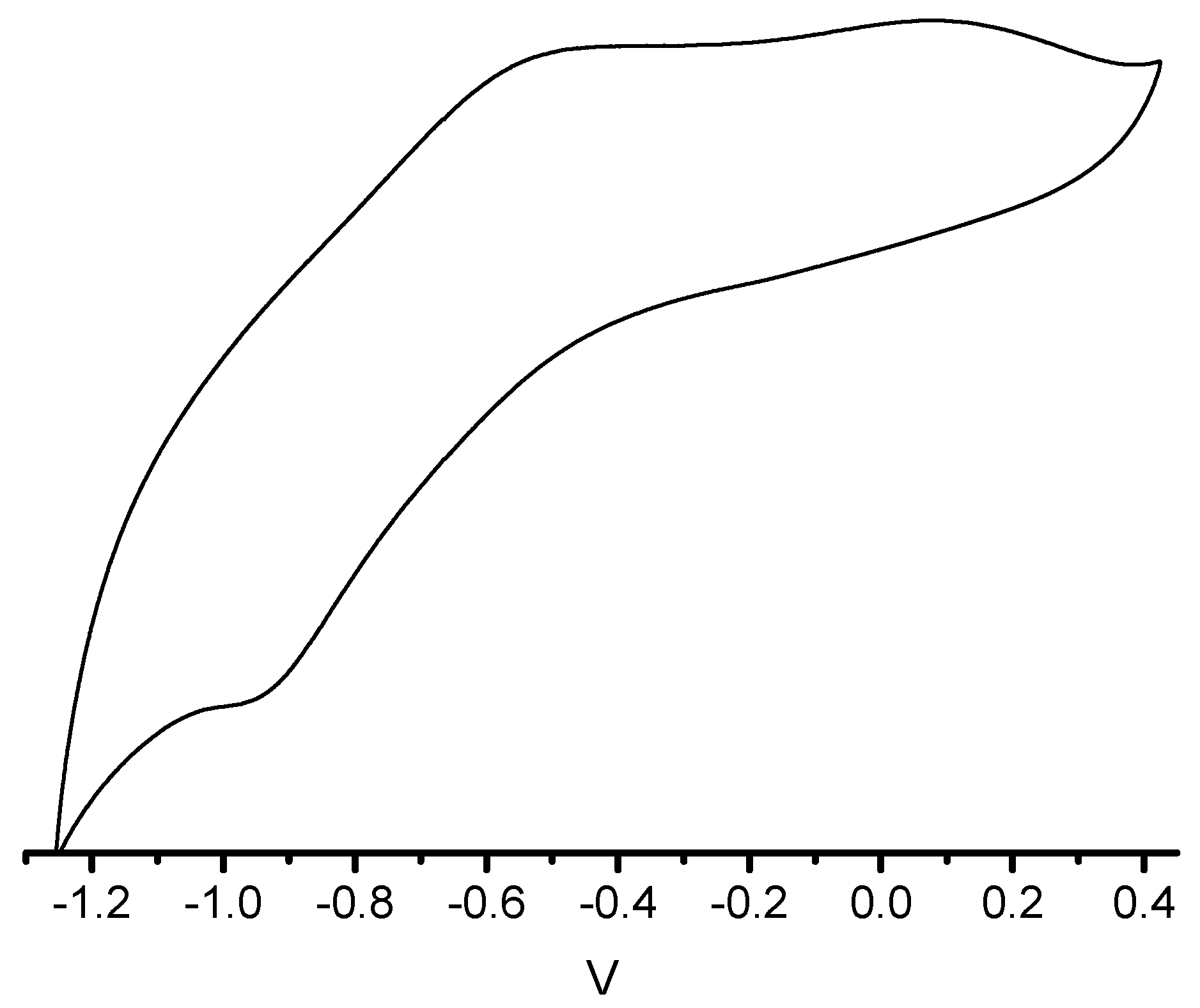{kind=link}
| Compound (Solvent) | λabs U=O (nm) | λem (nm) | E0-0 (cm−1) | τ (μs) | χ2 | Ref. |
|---|---|---|---|---|---|---|
| 1 (MeCN) | 460 | 514 | 20,267 | 1.30 | 1.40 | This work |
| [Et4N]3[UO2(NCS)5] (MeCN) | 440 | 520 | 20,072 | 1.40 | 1.02 | [11] |
| [UO2Cl2(OPPh3)2] (MeCN) | 440 | 515 | 20,325 | 1.08 | 1.07 | [8] |
| [UO2Cl4]2− (MeBu3N[Tf2N]) | 509 | 20,329 | 0.7 | [26] |
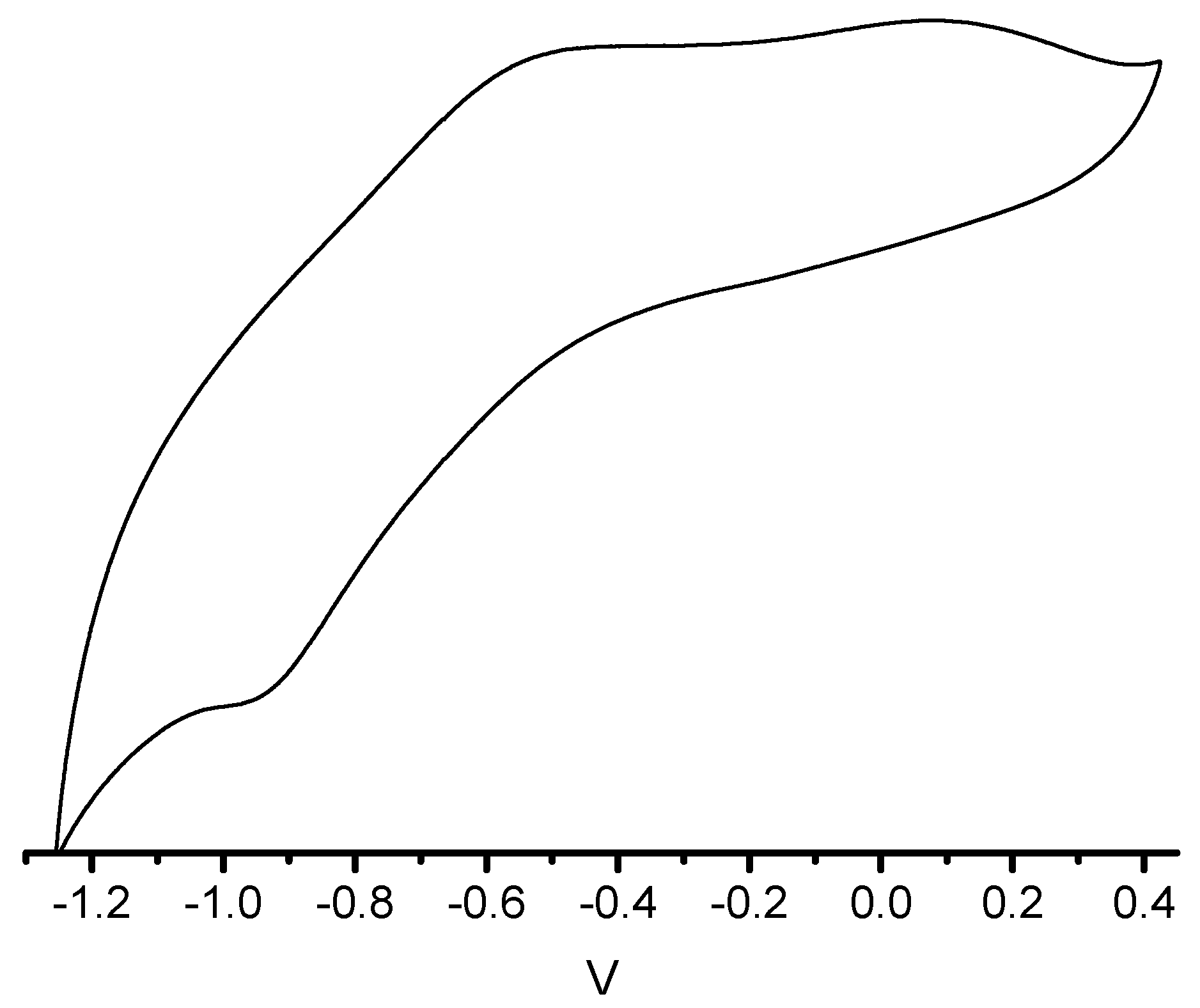
3. Experimental Section
Synthesis of 1
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Günther, R.T.; Manley, J.J. A mural glass mosaic from the Imperial Roman Villa near Naples. Archaeologia 1912, 63, 99–108. [Google Scholar] [CrossRef]
- Denning, R.G. Electronic structure and bonding in actinyl ions and their analogs. J. Phys. Chem. A 2007, 111, 4125–4143. [Google Scholar] [CrossRef] [PubMed]
- Liddle, S.T. The renaissance of non-aqueous uranium chemistry. Angew. Chem. Int. Ed. 2015, 54, 8604–8641. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.B.; Gaunt, A.J. Recent developments in synthesis and structural chemistry of nonaqueous actinide complexes. Chem. Rev. 2013, 113, 1137–1198. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.J. New reactivity of the uranyl ion. Chem. Eur. J. 2012, 18, 16258–16271. [Google Scholar] [CrossRef] [PubMed]
- Natrajan, L.S. Developments in the photophysics and photochemistry of actinide ions and their coordination compounds. Coord. Chem. Rev. 2012, 256, 1583–1603. [Google Scholar] [CrossRef]
- Drobot, B.; Steudtner, R.; Raff, J.; Geipel, G.; Brendler, V.; Tsushima, S. Combining luminescence spectroscopy, parallel factor analysis and quantum chemistry to reveal metal speciation—A case study of uranyl(VI) hydrolysis. Chem. Sci. 2015, 6, 964–972. [Google Scholar] [CrossRef]
- Hashem, E.; McCabe, T.; Schulzke, C.; Baker, R.J. Synthesis, structure and photophysical properties of [UO2X2(O=PPh3)2] (X = Cl, Br, I). Dalton Trans. 2014, 43, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Redmond, M.P.; Cornet, S.M.; Woodall, S.D.; Whittaker, D.; Collison, D.; Helliwell, M.; Natrajan, L.S. Probing the local coordination environment and nuclearity of uranyl(VI) complexes in non-aqueous media by emission spectroscopy. Dalton Trans. 2011, 40, 3914–3926. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, N.; Shimojo, K.; Brooks, N.R.; Nagaishi, R.; Naganawa, H.; van Hecke, K.; van Meervelt, L.; Binnemans, K.; Kimura, T. Thermochromic properties of low-melting ionic uranyl isothiocyanate complexes. Chem. Commun. 2011, 47, 4490–4492. [Google Scholar] [CrossRef] [PubMed]
- Hashem, E.; Platts, J.A.; Hartl, F.; Lorusso, G.; Evangelisti, M.; Schulzke, C.; Baker, R.J. Thiocyanate complexes of uranium in multiple oxidation states: A combined structural, magnetic, spectroscopic, spectroelectrochemical, and theoretical study. Inorg. Chem. 2014, 53, 8624–8637. [Google Scholar] [CrossRef] [PubMed]
- Crawford, M.-J.; Karaghiosoff, K.; Mayer, P. The homoleptic U(NCSe)84− anion in (Pr4N)4U(NCSe)8·2CFCl3 and Th(NCSe)4(OP(NMe2)3)4·0.5CH3CN·0.5H2O: First structurally characterized actinide isoselenocyanates. Z. Anorg. Allg. Chem. 2010, 636, 1903–1906. [Google Scholar] [CrossRef]
- Rowland, C.E.; Kanatzidis, M.G.; Soderholm, L. Tetraalkylammonium uranyl isothiocyanates. Inorg. Chem. 2012, 51, 11798–11804. [Google Scholar] [CrossRef] [PubMed]
- Straka, M.; Patzschke, M.; Pyykkö, P. Why are hexavalent uranium cyanides rare while U–F and U–O bonds are common and short? Theor. Chem. Acc. 2003, 109, 332–340. [Google Scholar] [CrossRef]
- Surbella, R.G.; Cahill, C.L. The exploration of supramolecular interactions stemming from the [UO2(NCS)4(H2O)]2− tecton and substituted pyridinium cations. CrystEngComm 2014, 16, 2352–2364. [Google Scholar] [CrossRef]
- Carter, T.J.; Wilson, R.E. Coordination chemistry of homoleptic actinide(IV)–thiocyanate complexes. Chem. Eur. J. 2015, 21, 15575–15582. [Google Scholar] [CrossRef] [PubMed]
- Fortier, S.; Hayton, T.W. Oxo ligand functionalization in the uranyl ion (UO22+). Coord. Chem. Rev. 2010, 254, 197–214. [Google Scholar] [CrossRef]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (IUPAC Recommendations 2011). J. Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Alvarez, S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [Google Scholar] [CrossRef] [PubMed]
- Michalczyk, R.; Schmidt, J.G.; Moody, E.; Li, Z.; Wu, R.; Dunlap, R.B.; Odom, J.D.; Silks, L.A., III. Unusual C–H···Se=C interactions in aldols of chiral N-acyl selones detected by gradient-selected 1H–77Se HMQC NMR spectroscopy and X-ray crystallography. Angew. Chem. Int. Ed. 2000, 39, 3067–3070. [Google Scholar] [CrossRef]
- Uhl, W.; Wegener, P.; Layh, M.; Hepp, A.; Wuerthwein, E.-U. Chalcogen capture by an Al/P-based frustrated lewis pair: Formation of Al-E-P bridges and intermolecular tellurium–tellurium interactions. Organometallics 2015, 34, 2455–2462. [Google Scholar] [CrossRef]
- Kobayashi, K.; Masu, H.; Shuto, A.; Yamaguchi, K. Control of face-to-face π–π stacked packing arrangement of anthracene rings via chalcogen–chalcogen interaction: 9,10-Bis(methylchalcogeno)anthracenes. Chem. Mater. 2005, 17, 6666–6673. [Google Scholar] [CrossRef]
- Bleiholder, C.; Gleiter, R.; Werz, D.B.; Koeppel, H. Theoretical investigations on heteronuclear chalcogen–chalcogen interactions: On the nature of weak bonds between chalcogen centers. Inorg. Chem. 2007, 46, 2249–2260. [Google Scholar] [CrossRef] [PubMed]
- Bleiholder, C.; Werz, D.B.; Koeppel, H.; Gleiter, R. Theoretical investigations on chalcogen–chalcogen interactions: What makes these nonbonded interactions bonding? J. Am. Chem. Soc. 2006, 128, 2666–2674. [Google Scholar] [CrossRef] [PubMed]
- Fazekas, Z.; Yamamura, T.; Tomiyasu, H. Deactivation and luminescence lifetimes of excited uranyl ion and its fluoro complexes. J. Alloys Compd. 1998, 271–273, 756–759. [Google Scholar] [CrossRef]
- Sornein, M.-O.; Cannes, C.; le Naour, C.; Lagarde, G.; Simoni, E.; Berthet, J.-C. Uranyl complexation by chloride ions. Formation of a tetrachlorouranium(VI) complex in room temperature ionic liquids [Bmim][Tf2N] and [MeBu3N][Tf2N]. Inorg. Chem. 2006, 45, 10419–10421. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, H.C.; Royal, D.S.; Helliwell, M.; Pope, S.J.A.; Ashton, L.; Goodacre, R.; Sharrad, C.A. Structural, spectroscopic and redox properties of uranyl complexes with a maleonitrile containing ligand. Dalton Trans. 2011, 40, 5939–5952. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.L.; Conradson, S.D.; Donohoe, R.J.; Keogh, D.W.; Morris, D.E.; Palmer, P.D.; Rogers, R.D.; Tait, C.D. Chemical speciation of the uranyl ion under highly alkaline conditions. Synthesis, structures, and oxo ligand exchange dynamics. Inorg. Chem. 1999, 38, 1456–1466. [Google Scholar] [CrossRef]
- Yaprak, D.; Spielberg, E.T.; Bäcker, T.; Richter, M.; Mallick, B.; Klein, A.; Mudring, A.-V. A roadmap to uranium ionic liquids: Anti-crystal engineering. Chem. Eur. J. 2014, 20, 6482–6493. [Google Scholar] [CrossRef] [PubMed]
- Ogura, T.; Takao, K.; Sasaki, K.; Arai, T.; Ikeda, Y. Spectroelectrochemical identification of a pentavalent uranyl tetrachloro complex in room-temperature ionic liquid. Inorg. Chem. 2011, 50, 10525–10527. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Hiroe, K.; Asanuma, N.; Shirai, A. Electrochemical studies on uranyl(VI) chloride complexes in ionic liquid, 1-butyl-3-methylimidazolium chloride. J. Nucl. Sci. Technol. 2009, 46, 158–162. [Google Scholar] [CrossRef]
- Takao, K.; Tsushima, S.; Ogura, T.; Tsubomura, T.; Ikeda, Y. Experimental and theoretical approaches to redox innocence of ligands in uranyl complexes: What is formal oxidation state of uranium in reductant of uranyl(VI)? Inorg. Chem. 2014, 53, 5772–5780. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, M.P.; Burns, C.J.; Paine, R.T.; Scott, B.L. Synthesis and crystal structure of UO2Cl2(THF)3: A simple preparation of an anhydrous uranyl reagent. Inorg. Chem. 1999, 38, 4156–4158. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nuzzo, S.; Browne, M.P.; Twamley, B.; Lyons, M.E.G.; Baker, R.J. A Structural and Spectroscopic Study of the First Uranyl Selenocyanate, [Et4N]3[UO2(NCSe)5]. Inorganics 2016, 4, 4. https://doi.org/10.3390/inorganics4010004
Nuzzo S, Browne MP, Twamley B, Lyons MEG, Baker RJ. A Structural and Spectroscopic Study of the First Uranyl Selenocyanate, [Et4N]3[UO2(NCSe)5]. Inorganics. 2016; 4(1):4. https://doi.org/10.3390/inorganics4010004
Chicago/Turabian StyleNuzzo, Stefano, Michelle P. Browne, Brendan Twamley, Michael E. G. Lyons, and Robert J. Baker. 2016. "A Structural and Spectroscopic Study of the First Uranyl Selenocyanate, [Et4N]3[UO2(NCSe)5]" Inorganics 4, no. 1: 4. https://doi.org/10.3390/inorganics4010004
APA StyleNuzzo, S., Browne, M. P., Twamley, B., Lyons, M. E. G., & Baker, R. J. (2016). A Structural and Spectroscopic Study of the First Uranyl Selenocyanate, [Et4N]3[UO2(NCSe)5]. Inorganics, 4(1), 4. https://doi.org/10.3390/inorganics4010004