Abstract
The Ce(III) hydrotris(3,5-dimethylpyrazolyl)borate complex [Ce(TpMe2)2(κ2-dmpz)] (1) (TpMe2 = {HB(dmpz)3}−; dmpz = 3,5-dimethylpyrazolide) was isolated in fair yield from the reaction of [Ce(I)3(THF)4] with two equivalents of [K(TpMe2)] via the facile decomposition of TpMe2. [Ce(TpMe2)2(bipy)] (2) was synthesized in poor yield by the “one-pot” reaction of [Ce(I)3(THF)4], bipy (bipy = 2,2′-bipyridine), KC8 and two equivalents of [K(TpMe2)] in tetrahydrofuran (THF). The reaction of 2 with N-methylmorpholine-N-oxide produced the known decomposition product [Ce(TpMe2)(μ-BOpMe2)]2 (3) (BOpMe2 = {HBO(dmpz)2}2−) in poor yield, presumably by N–O and B–N bond cleavage of a reactive intermediate. The reaction of 2 with trimethylsilylazide gave [Ce(TpMe2)2(N3)] (4) in poor yield; the fate of bipy and the trimethylsilyl group is unknown. Complexes 1–4 were characterized by single crystal XRD, NMR and FTIR spectroscopy and elemental analysis. Complex 2 was additionally probed by UV/Vis/NIR and Electron Paramagnetic Resonance (EPR) spectroscopies, Cyclic Voltammetry (CV) and magnetometry, which together indicate a formal 4f1 Ce(III) center coordinated by a bipy·− radical anion in this system.
1. Introduction
Complexes that exhibit terminal unsupported multiple bonds between a transition metal and a p-block element are legion, and interest in these species has surged in tandem with their increasing applicability in synthetic processes [1]. In contrast, the corresponding lanthanide (Ln) chemistry is underdeveloped, as the large orbital energy mis-match between Ln valence orbitals and the 2pz orbital of C, N and O makes these bonds highly polarized, making them prone to decomposition and oligomerisation pathways [2,3]. As such the isolation of Ln=CR2, Ln=NR and Ln=O bonds is a major synthetic challenge, with the first structurally characterized supported Ce(IV)=CR2 [4], terminal unsupported Ln=NR (Ln = Y, Lu) [5] and terminal supported Ce(IV)=O [6] complexes reported very recently, whilst the corresponding terminal unsupported Sc=NR chemistry has flourished since the first example was reported in 2010 by Chen [7].
Synthetic routes to terminal unsupported actinide (An) An=CR2, An=NR and An=O multiple bonds are far more developed than pathways to the corresponding Ln species [8,9]. Andersen [10], Bart [11,12,13] and Zi and Walter [14] have shown that U(IV) and Th(IV) oxo and imido complexes can be prepared by the respective addition of N-oxides or organoazides to An(II) synthons, which mimic the reactivity of An(II) complexes although the metal is formally in the +4 oxidation state. We set out to find if this methodology could be extended to cerium, which is unique amongst the Lns as it has a readily accessible +4 oxidation state [EӨ Ce(IV) → Ce(III) = 1.74 V] [15].
The An(II) synthon utilized by Bart that is of most relevance here is [U(TpMe2)2(bipy)] (TpMe2 = hydrotris(3,5-dimethylpyrazolyl)borate; bipy = 2,2′-bipyridine) [16], which formally contains a bipy dianion and a U(IV) center. Marques previously reported a Ln(II) synthon analogue, [La(TpMe2)2(bipy)] [17], which formally contains a monoreduced bipy·− radical anion and lanthanum in the +3 oxidation state. As TpMe2 ligands have been shown to stabilize a wide variety of interesting motifs in Ln chemistry [18], we reasoned that they could be suitable ancillary ligands to support terminal Ce(IV)=O or Ce(IV)=NR multiple bonds.
We envisaged that an analogous Ce(II) synthon, [Ce(TpMe2)2(bipy)], could react with N-oxides or organoazides to give complexes that contain terminal unsupported Ce(IV)=O or Ce(IV)=NR multiple bonds, provided the Ce(III) → Ce(IV) oxidation potential in these complexes could be overcome by these reagents. [Ce(TpMe2)2(bipy)] should contain a formal Ce(III) 4f1 center and the coupling of this unpaired spin with a ligand radical could lead to unusual physicochemical properties. It is noteworthy that interesting multiconfigurational behavior has previously been observed in [Yb(Cp*)2(bipy)] [19,20], where the electronic ground state contains contributions from Yb(III)/bipy·− (4f13) and Yb(II)/bipy (4f14) configurations, but the donor properties of Cp* ligands are not comparable to those of TpMe2 ligands. The synthesis and characterization of [Ce(TpMe2)2(bipy)] and its reactivity towards N-oxides and organoazides is described herein.
2. Results and Discussion
2.1. Preparation of a Ce(II) Synthon
2.1.1. Synthesis and Structural Characterization of [Ce(TpMe2)2(κ2-dmpz)] (1)
In an effort to prepare the heteroleptic complex [Ce(TpMe2)2(I)], [Ce(I)3(THF)4] [21] was reacted with two equivalents of [K(TpMe2)] [22] in THF by slight modifications of the procedures previously used to synthesize [Ln(TpMe2)2(X)] (Ln = La, X = Cl, I [23]; Ln = Nd, X = Cl [24]; Ln = Sm, X = Cl, Br [25]; Ln = Eu, X = Cl [25]). However, [Ce(TpMe2)2(I)] was not identified in the reaction mixture, and the only isolable product was [Ce(TpMe2)2(κ2-dmpz)] (1), which was obtained in fair crystalline yield from a saturated toluene solution (Scheme 1). The relatively high yield of 1 showed that this route is not suitable for the preparation of [Ce(TpMe2)2(I)] under the conditions we employed but the one-pot method used in Section 2.1.2 circumvented the need to isolate [Ce(TpMe2)2(X)] (see below).
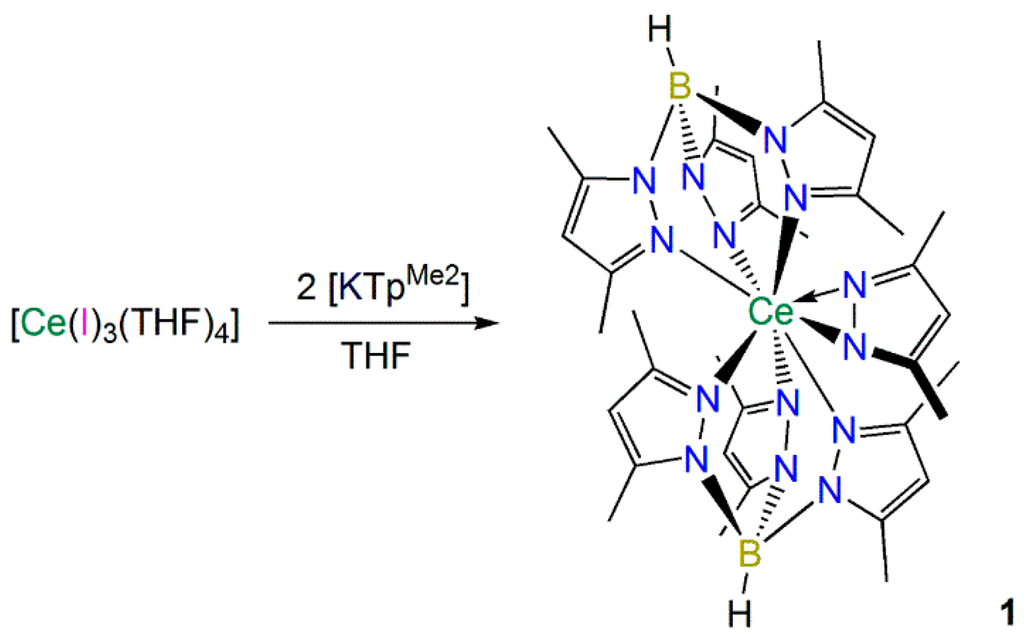
Scheme 1.
Synthesis of 1.
Complex 1 presumably forms by the decomposition of a TpMe2 ligand via the mechanism previously postulated by Kunrath et al., when [La(TpMe2)2(κ2-dmpz)] was isolated during the synthesis of [La(TpMe2)2(X)] (X = Cl, I) [23]. Coordination of TpMe2 to lanthanum was proposed to twist pyrazolyl rings and weaken B–N bonds, promoting bond cleavage to form (dmpz)− in situ, which reacts with [La(TpMe2)2(X)] to give [La(TpMe2)2(κ2-dmpz)]. This mechanism has been corroborated by a control experiment, where [La(TpMe2)2(X)] was treated with dmpzH and triethylamine, and [La(TpMe2)2(κ2-dmpz)] was detected in the 1H NMR spectrum [23].
It is noteworthy that we were not able to identify any traces of [Ce(TpMe2)2(I)] in the reaction mixture during the synthesis of 1, whereas [La(TpMe2)2(κ2-dmpz)] was a minor product during the preparation of [La(TpMe2)2(I)] [23], and no dmpz-containing byproducts were reported from the syntheses of [Ln(TpMe2)2(Cl)] (Ln = Sm [25]; Nd or Eu [24]), and [Sm(TpMe2)2][I] [25]. However, the preparation of [Ce(TpMe2)2(Cl)] from CeCl3 has previously been described as problematic by Sella [24], whereas the synthesis of [Ce(TpMe2)2(OTf)] from Ce(OTf)3 is more straightforward due to OTf− being a better leaving group than Cl− [26]. Kunrath et al., stated that it is difficult to coordinate two TpMe2 ligands to lanthanides due to steric crowding [23], therefore we conclude that when [Ce(I)3(THF)4] is used as a precursor for the synthesis of [Ce(TpMe2)2(I)] intermediates with high coordination numbers form, which promote TpMe2 decomposition by twisting and weakening B–N bonds.
The elemental analysis data for 1 correlated well with the predicted values, as with the other complexes (2–4, see below) reported in this paper. The 1H NMR spectrum of 1 exhibited resonances over a wide chemical shift range (δH: −7.99 to +18.26 ppm) due to the 4f1 formulation of Ce(III), and this precluded any interpretation of 13C{1H} NMR spectroscopic data. However, a single resonance was observed in the 11B{1H} NMR spectrum of 1 (δB: −0.66 ppm), which is downfield of that reported for [Ce(TpMe2)(COT)] (δB: −21.0 ppm) [27]. The solution magnetic susceptibility of 1 was measured by the Evans method (μeff = 2.33 µB) [28], and this correlates well with the free ion approximation for a Ce(III) 4f1, 2F5/2 ground term system (μeff: 2.54 µB [29]) considering weighing errors. The FTIR spectrum exhibited an absorption at 2547 cm−1 that is characteristic of the B–H stretching mode in tripodal Tp systems, and this was also seen in 2–4 (see below) [30,31].
The solid state structure of 1·C7H8 was determined by a single crystal XRD study and is depicted in Figure 1, with selected bond lengths and angles compiled in Table 1. The presence of toluene in the lattice causes 1·C7H8 to adopt a different space group to [La(TpMe2)2(κ2-dmpz)] [23], which was crystallized from cyclohexane. Due to the similar 8-coordinate ionic radii of La(III) [1.160 Å] and Ce(III) [1.143 Å] [32], variable crystal packing effects are likely the cause of two minor discrepancies between the solid state structures of 1 and [La(TpMe2)2(κ2-dmpz)]. Firstly, the dmpz ligand coordinates in a less symmetrical manner in 1 than in [La(TpMe2)2(κ2-dmpz)], as evidenced by the respective Ln–Ndmpz distances [2.385(5) and 2.505(5) Å vs. 2.468(4) and 2.514(3) Å] [23]. Secondly, the Ln–NTpMe2 distances in 1 [2.546(4)–2.667(4) Å] are within a narrower range than those found in [La(TpMe2)2(κ2-dmpz)] [2.601(3)–2.748(3) Å] [23]. All other metrical parameters for 1 are unremarkable.
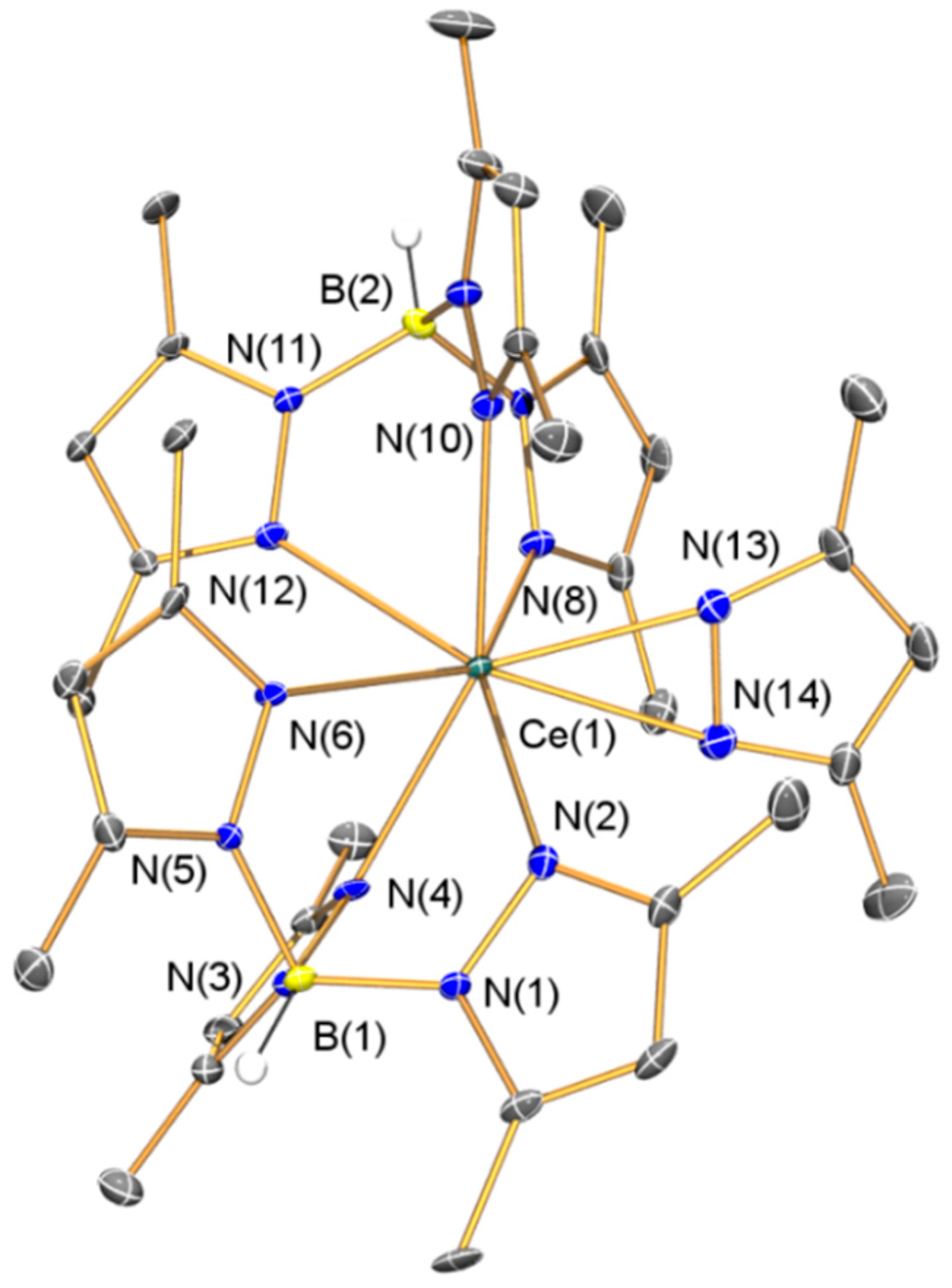
Figure 1.
Molecular structure of 1·C7H8 with selective atom labeling. Displacement ellipsoids set at 30% probability level and hydrogen atoms (except B–H) and lattice solvent omitted for clarity.
2.1.2. Synthesis and Structural Characterization of [Ce(TpMe2)(bipy)] (2)
Marques and co-workers previously reported that the one-pot reaction of [La(Cl)3(THF)1.5] with two equivalents of [K(TpMe2)] and one equivalent of bipy over a Na/Hg amalgam in THF gave [La(TpMe2)2(bipy)] in 90% yield [17]. We modified this procedure to prepare [Ce(TpMe2)2(bipy)] (2) using [Ce(I)3(THF)4] [21], two equivalents of [K(TpMe2)] [22], one equivalent of bipy and KC8 [33] in THF (Scheme 2), in an effort to avoid the use of mercury. A poor crystalline yield of 2 (22%) was reproducibly obtained using our method, likely a result of the difficulties encountered during the synthesis of 1 (see above), though no other products could be identified in the reaction mixtures. The intense dark red color of 2 indicated that the complex contained a bipy·− radical monoanion; therefore we collected a wide range of characterization data to probe this system.
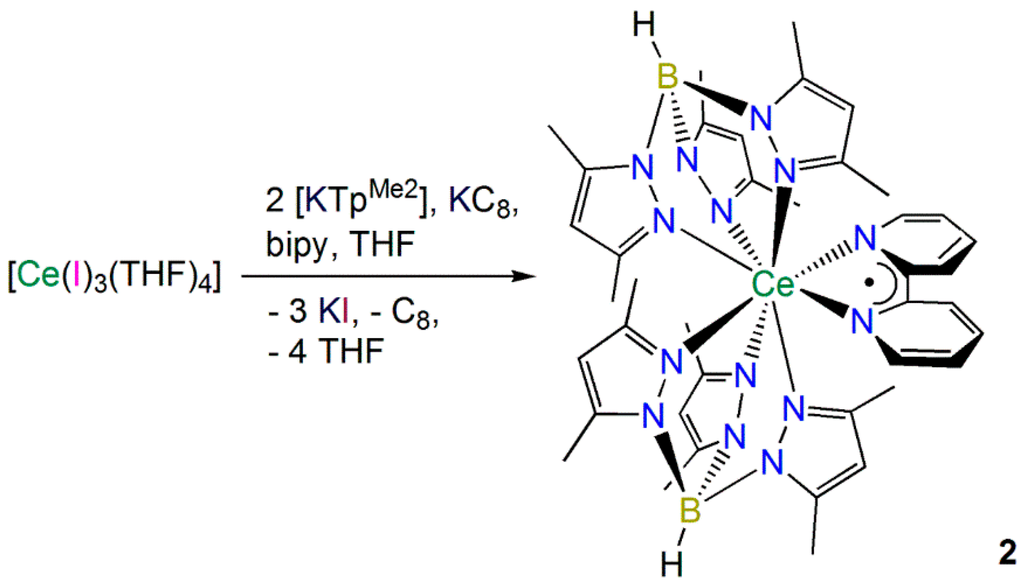
Scheme 2.
Synthesis of 2.
The 1H NMR spectrum of 2 exhibits a similar range of resonances to 1 [δH: −9.30 to 19.84 ppm], with a wide spectral window of +200 to −200 ppm evaluated as bipy resonances were observed at large negative chemical shifts for [Ce(Cp*)2(bipy)] (Cp* = C5Me5) [34]. The bipy proton resonances in 2 could only be tentatively assigned, as neutral and monoanionic bipy·− resonances may be difficult to distinguish in a paramagnetic spectrum. In Marques’ report of the 1H NMR spectrum of the lanthanum analogue [La(TpMe2)2(bipy)] these signals were not observed [17]. One signal was seen in the 11B{1H} NMR spectrum of 2 (δB: 10.48 ppm); however the 13C{1H} NMR spectrum could not be interpreted. The FTIR spectrum of 2 exhibited characteristic absorptions for a bipy radical anion (v = 1541, 1494 and 941 cm−1) with the lowest frequency peak correlating with that assigned for [La(TpMe2)2(bipy)] (v = 940 cm−1) [17]. The solution susceptibility of 2 (μeff = 1.59 µB) was reproducibly much lower than the value obtained for 1, which could be attributed to either partial decomposition of 2 or antiferromagnetic coupling between Ce(III) and bipy·−. To probe this result a variable temperature magnetic analysis was performed together with an EPR spectroscopy study (see below).
The solid state configuration of 2·(C4H8O)2 was confirmed by single crystal XRD (depicted in Figure 2, with selected bond lengths and angles compiled in Table 1). The metrical parameters of structurally analogous [La(TpMe2)2(bipy)]·(C4H8O)2 have been discussed previously [17], and as these are nearly identical to those in 2·(C4H8O)2 considering the small difference in ionic radii between lanthanum and cerium [32] these will not be commented on here for brevity. The salient point of the discussion of the structure of [La(TpMe2)2(bipy)] is that the distances throughout the bipy scaffold were used as evidence of its radical monoanionic formulation (the NMR and FTIR spectroscopy and magnetic data of [La(TpMe2)2(bipy)] concurred with this assignment) [17], and statistically identical intra-ligand bond distances were observed in the coordinated bipy framework in 2.
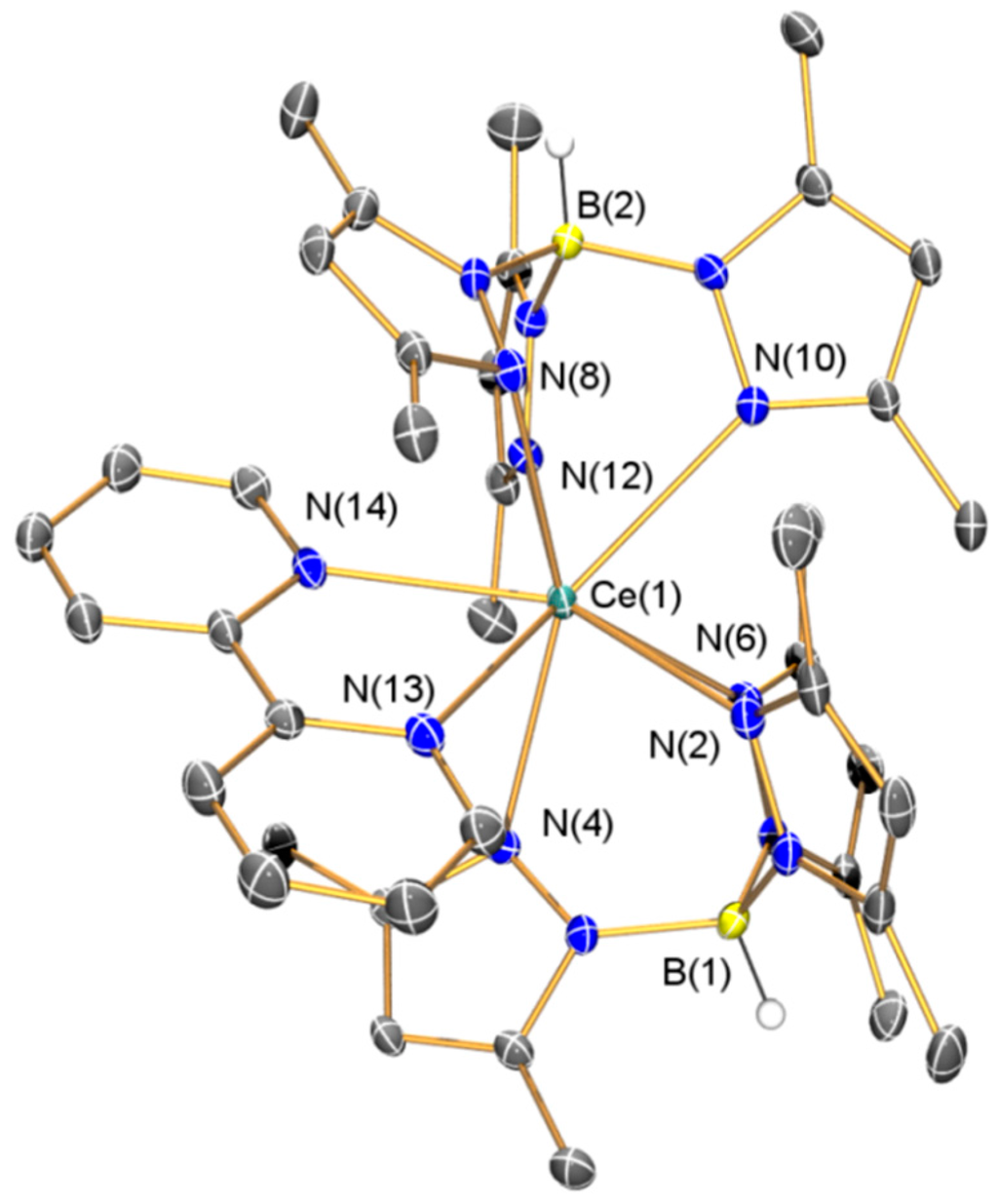
Figure 2.
Molecular structure of 2·(C4H8O)2 with selective atom labeling. Displacement ellipsoids set at 30% probability level and hydrogen atoms (except B–H) and lattice solvent omitted for clarity.
2.1.3. Further Characterization of [Ce(TpMe2)(bipy)] (2)
The electronic structure of 2 was probed by additional techniques in an effort to unequivocally establish a formal Ce(III)/bipy·− configuration. The UV/Vis/NIR spectrum of 2 exhibits a strong absorption at 387 nm (εmax ≈ 4100 M−1·cm−1) and a weaker, broad absorption at 812–972 (εmax ≈ 500 M−1·cm−1) nm (Figure 3). These absorptions are comparable to those observed for [Yb(Cp*)2(bipy)], which were assigned as π→π* and π*→π* transitions that arise from the unpaired electron in the bipy radical π-system [19].
Bipy has neutral, monoanionic and dianionic forms that can be easily interchanged and these processes are readily observed by electrochemistry [35]; therefore the cyclic voltammogram of 2 was obtained (Figure 4). Only one Nernstian quasi-reversible process was observed within the range limited by solvent and electrolyte experimental conditions (E = −2.34 V vs. Fc/Fc+, ΔE = 95 mV), which was assigned to a [Ce(TpMe2)2(bipy)]+/ [Ce(TpMe2)2(bipy·−)] process based on its similarity to the first reduction potential of bipy that we measured using identical conditions (E = −2.72 V vs. Fc/Fc+). The second reduction potential of bipy and the oxidation of Ce(III) to Ce(IV) could not be observed under the conditions employed, which indicates that strong oxidizing agents are required to form Ce(IV) complexes supported by TpMe2 ligands. This is unsurprising given both the considerable precedent of Tp ligands stabilizing metals in low oxidation states [18] and the dominance of electron-rich donor ligands in Ce(IV) chemistry [36].
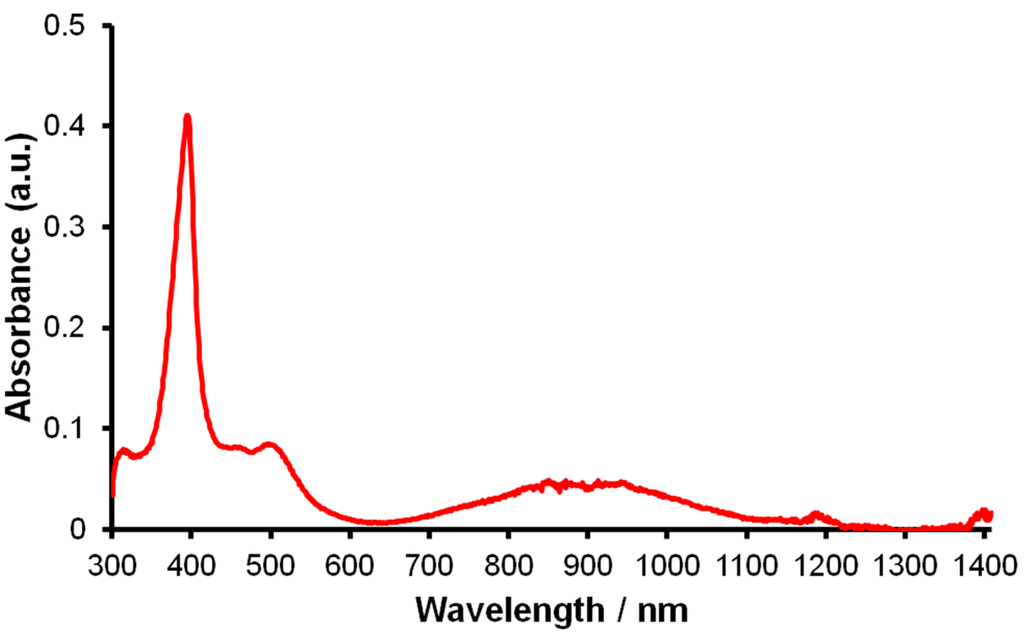
Figure 3.
UV/Vis/NIR spectrum of 2 (0.1 mM in THF).
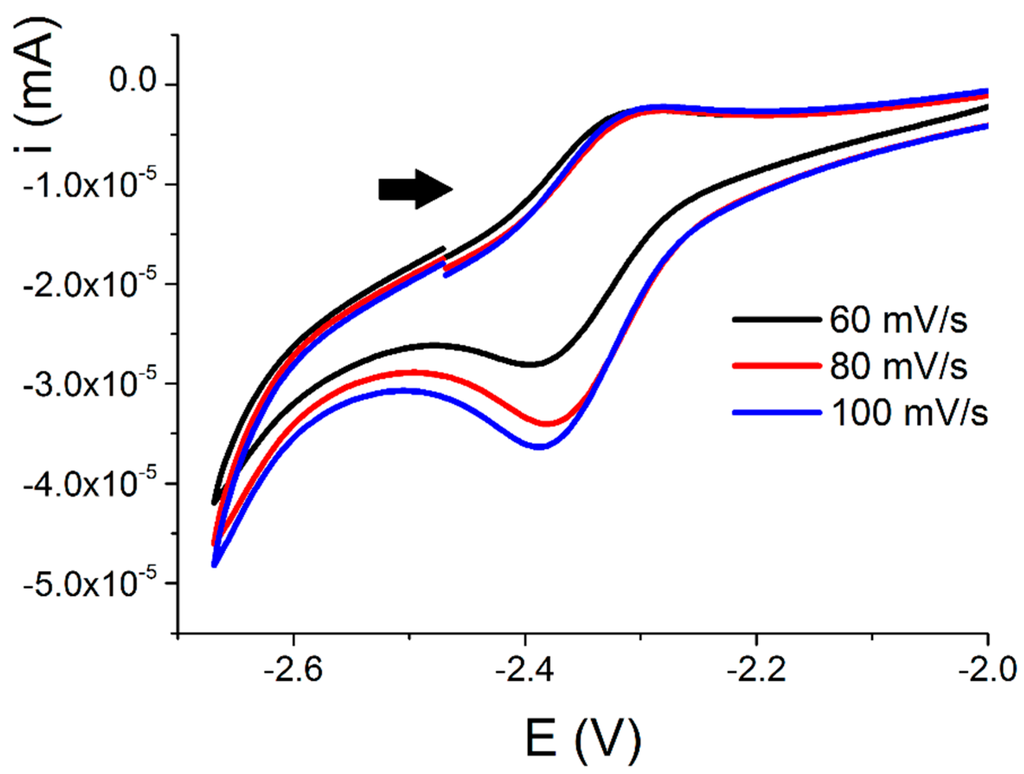
Figure 4.
Cyclic voltammogram of 2 (0.5 mM in THF, 0.1 M [NBun4][BF4]). Black arrow shows the direction of applied potential from the origin.
The powder X-band EPR spectrum of 2 was collected in an effort to observe resonances arising from coupling of the Ce(III) and bipy·− radical unpaired electrons. No signal was seen at room temperature but a highly anisotropic complex spectrum was obtained at 5 K. We performed a variable temperature study to track the thermal evolution of the spectrum (Figure 5). The bulk features at 5 K are almost fully maintained at 10 K, but at 15 K the spectrum is less distinct and at higher temperatures the fine structure cannot be discerned from the baseline. The 5 K spectrum could not be modeled and therefore we were not able to extract any parameters but this highlights the complex nature of the coupling between the bipy·− radical electron and the 2F5/2 ground state doublet deriving from Ce(III).
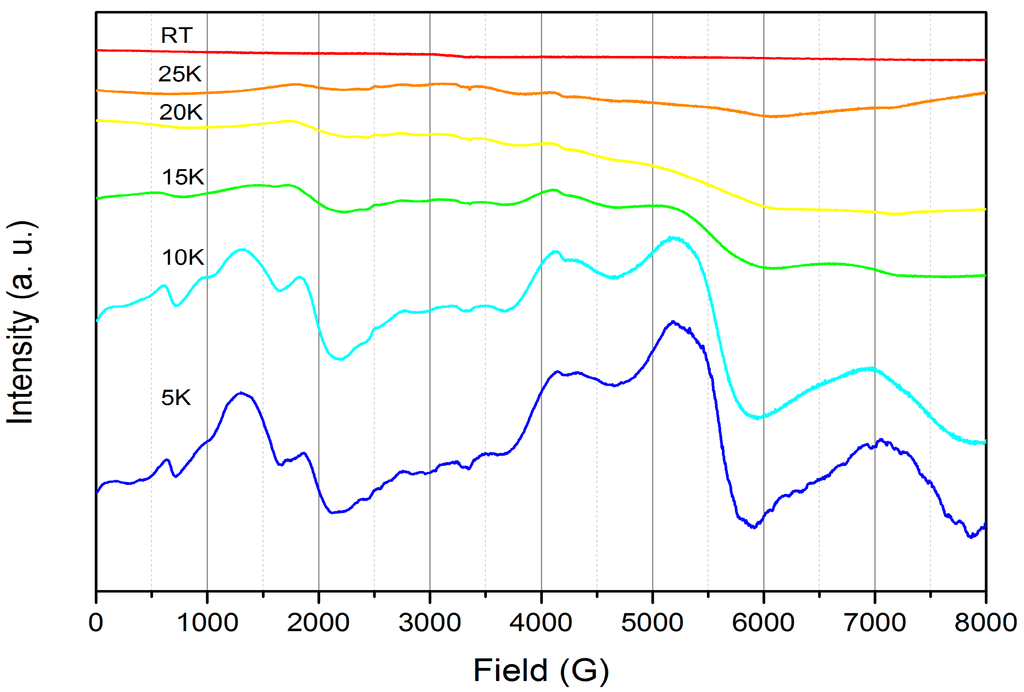
Figure 5.
Variable temperature powder X-band EPR spectra of 2 (v = 9.385 GHz).
Magnetic measurements were performed on a solid sample of 2 suspended in eicosane and the magnetic susceptibility product (χMT) was measured from 300 to 2 K in a 10,000 Oe applied dc field (Figure 6). At 300 K, χMT = 0.44 cm3·Kmol−1 (1.21 μB), which is comparable to the moment measured in solution at 298 K (0.58 cm3·Kmol−1), taking into account weighing errors. These values are much lower than is predicted for a system containing one organic radical and one Ce(III) center (1.1 cm3·Kmol−1) and are closer to a Ce(III)-only system. The discrepancy between predicted and observed χMT values at 300 K can be attributed to the presence of diamagnetic impurities, or more complex magnetic behavior in 2. The χMT value decreases to 0.15 cm3·Kmol−1 (0.41 μB) at 2 K. We analyzed 2 further by measuring the magnetization against variable applied dc field at 5 K from 0–70,000 Oe, finding a near-linear correlation of M vs. H, with M = 0.39 μB at 70,000 Oe (Figure 7). In contrast at 2 K the data forms a curve, with a linear dependence of M vs. H from 0 to around 20,000 Oe (M = 0.27 μB), but at higher fields the effect of field on M is reduced. At both temperatures saturation was not reached at the maximum obtainable field, and this was observed reproducibility on separate samples, consistent with a system containing two unpaired electrons. There is also an essentially linear relationship of M and H from 35,000–70,000 Oe (M = 0.50 μB at 70,000 Oe) with ∂M/∂H for this range of fields lower than for the corresponding data at 5 K.
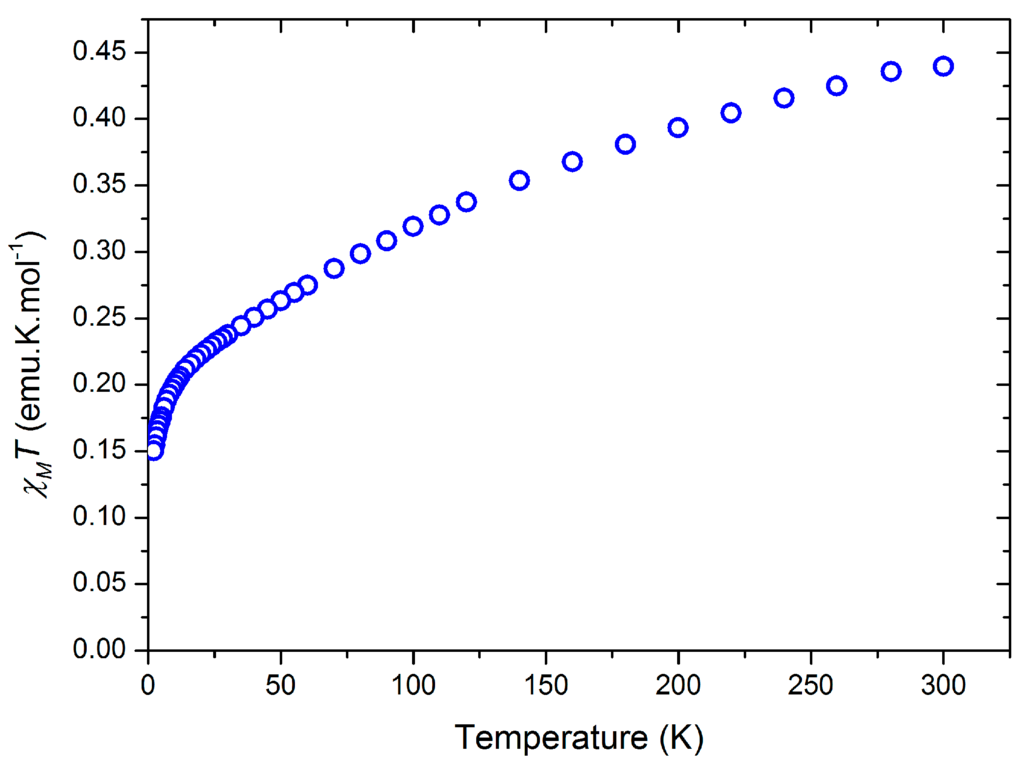
Figure 6.
Variation of the product of the molar magnetic susceptibility product (χMT, cm3·Kmol−1) with temperature (K) of 2.
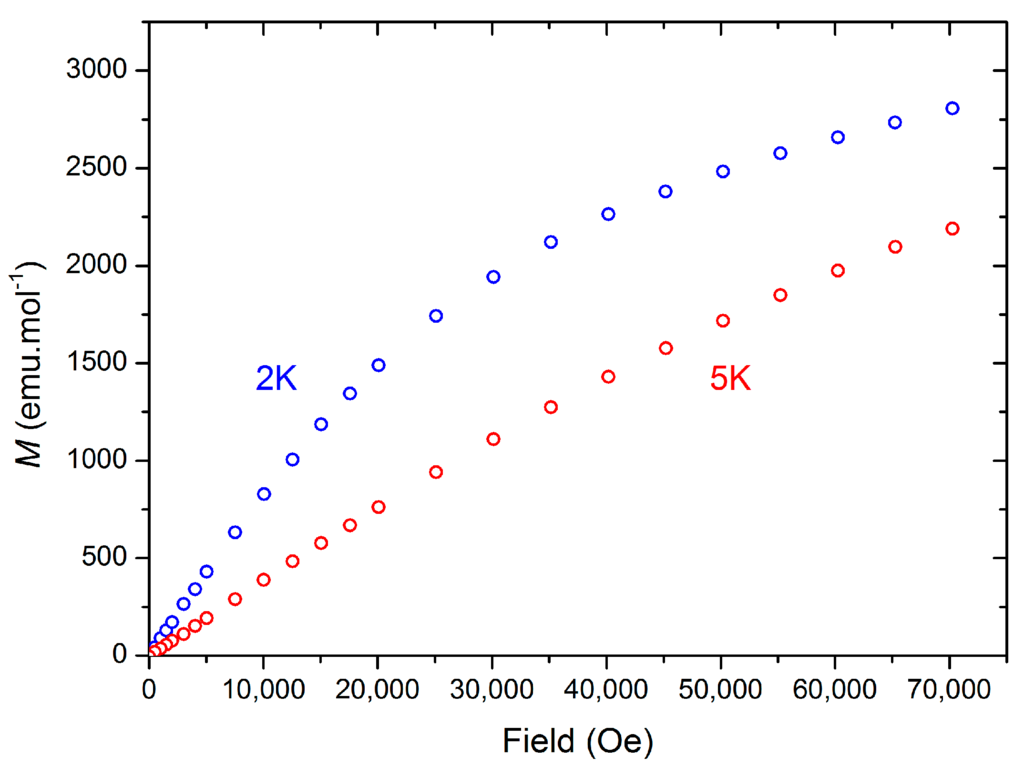
Figure 7.
Magnetization (M, μB) vs. Field (Oe) of 2 at 2 K and 5 K.
2.2. Reactivity of a Ce(II) Synthon
2.2.1. Synthesis and Structural Characterization of [Ce(TpMe2)(μ-BOpMe2)]2 (3)
We were interested in finding out if 2 could react with 2e− oxidizing agents as a Ce(II) synthon, mimicking the reactivity profile of a Ce(II) complex, even though the measurements in Section 2.1.3 indicate that Ce is in the +3 oxidation state in 2. The reactions of 2 with N-oxides were investigated as Bart previously showed that the treatment of [U(TpMe2)(bipy)] with pyridine-N-oxide gave the U(IV) terminal oxo complex [U(TpMe2)(O)] [11]. In contrast the reaction of 2 with N-methylmorpholine-N-oxide, pyridine-N-oxide or TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl) all gave the Ce(III) complex [Ce(TpMe2)(μ-BOpMe2)]2 (3) as the only isolable product in ca. 20% crystalline yield in each case (Scheme 3). Complex 3 was previously reported by Sella and co-workers to form in trace amounts via the partial hydrolysis of a range of heteroleptic complexes, [Ce(TpMe2)2(X)] (X = anionic ligand), and a mechanism for this decomposition was postulated whereby dmpzH was generated [37]. As no other products could be identified during the formation of 3 it is not certain if the oxygen atom derives from initial coordination of the N-oxide or adventitious O2/H2O; however the isolation of 3 was reproducible, performed with the strict exclusion of oxygen and water, and was formed in higher yields than previously reported.
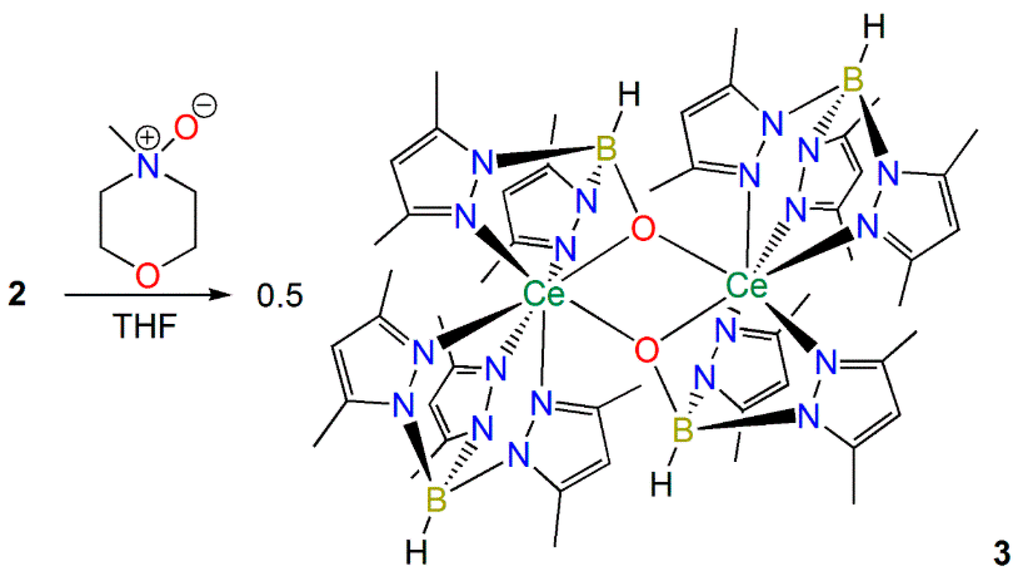
Scheme 3.
Synthesis of 3.
Whilst the solid state structure of 3 was obtained previously by Sella, no other characterization data were reported [37]; therefore we acquired elemental analysis and collected spectroscopic data for 3. Unusually in the original report 3 was reported to exhibit a dark red color [37], whilst the samples we obtained were pale yellow. The 1H NMR and FTIR spectra of 3 were unremarkable and as with 1 and 2 the 13C{1H} NMR spectrum could not be interpreted, though the 11B{1H} NMR spectrum of 3 contained one resonance (δB: 0.60 ppm). The Evans method susceptibility was also determined (μeff: 2.71 µB). Although the solid state structure of 3 has been reported previously [37], we include this data here for completeness as we obtained a polymorph (depicted in Figure 8, with selected bond lengths and angles compiled in Table 1).
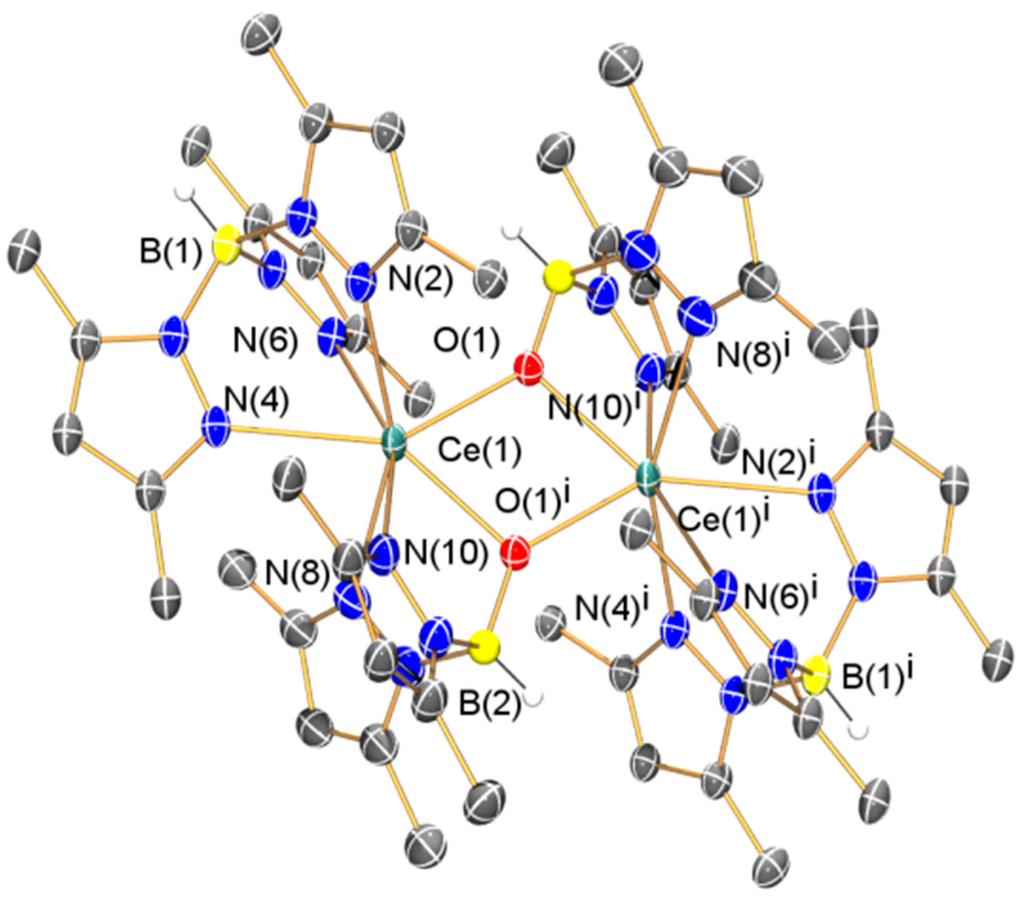
Figure 8.
Molecular structure of 3·(C4H8O)2 with selective atom labeling. Displacement ellipsoids set at 30% probability level and hydrogen atoms (except B–H) and lattice solvent omitted for clarity. Symmetry operation to generate equivalent atoms: i = 1 − x, 1 − y, 1 − z.

Table 1.
Selected bond lengths (Å) and angles (°) for 1–4.
| 1·C7H8 | |||
| Ce(1)–N(2) | 2.653(5) | Ce(1)–N(4) | 2.582(4) |
| Ce(1)–N(6) | 2.631(5) | Ce(1)–N(8) | 2.624(5) |
| Ce(1)–N(10) | 2.546(4) | Ce(1)–N(12) | 2.667(4) |
| Ce(1)–N(13) | 2.385(5) | Ce(1)–N(14) | 2.505(5) |
| N(13)–N(14) | 1.368(6) | Ce(1)–N(13)–N(14) | 78.7(3) |
| Ce(1)–N(14)–N(13) | 69.0(3) | N(13)–Ce(1)–N(14) | 32.38(14) |
| 2·(C4H8O)2 | |||
| Ce(1)–N(2) | 2.591(4) | Ce(1)–N(4) | 2.650(3) |
| Ce(1)–N(6) | 2.711(4) | Ce(1)–N(8) | 2.648(4) |
| Ce(1)–N(10) | 2.719(3) | Ce(1)–N(12) | 2.577(4) |
| Ce(1)–N(13) | 2.592(4) | Ce(1)–N(14) | 2.612(4) |
| C(35)–C(36) | 1.431(6) | C(35)–N(13) | 1.373(6) |
| C(36)–N(14) | 1.379(5) | N(13)–Ce(1)–N(14) | 62.40(12) |
| Ce(1)–N(13)–C(35) | 122.1(3) | Ce(1)–N(14)–C(36) | 120.7(3) |
| N(13)–C(35)–C(36) | 117.0(4) | N(14)–C(36)–C(35) | 117.8(4) |
| 3·(C4H8O)2 | |||
| Ce(1)–N(2) | 2.668(4) | Ce(1)–N(4) | 2.584(4) |
| Ce(1)–N(6) | 2.561(4) | Ce(1)–N(7) | 2.591(4) |
| Ce(1)–N(9) | 2.578(4) | Ce(1)–O(1) | 2.292(3) |
| Ce(1)–O(1i) | 2.378(3) | B(1)–O(1) | 1.407(6) |
| Ce(1)···Ce(1i) | 3.7987(5) | Ce(1)–O(1)–Ce(1i) | 108.84(12) |
| O(1)–Ce(1)–O(1i) | 71.16(12) | ||
| 4·C7H8 | |||
| Ce(1)–N(2) | 2.656(5) | Ce(1)–N(4) | 2.655(5) |
| Ce(1)–N(6) | 2.565(4) | Ce(1)–N(8) | 2.665(5) |
| Ce(1)–N(10) | 2.539(5) | Ce(1)–N(12) | 2.646(5) |
| Ce(1)–N(13) | 2.340(6) | N(13)–N(14) | 1.188(8) |
| N(14)–N(15) | 1.165(8) | Ce(1)–N(13)–N(14) | 165.5(5) |
| N(13)–N(14)–N(15) | 178.4(8) | ||
2.2.2. Synthesis and Structural Characterization of [Ce(TpMe2)2(N3)] (4)
Andersen [10], Bart [11] and Walter and Zi [13] have all shown that azides react with Th(II) synthons to yield Th(IV) imido complexes. Therefore we added trimethylsilyl azide to 2, and when no reaction was observed at room temperature over three days the reaction mixture was refluxed for 16 h and the Ce(III) complex [Ce(TpMe2)2(N3)] (4) was obtained as the only isolable product (Scheme 4). Whilst the mechanism for the formation of 4 and the fate of the silyl group is unclear, N–Si bond cleavage is relatively facile and Bart recently reported a range of reductive heterocouplings of the coordinated reduced bipy ligand in [U(Cp*)2(bipy)] with ketones [38], though the electronics of the spectator ligands in this system are vastly different to 2. Germane to this, no reaction was observed between 2 and mesityl azide, even after prolonged heating, showing that the preparation of Ce(IV) imido complexes by a formal two electron oxidation of 2 by silyl and aryl azides is not favored.
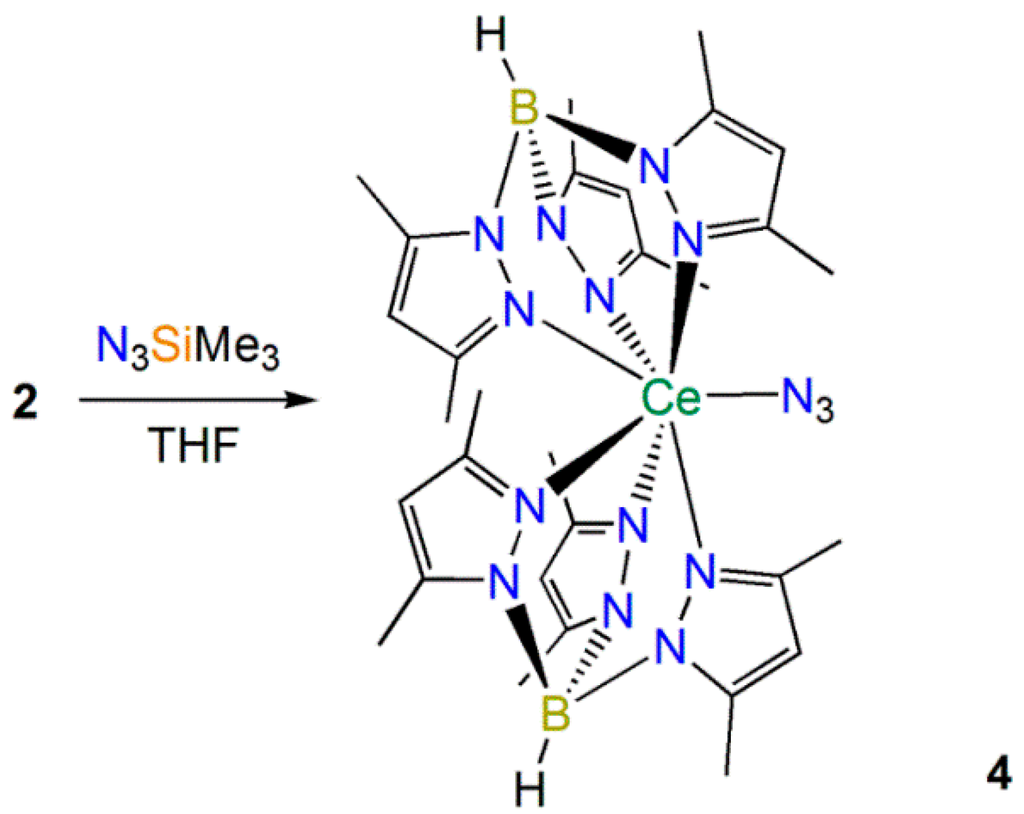
Scheme 4.
Synthesis of 4.
Gratifyingly, the 1H and 13C{1H} NMR spectra of 4 could be fully assigned, with the 13C signals all observed between 0 and 200 ppm. The Evans method susceptibility determined for 4 (μeff = 1.94 µB) was higher than that obtained for 2. Only one signal was observed in the 11B{1H} NMR spectrum of 4 (δB: −25.29 ppm), albeit at a higher field than 1–3, which we attribute to variable paramagnetic shift effects in the four complexes. The FTIR spectrum of 4 exhibited the diagnostic B–H stretching mode at 2555 cm−1, together with characteristic absorptions for an azide group (v = 2083 and 2065 cm−1) that are comparable to the absorption observed for [Ce(TRENDSAL)(N3)] (TRENDSAL = [N{CH2CH2N=CH(C6H2But2-3,5-O-2}3]3−) (v = 2044 cm−1) [39]. The composition of 4 was determined by single crystal XRD (depicted in Figure 9, with selected bond lengths and angles compiled in Table 1). The Ce(III)–Nazide distance in 4 [2.340(6) Å] is shorter than the Ce(IV)–Nazide distance observed in [Ce(TRENDSAL)(N3)] [2.437(3) Å] [39] and the azide in 4 binds with a less bent geometry [Ce(1)–N(13)–N(14) 165.5(5)°] than the azide in [Ce(TRENDSAL)(N3)] [Ce–N–N 133.4(2)°] [39], which is likely caused by the bulky TpMe2 ligands in 4. All other metrical parameters in 4 are unremarkable.
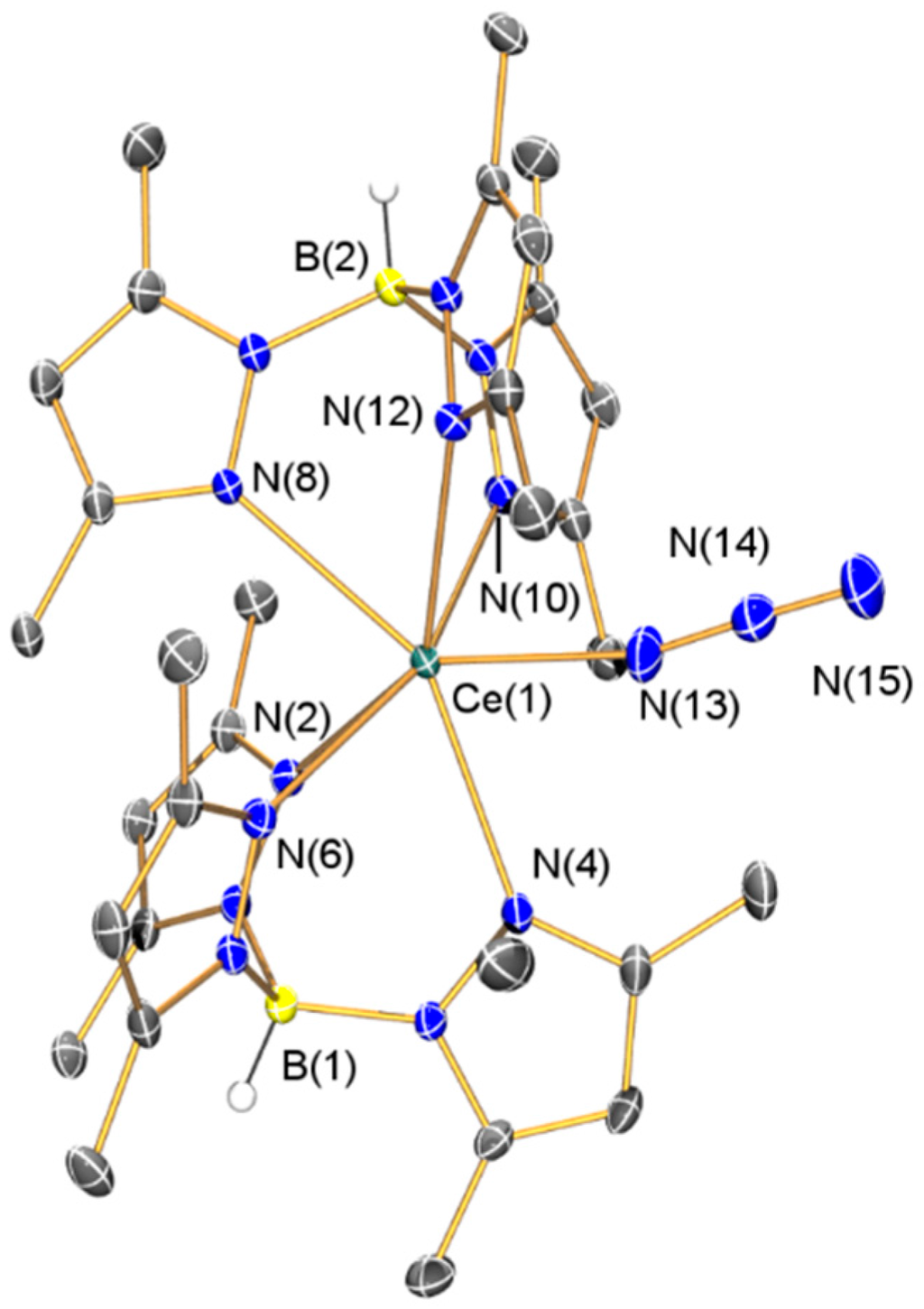
Figure 9.
Molecular structure of 4·C7H8 with selective atom labeling. Displacement ellipsoids set at 30% probability level and hydrogen atoms (except B–H) and lattice solvent omitted for clarity.
3. Experimental Section
3.1. General Procedures
All manipulations were carried out using standard Schlenk and glove box techniques under an atmosphere of dry argon. Solvents were dried by refluxing over potassium and degassed before use. All solvents were stored over potassium mirrors (with the exception of THF which was stored over activated 4 Å molecular sieves). Deuterated solvents were distilled from potassium, degassed by three freeze-pump-thaw cycles and stored under argon. [Ce(I)3(THF)4] [21], [K(TpMe2)] [22] and KC8 [33] were prepared according to published procedures. Most solid reagents were dried under vacuum for 4 h and most liquid reagents were dried over 4 Å molecular sieves and distilled before use. 1H, 13C{1H} and 11B{1H} NMR spectra were recorded on a spectrometer operating at 400.2, 100.6 and 128.3 MHz, respectively; chemical shifts are quoted in ppm and are relative to TMS (1H, 13C) or external BF3·Et2O (1.0 M in CDCl3) (11B). Magnetic susceptibility was determined by the Evans method [27]. FTIR spectra were recorded as Nujol mulls in KBr discs. Elemental microanalyses were carried out by Stephen Boyer at the Microanalysis Service, London Metropolitan University, UK. UV/Vis spectra were recorded in sealed 10 mm pathlength cuvettes. Redox potentials are referenced to the [Fe(Cp)2]+/[Fe(Cp)2] couple, which was used as an internal standard. Cyclic voltammetry was carried out using a sealed cell and a three-electrode arrangement, with a Pt wire working electrode, Pt flag secondary electrode and an AgCl/Ag wire pseudo-reference electrode. The susceptibility and magnetization curves were obtained using a SQUID magnetometer (Quantum Design, San Diego, CA, USA). The powdered sample was suspended in eicosane and sealed in a borosilicate glass NMR tube under vacuum, with diamagnetic contributions subtracted from the data. X-band EPR spectra were recorded using a MD5 dielectric resonator (Bruker, Billerica, MA, USA). The spectrometer is equipped with a CF935 cryostat connected to an Intelligent Temperature Controller (Oxford Instruments, Abingdon, UK).
3.2. Synthesis
[Ce(TpMe2)2(κ2-dmpz)] (1). THF (30 mL) was added to a Schlenk containing [Ce(I)3(THF)4] (1.62 g, 2 mmol) and [K(TpMe2)] (1.35 g, 4 mmol) at −78 °C. The yellow suspension was allowed to slowly warm to room temperature and was stirred for 72 h. Volatiles were removed in vacuo and the solid was extracted with toluene (13 mL), filtered and stored at −25 °C overnight to give yellow crystals of 1. A second crop was obtained (1.06 g, 64%). Analysis Calculated (Anal. Calcd) for C35H51B2CeN14: C 50.65, H 6.19, N 23.62. Found: C 50.77, H 6.28, N, 23.55. 1H NMR (d6-benzene, 298 K): δ −7.60 (s, 6H, dmpz-CH3), 3.61 (br, 36H, TpMe2-CH3), 8.33 (br, 1H, dpmz-CH), 8.93 (br, 6H, TpMe2-CH), 18.49 (s, 3H, TpMe2-CH). 11B{1H} NMR (d6-benzene, 298 K): δ −0.66 (s, BH). Evans method susceptibility (d6-benzene, 298 K) μeff: 2.33 µB. FTIR v/cm−1 (Nujol): 2547 (w, B–H str), 1538 (m), 1438 (m), 1200 (m), 1067 (m), 1030 (m), 982 (m), 809 (m), 732 (w), 697 (m), 648 (m).
[Ce(TpMe2)2(bipy)] (2). [Ce(I)3(THF)4] (5.67 g, 7.0 mmol) and [K(TpMe2)] (4.71 g, 14.0 mmol) were suspended in THF (40 mL) at −78 °C. The mixture was stirred for 20 min at −78 °C and bipy (1.09 g, 7.0 mmol) in THF (5 mL) was added dropwise. The mixture was stirred for 20 min at −78 °C and the reaction mixture was transferred into a Schlenk containing KC8 (1.00 g, 7.4 mmol) and THF (10 mL) at −78 °C using a wide bore cannula. The dark brown reaction mixture was allowed to warm to room temperature and was stirred for 96 h. The reaction mixture was concentrated to ca. 30 mL, filtered and stored at −25 °C for 16 h to give dark red crystals of 2. Several more crops were obtained (1.37 g, 22%). Anal. Calcd for C40H52B2CeN14: C, 53.92; H, 5.88; N, 22.00. Found: C, 53.86; H, 5.71; N, 22.25. 1H NMR (d6-benzene, 298 K): δ −9.30 (br, 18H, TpMe2-CH3), −6.14 (br, 1H, bipy-CH), 3.16 (br, 2H, bipy-CH), 3.44 (br, 1H, bipy-CH), 5.05 (br, 18H, TpMe2-CH3), 6.13 (br, 6H, TpMe2-CH), 12.86 (br, 3H, bipy-CH), 19.84 (br, 1H, bipy-CH). 11B{1H} NMR (d6-benzene, 298 K): δ 10.48 (s, BH). Evans method susceptibility (d6-benzene, 298 K) μeff: 1.59 µB. FTIR v/cm−1 (Nujol): 2513 (w, B–H str), 1541 (m, bipy radical anion), 1494 (m, bipy radical anion), 1410 (m), 1286 (m), 1265 (m), 1208 (m), 1168 (m), 1146 (m), 1115 (m), 1072 (m), 1028 (m), 992 (m), 977 (m), 941 (m, bipy radical anion), 829 (m), 773 (m), 641 (m).
[Ce(TpMe2)(μ-BOpMe2)]2 (3). THF (20 mL) was added to 2 (1.71 g, 1.9 mmol) and N-methylmorpholine-N-oxide (0.225 g, 2.0 mmol) at −78 °C. The orange reaction mixture was allowed to slowly warm to room temperature and was stirred for 16 h, forming a white precipitate. Volatiles were removed in vacuo and the solid was extracted with toluene (2 mL), filtered, and stored at −25 °C for 16 h to give pale yellow crystals of 3 (0.25 g, 21%). Anal. Calcd for C50H74B4Ce2N20O2: C 45.82, H 5.69, N 21.60. Found: C 45.63, H 5.43, N 21.36. 1H NMR (d6-benzene, 298 K): δ 3.55 (br, 36H, TpMe2-CH3), 5.85 (s, 12H, BOpMe2-CH3), 8.15 (s, 6H, TpMe2-CH), 8.78 (s, 12H, BOpMe2-CH3), 18.51 (s, 4H, BOpMe2-CH). 11B{1H} NMR (d6-benzene, 298 K): δ 0.60 (s, BH). Evans method susceptibility (d6-benzene, 298 K) μeff: 2.71 µB. FTIR v/cm−1 (Nujol): 2551 (w, B–H str), 1540 (m), 1415 (m), 1198 (m), 1076 (m), 1037 (m), 694 (m), 649 (m).
[Ce(TpMe2)2(N3)] (4). Me3SiN3 (0.12 g, 1.0 mmol) in THF (10 mL) was added dropwise to a suspension of 2 (0.89 g, 1.0 mmol) in THF (10 mL). The reaction mixture was stirred for 3 days and refluxed at 80 °C for 16 h. Volatiles were removed in vacuo and the solid was extracted with toluene (10 mL), filtered, reduced in volume to ca. 2 mL and stored at −25 °C overnight to give yellow crystals of 4 (0.22 g, 29%). Anal. Calcd for C30H44B2CeN15: C, 46.38; H, 5.71; N, 27.04. Found: C, 46.49; H, 5.63; N, 26.86. 1H NMR (d6-benzene, 298 K): δ: −19.32 (s, 2H, TpMe2-CH), −19.09 (s, 6H, TpMe2-CH3), −2.19 (s, 6H, TpMe2-CH3), −0.07 (s, 6H, TpMe2-CH3), 2.42 (s, 6H, TpMe2-CH3), 5.52 (s, 2H, TpMe2-CH), 7.00 (s, 6H, TpMe2-CH3), 10.02 (s, 2H, TpMe2-CH), 12.63 (s, 2H, TpMe2-CH), 18.93 (s, 6H, TpMe2-CH3). 13C{1H} NMR (d6-benzene, 298 K): δ: 11.08 (TpMe2-CH3), 14.91 (TpMe2-CH3), 101.76 (TpMe2-CH), 104.06 (TpMe2-CH), 120.78 (TpMe2-C), 123.24 (TpMe2-CH), 140.12 (TpMe2-C), 141.65 (TpMe2-C), 153.88 (TpMe2-C), 154.89 (TpMe2-C), 189.32 (TpMe2-C). 11B{1H} NMR (d6-benzene, 298 K): δ −25.29 (s, BH). Evans method susceptibility (d6-benzene, 298 K) μeff: 1.94 µB. FTIR v/cm−1 (Nujol): 2555 (w, B–H str), 2083 (s, N3), 2065 (s, N3), 1537 (m), 1201 (m), 1169 (m), 1072 (m), 1031 (m), 978 (w), 936 (w), 640 (w).
3.3. X-ray Crystallography
The crystal data for complexes 1–4 are compiled in Table 2 and Table 3. Crystals were examined on CCD area detector diffractometers using graphite- or mirror-monochromated Mo Kα (λ = 0.71073 Å) or Cu Kα (λ = 1.54184 Å) radiation. Intensities were integrated from data recorded on 1° frames by ω (1–3) or φ and ω rotation (4). Cell parameters were refined from the observed positions of all strong reflections in each data set. A Gaussian grid face-indexed (1–3) or multi-scan (4) absorption correction with a beam profile correction was applied. The structures were solved variously by direct and heavy atom methods and were refined by full-matrix least-squares on all unique F2 values, with anisotropic displacement parameters for all non-hydrogen atoms, and with constrained riding hydrogen geometries; Uiso(H) was set at 1.2 (1.5 for methyl groups) times Ueq of the parent atom. The largest features in final difference syntheses were close to heavy atoms and were of no chemical significance. CrysAlisPro [40] was used for control and integration, SHELXTL [41] and OLEX2 [42] were employed for structure solution and refinement and POVRAY [43] was used for molecular graphics. Highly disordered lattice solvent in 3 could not be modelled and Platon SQUEEZE was used to resolve this component [44]. CCDC 1423263–1423266 (1–4) contain the supplementary crystal data for this article. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Table 2.
Crystallographic data for 1–2.
| Parameter | 1·C7H8 | 2·(C4H8O)2 |
|---|---|---|
| Formula | C42H59B2CeN14 | C48H68B2CeN14O2 |
| Fw | 921.77 | 1034.90 |
| cryst size, mm | 0.171 × 0.152 × 0.095 | 0.116 × 0.110 × 0.095 |
| cryst syst | Triclinic | Monoclinic |
| space group | P–1 | P21/c |
| a, Å | 11.1262(9) | 14.2034(4) |
| b, Å | 14.6920(14) | 20.6250(7) |
| c, Å | 14.7874(14) | 17.1106(6) |
| α, ° | 70.037(9) | 90 |
| β, ° | 85.302(8) | 95.066(3) |
| γ, ° | 81.302(7) | 90 |
| V, Å3 | 2244.7(4) | 4992.9(3) |
| Z | 2 | 4 |
| ρcalcd, g cm−3 | 1.364 | 1.377 |
| μ, mm−1 | 1.061 | 0.965 |
| no. of reflections measd | 15541 | 22174 |
| no. of unique reflns, Rint | 8201, 0.0954 | 9125, 0.0630 |
| no. of reflns with F2 > 2σ(F2) | 6502 | 7004 |
| transmn coeff range | 0.806–1.000 | 0.965–1.000 |
| R, Rw a (F2 > 2σ(F2)) | 0.0642, 0.1312 | 0.0523, 0.1058 |
| R, Rw a (all data) | 0.0845, 0.1414 | 0.0746, 0.1163 |
| S a | 1.046 | 1.029 |
| Number of parameters | 547 | 708 |
| max., min. diff map, e Å−3 | 3.59, −0.17 | 1.55, −1.61 |
a Conventional R = Ʃ||Fo| − |Fc||/Ʃ|Fo|; Rw = [Ʃw(Fo2 − Fc2)2/Ʃw(Fo2)2]1/2; S = [Ʃw(Fo2 − Fc2)2/no. data—no. params)]1/2 for all data.
Table 3.
Crystallographic data for 3–4.
| Parameter | 3·(C4H8O)2 | 4·C7H8 |
|---|---|---|
| Formula | C62H90B4Ce2N20O2 | C37H52B2CeN15 |
| Fw | 1447.62 | 868.67 |
| cryst size, mm | 0.320 × 0.260 × 0.164 | 0.279 × 0.204 × 0.129 |
| cryst syst | Triclinic | Triclinic |
| space group | P–1 | P–1 |
| a, Å | 10.0579(4) | 11.2609(3) |
| b, Å | 13.3606(6) | 12.5209(3) |
| c, Å | 13.6905(7) | 16.7464(4) |
| α, ° | 87.438(4) | 105.035(2) |
| β, ° | 73.681(4) | 96.314(2) |
| γ, ° | 71.869(4) | 112.313(3) |
| V, Å3 | 1676.10(14) | 2051.38(10) |
| Z | 1 | 2 |
| ρcalcd, g cm−3 | 1.291 | 1.406 |
| μ, mm−1 | 10.730 | 1.157 |
| no. of reflections measd | 13,249 | 28,373 |
| no. of unique reflns, Rint | 5891, 0.0581 | 28,373, 0.060 |
| no. of reflns with F2 > 2σ(F2) | 5342 | 22,381 |
| transmn coeff range | 0.197–1.000 | 0.687–1.000 |
| R, Rw a (F2 > 2σ(F2)) | 0.0448, 0.1119 | 0.0515, 0.1456 |
| R, Rw a (all data) | 0.0496, 0.1153 | 0.0654, 0.0515 |
| S a | 1.015 | 1.097 |
| Number of parameters | 368 | 510 |
| max., min. diff map, e Å−3 | 1.53, −2.10 | 1.63, −1.08 |
a Conventional R = Ʃ||Fo| − |Fc||/Ʃ|Fo|; Rw = [Ʃw(Fo2 − Fc2)2/Ʃw(Fo2)2]1/2; S = [Ʃw(Fo2 − Fc2)2/no. data—no. params)]1/2 for all data.
4. Conclusions
We have investigated novel synthetic routes to prepare heteroleptic cerium tris(pyrazolyl)borate complexes, including a Ce(II) synthon, 2, that formally contains a monoreduced bipy·− ligand and a Ce(III) center. We used a wide range of techniques to probe the electronic structure of 2 and conclude that the coordinated bipy exhibits features of a monoreduced radical. However, the EPR spectrum and magnetic measurements indicate that the coupling of this radical with the formal 4f1 electron is complex, and future analysis of a more extensive library of Ce(II) synthons containing radical ligands would facilitate a deeper understanding of this coupling. A selected reactivity study of 2 with potential 2e− oxidants was performed and whilst bipy was eliminated in each isolated product cerium was not found in the +4 oxidation state. The formation of 3 and 4 suggests that the reaction of N-oxides and azides with Ce(II) synthons supported by bipy·− and TpMe2 is not a favored route to Ce(IV) complexes exhibiting Ce=O or Ce=NR multiple bonds under the conditions we employed.
Supplementary Materials
Supplementary materials can be found at http://www.mdpi.com/2304-6740/3/4/0534/s1.
Acknowledgments
We thank the Engineering and Physical Sciences Research Council (Grant No EP/K039547/1, EP/L018470/1 and EP/L014416/1), including the national Electron Paramagnetic Resonance Facility, and The University of Manchester for funding this work. Additional research data supporting this publication are available from The University of Manchester eScholar repository at DOI:10.15127/1.280046.
Author Contributions
David P. Mills conceived and designed the experiments. Hao Zhu, Fabrizio Ortu and Marie-Emmanuelle Boulon performed the experiments. David P. Mills, Fabrizio Ortu, Marie-Emmanuelle Boulon and Hao Zhu analyzed the data. David P. Mills wrote the paper, with contributions from all co-authors.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nugent, W.A.; Mayer, J.M. Metal-Ligand Multiple Bonds; Wiley-Interscience: New York, NY, USA, 1988. [Google Scholar]
- Giesbrecht, G.R.; Gordon, J.C. Lanthanide alkylidene and imido complexes. Dalton Trans. 2004, 2387–2393. [Google Scholar] [CrossRef] [PubMed]
- Summerscales, O.T.; Gordon, J.C. Complexes containing multiple bonding interactions between lanthanoid elements and main-group fragments. RSC Adv. 2013, 3, 6682–6692. [Google Scholar] [CrossRef]
- Gregson, M.; Lu, E.; McMaster, J.; Lewis, W.; Blake, A.J.; Liddle, S.T. A cerium(IV)–carbon multiple bond. Angew. Chem. Int. Ed. 2013, 52, 13016–13019. [Google Scholar] [CrossRef] [PubMed]
- Schädle, D.; Meermann-Zimmerman, M.; Schädle, C.; Maichle-Mössmer, C.; Anwander, R. Rare-earth metal complexes with terminal imido ligands. Eur. J. Inorg. Chem. 2015, 2015, 1334–1339. [Google Scholar]
- So, Y.-M.; Wang, G.-C.; Li, Y.; Sung, H.H.-Y.; Williams, I.D.; Lin, Z.; Leung, W.-H. A tetravalent cerium complex containing a Ce=O bond. Angew. Chem. Int. Ed. 2014, 53, 1626–1629. [Google Scholar] [CrossRef] [PubMed]
- Lu, E.; Li, Y.; Chen, Y. A scandium terminal imido complex: Synthesis, structure and DFT studies. Chem. Commun. 2010, 46, 4469–4471. [Google Scholar] [CrossRef] [PubMed]
- Hayton, T.W. Metal-ligand multiple bonding in uranium: Structure and reactivity. Dalton Trans. 2010, 39, 1129–1404. [Google Scholar] [CrossRef] [PubMed]
- Hayton, T.W. Recent developments in actinide-ligand multiple bonding. Chem. Commun. 2013, 49, 2956–2973. [Google Scholar] [CrossRef] [PubMed]
- Zi, G.; Jia, L.; Werkema, E.L.; Walter, M.D.; Gottfriedsen, J.P.; Andersen, R.A. Preparation and reactions of base-free bis(1,2,4-tri-tert-butylcyclopentadienyl)uranium oxide, Cp'2UO. Organometallics 2005, 24, 4251–4264. [Google Scholar] [CrossRef]
- Kraft, S.J.; Walensky, J.; Fanwick, P.E.; Hall, M.B.; Bart, S.C. Crystallographic evidence of a base-free uranium(IV) terminal oxo species. Inorg. Chem. 2010, 49, 7620–7622. [Google Scholar] [CrossRef] [PubMed]
- Matson, E.M.; Kiemicki, J.J.; Anderson, N.H.; Fanwick, P.E.; Bart, S.C. Isolation of a uranium(III) benzophenone ketyl radical that displays redox-active ligand behavior. Dalton Trans. 2014, 43, 17885–17888. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.H.; Odoh, S.O.; Yao, Y.; Williams, U.J.; Schaefer, B.A.; Kiernicki, J.J.; Lewis, A.J.; Goshert, M.D.; Fanwick, P.E.; Schelter, E.J.; et al. Harnessing redox activity for the formation of uranium tris(imido) compounds. Nat. Chem. 2014, 6, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Zi, G.; Walter, M.D. Synthesis, structure and reactivity of a thorium metallocene containing a 2,2ʹ-bipyridyl ligand. Organometallics 2012, 31, 672–679. [Google Scholar] [CrossRef]
- Nugent, L.J.; Baybarz, R.D.; Burnett, J.L.; Ryan, J.L. Electron-transfer and f-d absorption bands of some lanthanide and actinide complexes and the standard (II–III) oxidation potential for each member of the lanthanide and actinide series. J. Phys. Chem. 1973, 77, 1528–1539. [Google Scholar] [CrossRef]
- Kraft, S.J.; Fanwick, P.E.; Bart, S.C. Synthesis and characterization of a uranium(III) complex containing a redox-active 2,2ʹ-bipyridine ligand. Inorg. Chem. 2010, 49, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Roitershtein, D.; Domingos, A.; Pereira, L.C.J.; Ascenso, J.R.; Marques, N. Coordination of 2,2ʹ-bipyridyl and 1,10-phenanthroline to yttrium and lanthanum complexes based on a scorpionate ligand. Inorg. Chem. 2003, 42, 7666–7673. [Google Scholar] [CrossRef] [PubMed]
- Marques, N.; Sella, A.; Takats, J. Chemistry of the lanthanides using pyrazolylborate ligands. Chem. Rev. 2002, 102, 2137–2159. [Google Scholar] [CrossRef] [PubMed]
- Booth, C.H.; Walter, M.D.; Kazhdan, D.; Hu, Y.-J.; Lukens, W.W.; Bauer, E.D.; Maron, L.; Eisenstein, O.; Andersen, R.A. Decamethylytterbocene complexes of bipyridines and diazabutadienes: Multiconfigurational ground states and open-shell singlet formation. J. Am. Chem. Soc. 2009, 131, 6480–6491. [Google Scholar] [CrossRef] [PubMed]
- Nocton, G.; Booth, C.H.; Maron, L.; Andersen, R.A. Influence of the torsion angle in 3,3ʹ-dimethyl-2,2ʹ-bipyridine on the intermediate valence of Yb in (C5Me5)2Yb(3,3ʹ-Me2-bipy). Organometallics 2013, 32, 5305–5312. [Google Scholar] [CrossRef]
- Izod, K.; Liddle, S.T.; Clegg, W. A convenient route to lanthanide triiodide THF solvates. Crystal structures of LnI3(THF)4 [Ln = Pr] and LnI3(THF)3.5 [Ln = Nd, Gd, Y]. Inorg. Chem. 2004, 43, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Trofimenko, S. Boron-pyrazole chemistry. IV. Carbon- and boron-substituted poly(1-pyrazolyl)borates. J. Am. Chem. Soc. 1967, 89, 6288–6294. [Google Scholar] [CrossRef]
- Kunrath, F.A.; Casagrande, O.L., Jr.; Toupet, L.; Carpentier, J.-F. Synthesis and reactivity in salt metathesis reactions of trivalent [La(TpMe2)2X] (X = Cl, I) complexes: Crystal structures of [La(TpMe2)2Cl] and [La(TpMe2)2(κ2-pzMe2)]. Polyhedron 2004, 23, 2437–2445. [Google Scholar] [CrossRef]
- Galler, J.L.; Goodchild, S.; Gould, J.; McDonald, R.; Sella, A. Tris-pyrazolylborate complexes of redox inactive lanthanides—The structures of [(TpMe2)2NdX] (X = Cl, NPh2, dpm, H2BEt2). Polyhedron 2004, 23, 253–262. [Google Scholar] [CrossRef]
- Hillier, A.C.; Zhang, X.; Maunder, G.H.; Liu, S.Y.; Eberspacher, T.A.; Metz, M.V.; McDonald, R.; Domingos, A.; Marques, N.; Day, V.W.; et al. Synthesis and structural comparison of a series of divalent Ln(TpR,Rʹ)2 (Ln = Sm, Eu, Yb) and trivalent Sm(TpMe2)2X (X = F, Cl, I, BPh4) complexes. Inorg. Chem. 2001, 40, 5106–5116. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-Y.; Maunder, G.H.; Sella, A.; Stevenson, M.; Tocher, D.A. Synthesis and molecular structures of hydrotris(dimethylpyrazolyl)borate complexes of the lanthanides. Inorg. Chem. 1996, 35, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Amberger, H.-D.; Edelmann, F.T.; Gottfriedsen, J.; Herbst-Irmer, R.; Jank, S.; Kilimann, M.; Reddmann, H.; Schäfer, M. Synthesis, molecular, and electronic structure of (η8-C8H8)Ln(scorpionate) half-sandwich complexes: An experimental key to a better understanding of f-element-cyclooctatetraenyl bonding. Inorg. Chem. 2009, 48, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.F. The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. J. Chem. Soc. 1959, 2003–2005. [Google Scholar] [CrossRef]
- Van Vleck, J.H. Theory of Electric and Magnetic Susceptibilities; Oxford University Press: Oxford, UK, 1932. [Google Scholar]
- Akita, M.; Otha, K.; Takahashi, Y.; Hikichi, S.; Moro-oka, Y. Synthesis and structure determination of Rh–diene complexes with the hydridotris(3,5-diisopropylpyrazolyl)borate ligand, TpiPrRh(diene) (diene = cod, nbd): Dependence of the v(B–H) values on the hapticity of the TpiPr ligand (κ2 vs. κ3). Organometallics 1997, 16, 4121–4128. [Google Scholar] [CrossRef]
- Lopes, I.; Lin, G.Y.; Domingos, A.; Marques, N.; Takats, J. Unprecedented transformation of a hydrotris(pyrazolyl)borate ligand at a metal center: Synthesis and rearrangement of the first mixed Tp/Cp lanthanide complex, Sm(TpMe2)2(Cp). J. Am. Chem. Soc. 1999, 121, 8110–8111. [Google Scholar] [CrossRef]
- Shannon, R.D.; Prewitt, C.T. Revised values of effective ionic radii. Acta Crystallogr. Sect. B 1970, 26, 1046–1048. [Google Scholar] [CrossRef]
- Bergbreiter, D.E.; Killough, J.M. Reactions of potassium-graphite. J. Am. Chem. Soc. 1978, 100, 2126–2134. [Google Scholar] [CrossRef]
- Mehdoui, T.; Berthet, J.-C.; Thuéry, P.; Salmon, L.; Rivière, E.; Ephritikhine, M. Lanthanide(III)/actinide(III) differentiation in the cerium and uranium complexes [M(C5Me5)2(L)]0,+ (L = 2,2ʹ-bipyridine, 2,2ʹ:6ʹ,2ʹʹ-terpyridine): Structural magnetic and reactivity studies. Chem. Eur. J. 2005, 11, 6994–7006. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, C.V.; Creutz, C.; Schwarz, H.A.; Sutin, N. Reduction potentials for 2,2ʹ-bipyridine and 1,10ʹ-phenanthroline couples in aqueous solutions. J. Am. Chem. Soc. 1983, 105, 5617–5623. [Google Scholar] [CrossRef]
- Schelter, E.J. Cerium under the lens. Nat. Chem. 2013, 5, 348. [Google Scholar] [CrossRef] [PubMed]
- Domingos, A.; Elsegood, M.R.J.; Hillier, A.C.; Lin, G.; Liu, S.Y.; Lopes, I.; Marques, N.; Maunder, G.H.; McDonald, R.; Sella, A.; et al. Facile pyrazolylborate ligand degradation at lanthanide centers: X-ray crystal structures of pyrazolylborinate-bridged bimetallics. Inorg. Chem. 2002, 41, 6761–6768. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, A.; Cladis, D.P.; Forrest, W.P.; Fanwick, P.E.; Bart, S.C. Reductive heterocoupling mediated by Cp*2U(2,2ʹ-bpy). Chem. Commun. 2012, 48, 1671–1673. [Google Scholar] [CrossRef] [PubMed]
- Dröse, P.; Gottfriedsen, J.; Hrib, C.G.; Jones, P.G.; Hilfert, L.; Edelmann, F.T. The first cationic complex of tetravalent cerium. Z. Anorg. Allg. Chem. 2011, 637, 369–373. [Google Scholar] [CrossRef]
- CrysAlis PRO, version 37; Agilent Technologies: Yarnton, UK, 2010.
- Sheldrick, G.M. Crystal structure refinement with SHELX. Acta Cryst. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- POVRAY, version 3.7.0. Persistence of Vision Pty. Ltd.: Williamstown, Australia, 2004. Available online: http://www.povray.org/ (Accessed on 16 August 2015).
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).