
Fluorene-Containing β-Diketonato Ligands and Their Rhodium(I) Complexes—A Characterization and Crystallographic Study


Abstract

1. Introduction
2. Results and Discussion
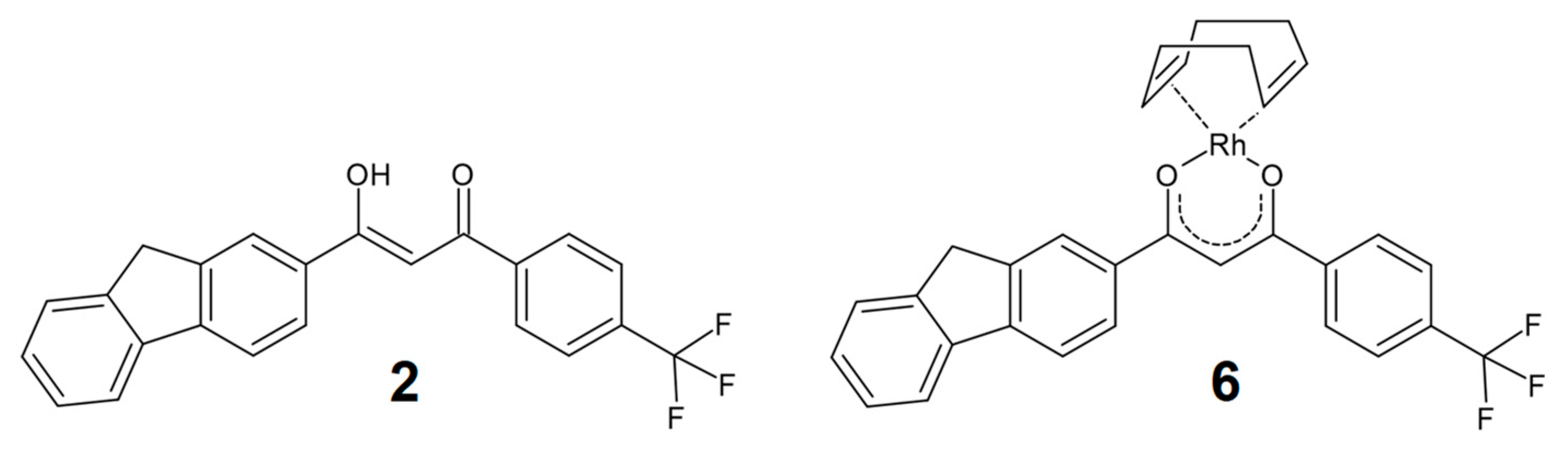
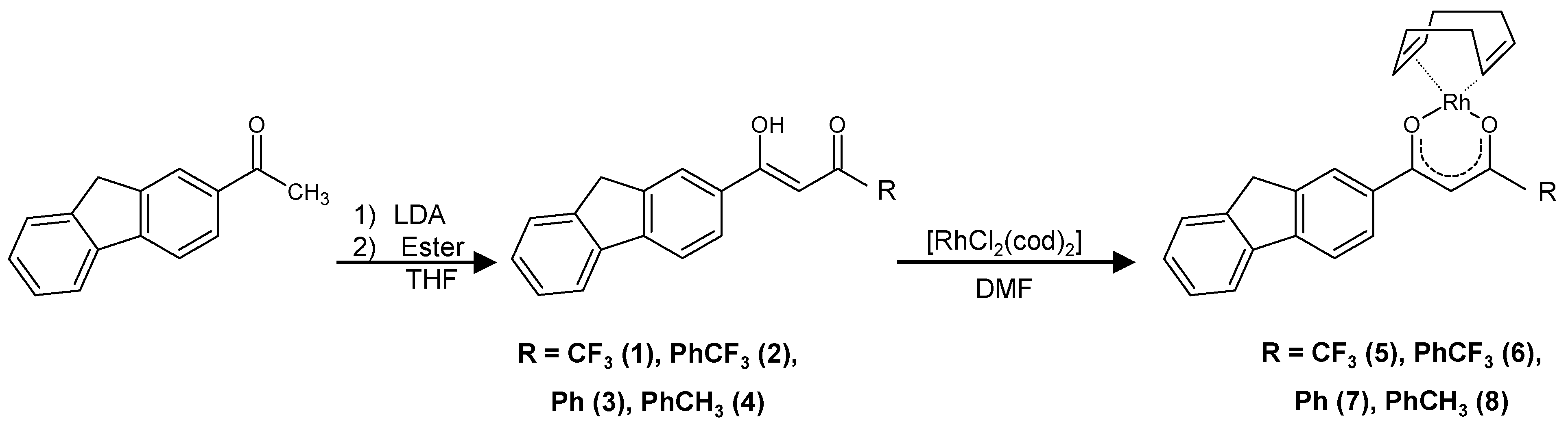
2.1. Synthesis and Characterization
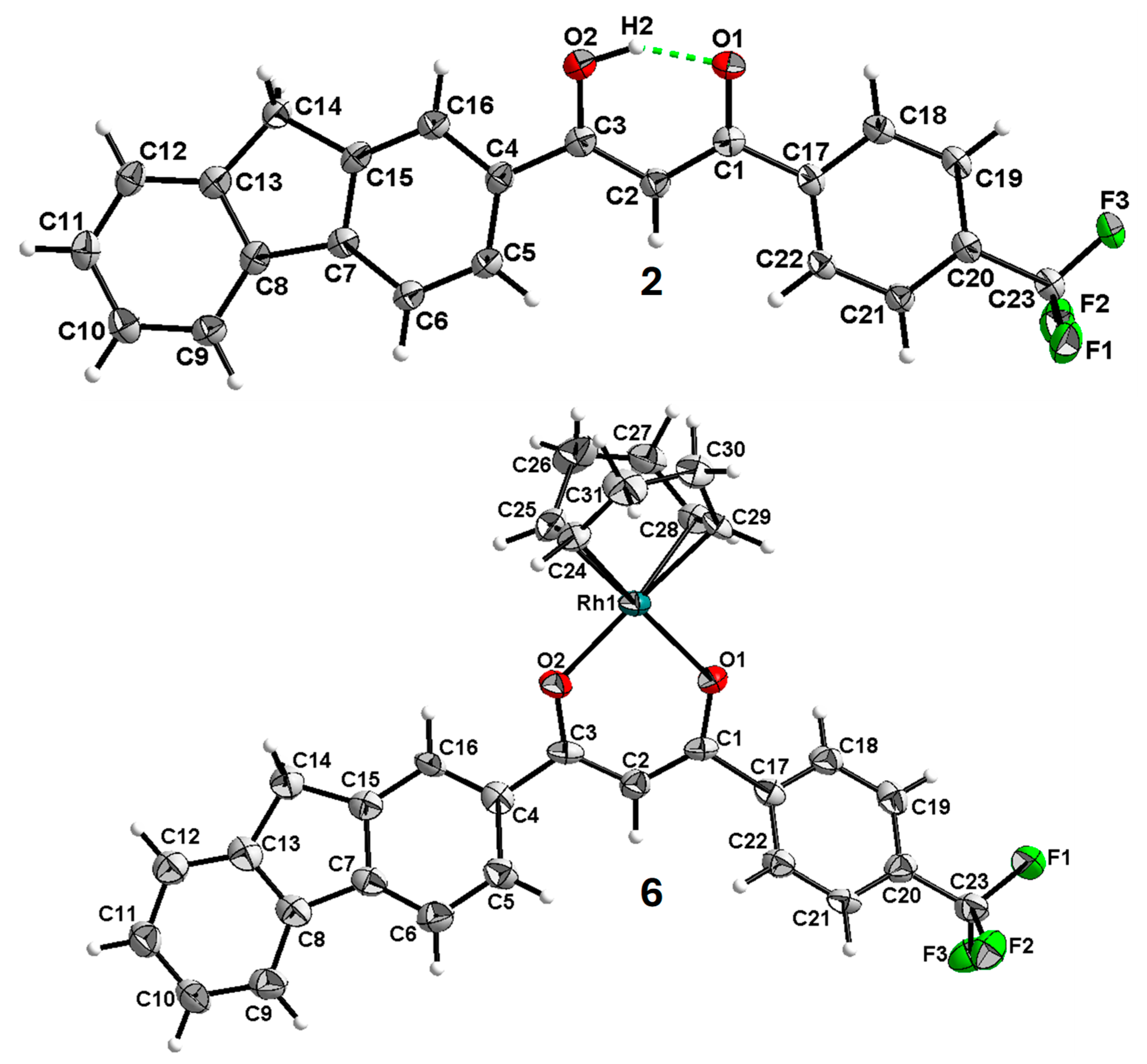
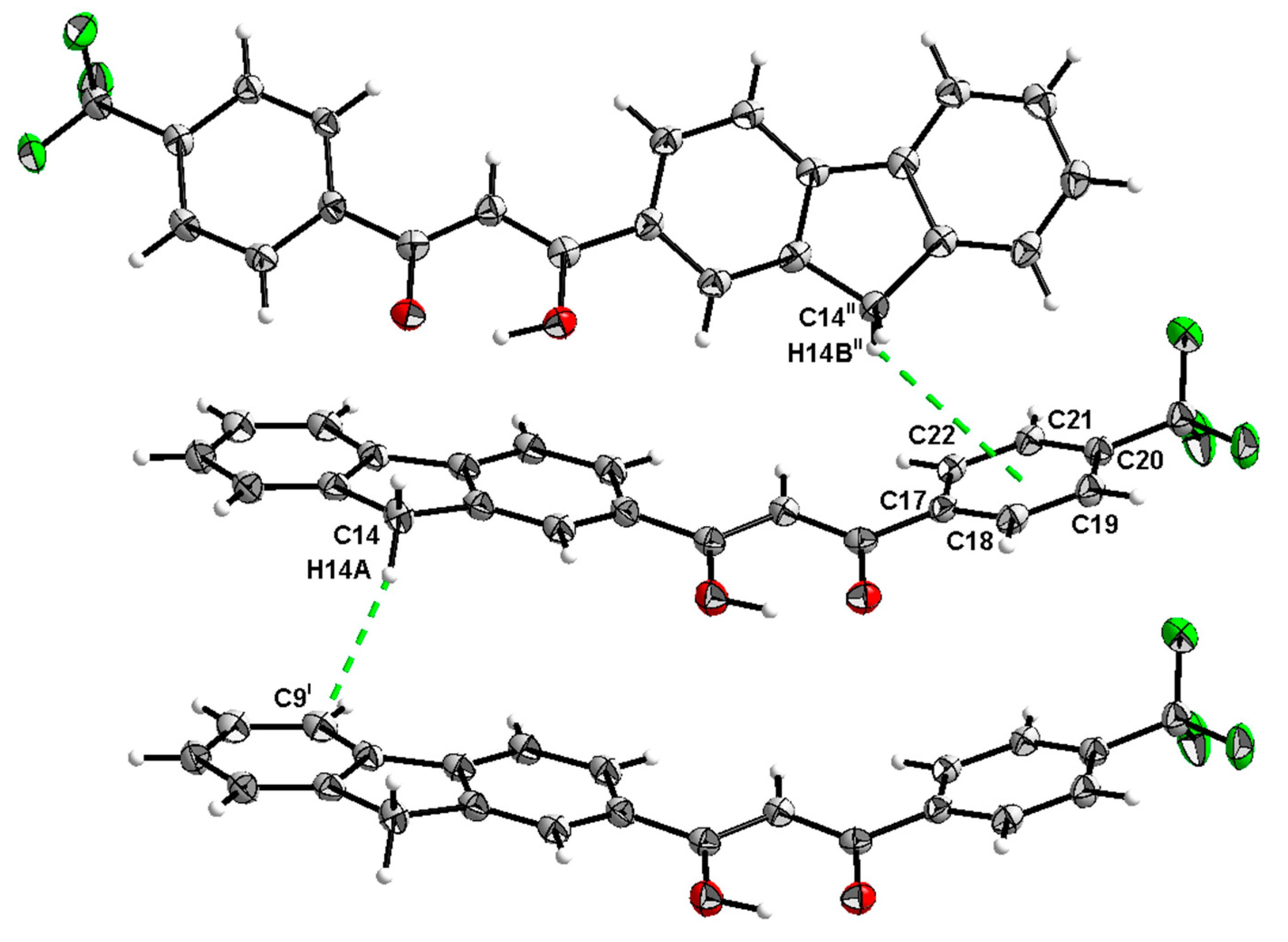
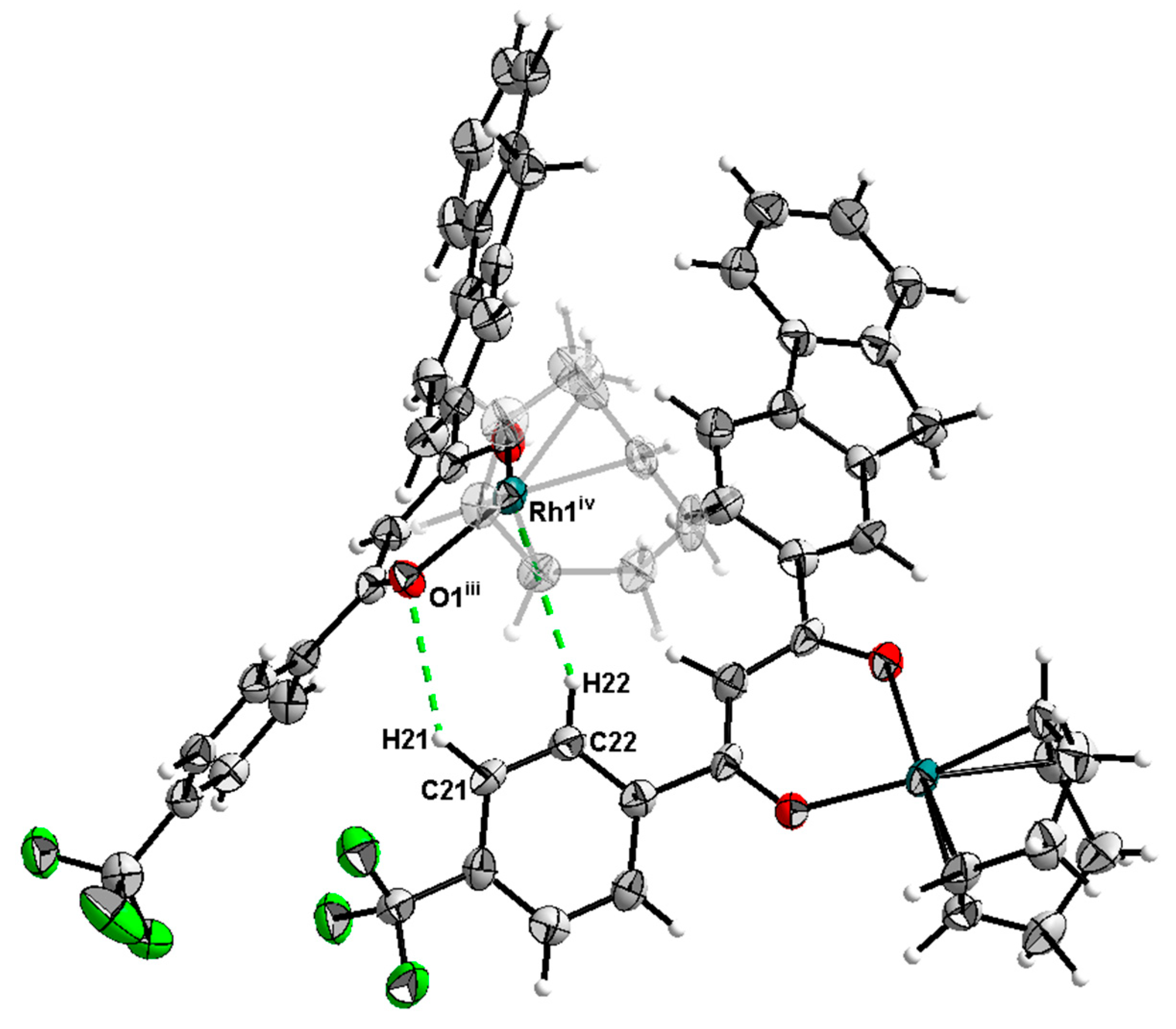
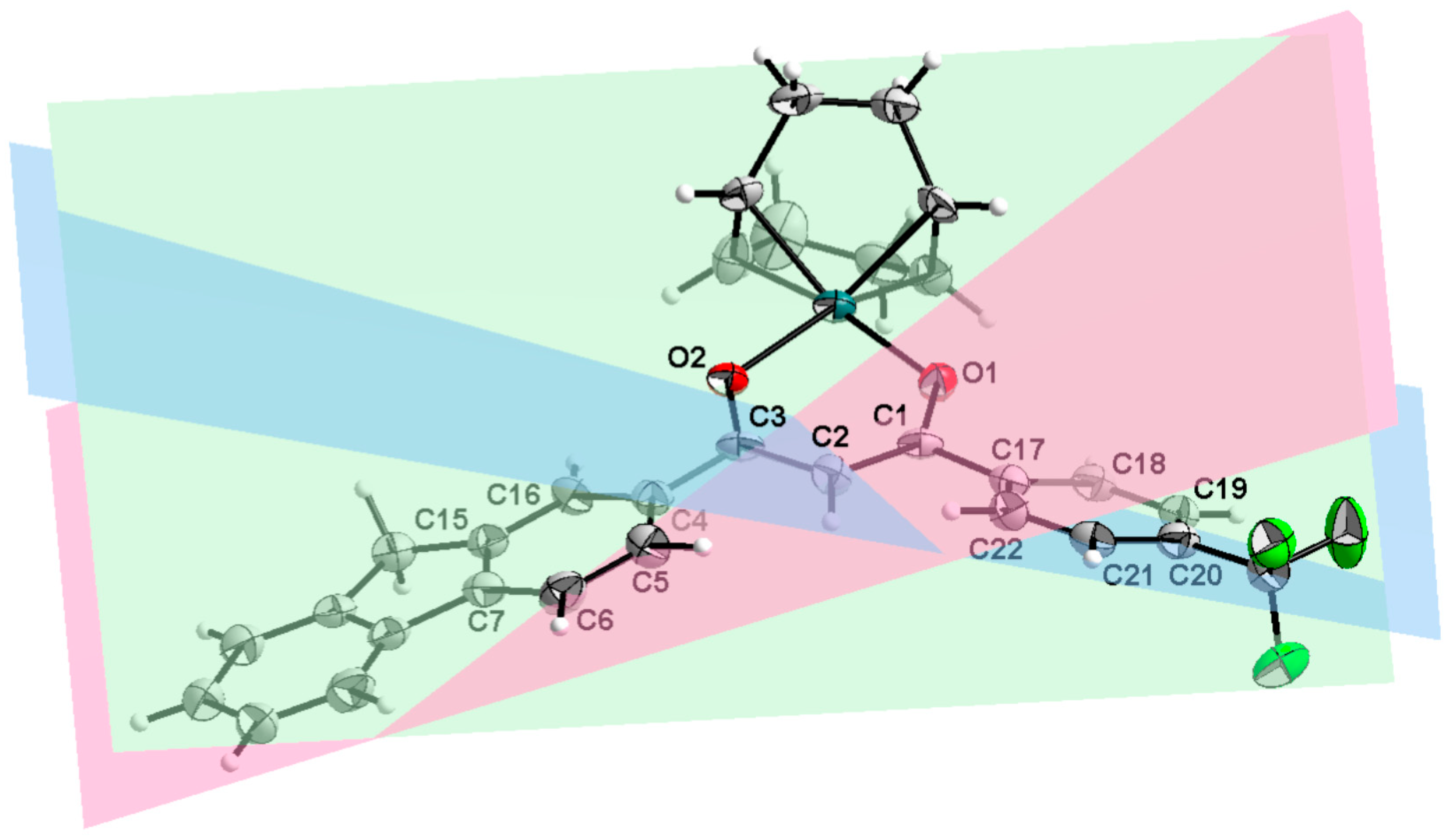
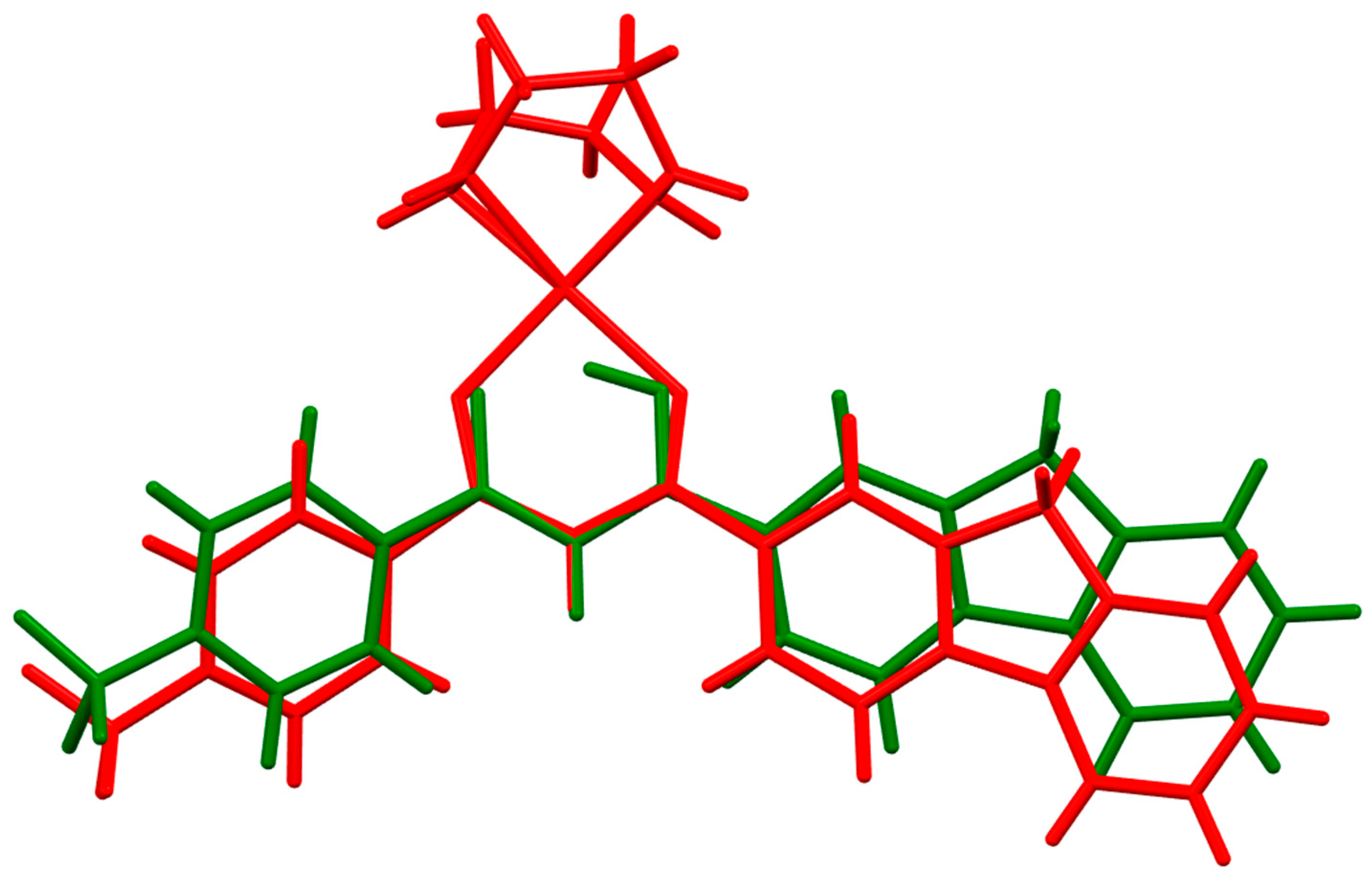
2.2. X-Ray Structure Determination
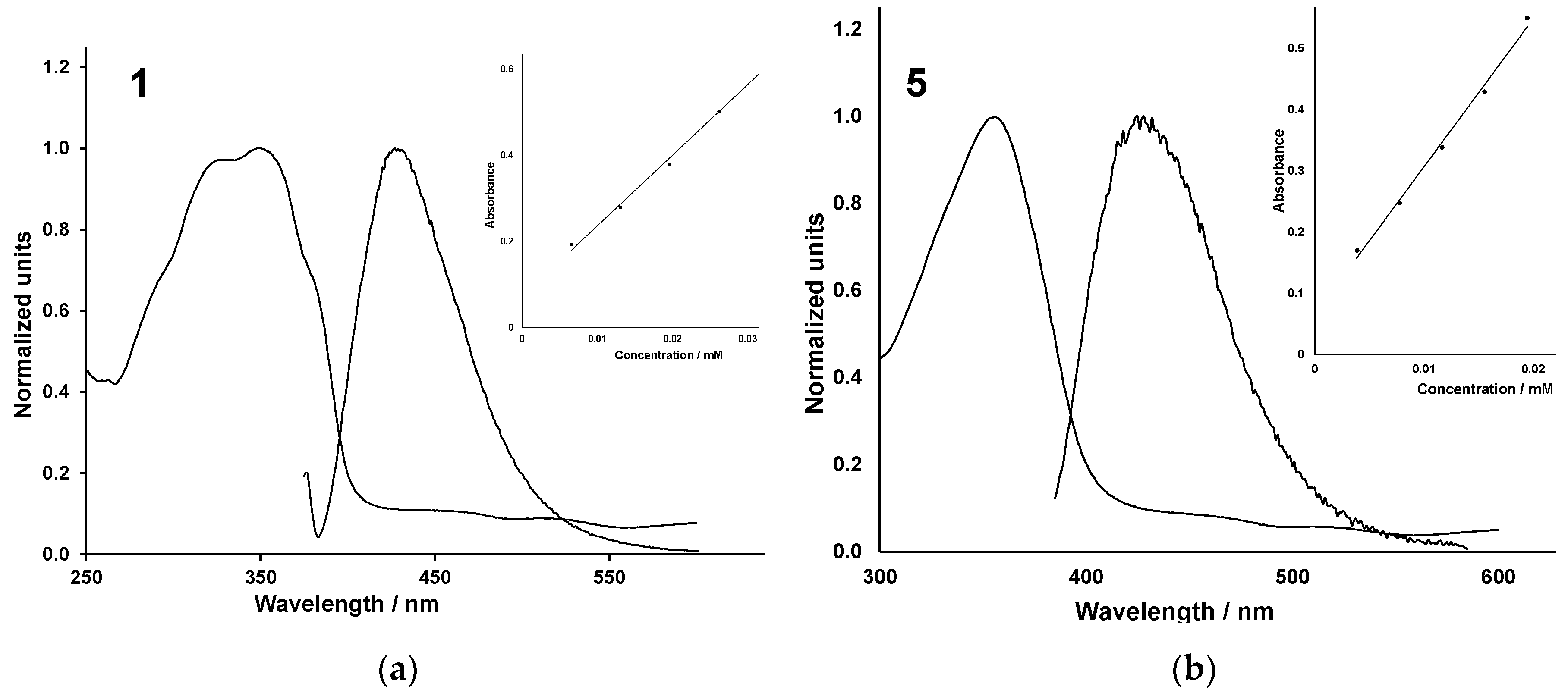
2.3. Spectroscopy
2.4. Electrochemistry
3. Materials and Methods
3.1. General
3.2. Synthesis
3.2.1. β-Diketone Syntheses
3.2.2. Rh Complexation
3.3. X-Ray Structure Determination and Refinement of 2 and 6
3.4. Electrochemistry of 1–8
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| cod | cyclooctadiene |
| LDA | Lithium diisopropylamine |
| NMR | Nuclear magnetic resonance |
| FTIR | Fourier transform infrared |
| CSD | Cambridge structural database |
| CV | Cyclic voltammetry |
| FcH | Ferrocene |
| Fc* | Decamethylferrocene |
| THF | tetrahydrofuran |
References
- Shaya, J.; Corridon, P.R.; Al-Omari, B.; Aoudi, A.; Shunnar, A.; Mohideen, M.I.H.; Qurashi, A.; Michel, B.Y.; Burger, A. Design, photophysical properties, and applications of fluorene-based fluorophores in two-photon fluorescence bioimaging: A review. J. Photochem. Photobiol. C Photochem. Rev. 2022, 52, 100529. [Google Scholar] [CrossRef]
- Liu, J.-W.; Zhou, H.-C.; Wang, Z.-K.; Tang, X.; Wu, H.-Y.; Wang, S.; Lai, W.-Y.; Li, Y.-H. Distinct Ir(III) complexes containing unsymmetric ligands with fluorene-oxadiazole groups and their performance of organic light-emitting diodes. Dye. Pigment. 2022, 202, 110252. [Google Scholar] [CrossRef]
- Tahir Waseem, M.; Muhammad Junaid, H.; Gul, H.; Ali Khan, Z.; Yu, C.; Anjum Shahzad, S. Fluorene based fluorescent and colorimetric sensors for ultrasensitive detection of nitroaromatics in aqueous medium. J. Photochem. Photobiol. A Chem. 2022, 425, 113660. [Google Scholar] [CrossRef]
- Bezgin Carbas, B. Fluorene based electrochromic conjugated polymers: A review. Polymer 2022, 254, 125040. [Google Scholar] [CrossRef]
- Li, H.; Yu, M.; Luo, M.; Li, Y.; Chen, Z.; He, J.; Wu, M.; Liu, Y.; Wang, K.; Zhan, X.; et al. Reconfigurable amplified circularly polarized photo/electro-luminescence from simple solution self-assembly of a chiral deep-blue fluorene-based conjugated polymer. J. Mater. Chem. C 2025, 13, 13731–13741. [Google Scholar] [CrossRef]
- Zhu, S.; Pan, Q.; Li, Y.; Liu, W.; Liu, R.; Zhu, H. Fluorene-decorated Ir(iii) complexes: Synthesis, photophysics and tunable triplet excited state properties in aggregation. Dalton Trans. 2022, 51, 13322–13330. [Google Scholar] [CrossRef]
- Pham, H.D.; Gil-Escrig, L.; Feron, K.; Manzhos, S.; Albrecht, S.; Bolink, H.J.; Sonar, P. Boosting inverted perovskite solar cell performance by using 9,9-bis(4-diphenylaminophenyl)fluorene functionalized with triphenylamine as a dopant-free hole transporting material. J. Mater. Chem. A 2019, 7, 12507–12517. [Google Scholar] [CrossRef]
- Zuo, X.; Zhang, S.; Bai, H.; Yu, Q.; Zhao, Q.; Sun, M.; Zhao, X.; Feng, X. Effects of fluorene-9-bisphenol exposure on anxiety-like and social behavior in mice and protective potential of exogenous melatonin. Environ. Sci. Pollut. Res. 2024, 31, 29385–29399. [Google Scholar] [CrossRef] [PubMed]
- Hussein, E.M.; Alsantali, R.I.; Morad, M.; Obaid, R.J.; Altass, H.M.; Sayqal, A.; Abourehab, M.A.S.; Elkhawaga, A.A.; Aboraia, A.S.M.; Ahmed, S.A. Bioactive fluorenes. Part III: 2,7-dichloro-9H-fluorene-based thiazolidinone and azetidinone analogues as anticancer and antimicrobial against multidrug resistant strains agents. BMC Chem. 2020, 14, 42. [Google Scholar] [CrossRef]
- Urade, R.; Chang, W.-T.; Ko, C.-C.; Li, R.-N.; Yang, H.-M.; Chen, H.-Y.; Huang, L.-Y.; Chang, M.-Y.; Wu, C.-Y.; Chiu, C.-C. A fluorene derivative inhibits human hepatocellular carcinoma cells by ROS-mediated apoptosis, anoikis and autophagy. Life Sci. 2023, 329, 121835. [Google Scholar] [CrossRef]
- Feng, Q.; Liu, Y.; Zou, L.; Lei, M.; Zhu, C.; Xia, W. Fluorene-9-bisphenol exposure damages the testis in mice through a novel mechanism of ferroptosis. Food Chem. Toxicol. 2024, 184, 114385. [Google Scholar] [CrossRef]
- Kumbar, M.; Patil, S.A.; Toragalmath, S.S.; Jawoor, S.S.; Shettar, A. Synthesis of novel metal (II) complexes tailored from 9-oxo-9H-fluorene-1-carboxylic acid via green protocol: DNA cleavage and anticancer studies. Inorganica Chim. Acta 2020, 500, 119210. [Google Scholar] [CrossRef]
- Marufa, S.S.; Rahman, T.; Rahman, M.M.; Rahman, M.M.; Khan, S.J.; Jahan, R.; Nishino, H.; Alam, M.S.; Haque, M.A. Design, synthesis, molecular docking, and dynamics studies of novel thiazole-Schiff base derivatives containing a fluorene moiety and the assessment of their antimicrobial and antioxidant activity. RSC Adv. 2024, 14, 35198–35214. [Google Scholar] [CrossRef]
- Haghdoost, M.M.; Golbaghi, G.; Guard, J.; Sielanczyk, S.; Patten, S.A.; Castonguay, A. Synthesis, characterization and biological evaluation of cationic organoruthenium(ii) fluorene complexes: Influence of the nature of the counteranion. Dalton Trans. 2019, 48, 13396–13405. [Google Scholar] [CrossRef]
- Popa, A.D.; Mădălan, A.M. Silver(I) complexes with a luminescent tripodal Schiff base ligand derived from fluorene-2-carboxaldehyde. Polyhedron 2022, 220, 115849. [Google Scholar] [CrossRef]
- Zhao, Z.; Han, X.; Liu, K.; Zhao, L.; Liu, Q. Six Fluorene-Based N-Heterocyclic Carbene Silver(I) Complexes: Structural Study and Recognition for Cu2+. Organometallics 2022, 41, 2394–2405. [Google Scholar] [CrossRef]
- Tian, Z.; Yang, X.; Liu, B.; Zhong, D.; Zhou, G. Photophysical properties and optical power limiting ability of Pt(II) polyynes bearing fluorene-type ligands with ethynyl units at different positions. J. Organomet. Chem. 2019, 895, 28–36. [Google Scholar] [CrossRef]
- Fourie, E. Fluorene-containing β-diketonato ligands and their Rhodium(I) complexes. Inorganica Chim. Acta 2021, 517, 120209. [Google Scholar] [CrossRef]
- de Gonzalo, G.; Alcántara, A.R. Recent Developments in the Synthesis of β-Diketones. Pharmaceuticals 2021, 14, 1043. [Google Scholar] [CrossRef]
- Woodward, A.W.; Frazer, A.; Morales, A.R.; Yu, J.; Moore, A.F.; Campiglia, A.D.; Jucov, E.V.; Timofeeva, T.V.; Belfield, K.D. Two-photon sensitized visible and near-IR luminescence of lanthanide complexes using a fluorene-based donor–π-acceptor diketonate. Dalton Trans. 2014, 43, 16626–16639. [Google Scholar] [CrossRef]
- Suwa, Y.; Yamaji, M.; Okamoto, H. Synthesis and photophysical properties of difluoroboronated β-diketones with the fluorene moiety that have high fluorescence quantum yields. Tetrahedron Lett. 2016, 57, 1695–1698. [Google Scholar] [CrossRef]
- Kemp, K.C.; Fourie, E.; Conradie, J.; Swarts, J.C. Ruthenocene-Containing β-Diketones: Synthesis, pKa′ Values, Keto–Enol Isomerization Kinetics, and Electrochemical Aspects. Organometallics 2008, 27, 353–362. [Google Scholar] [CrossRef]
- Conradie, J.; Lamprecht, G.J.; Otto, S.; Swarts, J.C. Synthesis and characterisation of ferrocene-containing β-diketonato complexes of rhodium(I) and rhodium(III). Inorganica Chim. Acta 2002, 328, 191–203. [Google Scholar] [CrossRef]
- Brandenburg, K.; Putz, H. Diamond, version 4.0; 53002; Crystal Impact GbR: Bonn, Germany, 2005. [Google Scholar]
- Wang, Q.; Bian, Y.; Dhawan, G.; Zhang, W.; Sorochinsky, A.E.; Makarem, A.; Soloshonok, V.A.; Han, J. FDA approved fluorine-containing drugs in 2023. Chin. Chem. Lett. 2024, 35, 109780. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Wu, K.; Zhang, T.; Wang, Z.; Wang, L.; Zhan, L.; Gong, S.; Zhong, C.; Lu, Z.-H.; Zhang, S.; Yang, C. De Novo Design of Excited-State Intramolecular Proton Transfer Emitters via a Thermally Activated Delayed Fluorescence Channel. J. Am. Chem. Soc. 2018, 140, 8877–8886. [Google Scholar] [CrossRef] [PubMed]
- Dalal, A.; Kamboj, R.C.; Kumar, D.; Sharma, M.K.; Selvarajan, N. Crystal structure of (Z)-3-[5-chloro-2-(prop-2-ynyloxy)phenyl]-3-hydroxy-1-[4-(trifluoromethyl)phenyl]prop-2-en-1-one. Acta Crystallogr. Sect. E 2015, 71, o556–o557. [Google Scholar] [CrossRef]
- Sato, K.; Sandford, G.; Shimizu, K.; Akiyama, S.; Lancashire, M.J.; Yufit, D.S.; Tarui, A.; Omote, M.; Kumadaki, I.; Harusawa, S.; et al. Synthesis of fluorinated isoxazoles using Selectfluor™: Preparation and characterization of 4-fluoroisoxazole, 4,4,5-trifluoroisoxazoline and 4,4-difluoro-5-hydroxyisoxazoline systems from one-pot and multi-step processes. Tetrahedron 2016, 72, 1690–1698. [Google Scholar] [CrossRef]
- Butler, T.; Mathew, A.S.; Sabat, M.; Fraser, C.L. Camera Method for Monitoring a Mechanochromic Luminescent β-Diketone Dye with Rapid Recovery. ACS Appl. Mater. Interfaces 2017, 9, 17603–17612. [Google Scholar] [CrossRef]
- Jecny, J.; Huml, K. The crystal and molecular structure of 1,3-diphenyl-1,3-propanedione-(1,6-dichloro-1,5-cyclooctadiene)rhodium(I). Acta Crystallogr. Sect. B 1974, 30, 1105–1110. [Google Scholar] [CrossRef]
- Mang, A.; Rotthowe, N.; Beltako, K.; Linseis, M.; Pauly, F.; Winter, R.F. Single-molecule conductance studies on quasi- and metallaaromatic dibenzoylmethane coordination compounds and their aromatic analogs. Nanoscale 2023, 15, 5305–5316. [Google Scholar] [CrossRef]
- Jecny, J.; Huml, K. Structure of [1,3-bis(4-methoxyphenyl)-1,3-propanedionato](1,6-dichloro-1,5-cyclooctadiene)rhodium(I), C25H25Cl2O4Rh. Acta Crystallogr. Sect. C 1985, 41, 503–504. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Pidluzhna, A.; Vembris, A.; Grzibovskis, R.; Zommere, M.A.; Bezvikonnyi, O.; Simokaitiene, J.; Baronaite, M.; Volyniuk, D.; Grazulevicius, J.V.; Ali, A.; et al. The Effect of Molecular Structure on the Properties of Fluorene Derivatives for OLED Applications. Molecules 2024, 29, 4918. [Google Scholar] [CrossRef]
- Kuhn, A.; von Eschwege, K.G.; Conradie, J. Electrochemical and density functional theory modeled reduction of enolized 1,3-diketones. Electrochim. Acta 2011, 56, 6211–6218. [Google Scholar] [CrossRef]
- Conradie, J.; Swarts, J.C. Relationship between electrochemical potentials and substitution reaction rates of ferrocene-containing β-diketonato rhodium(i) complexes; Cytotoxicity of [Rh(FcCOCHCOPh)(cod)]. Dalton Trans. 2011, 40, 5844–5851. [Google Scholar] [CrossRef] [PubMed]
- Fourie, E.; Niemantsverdriet, J.W.; Swarts, J.C. Synthesis Comparative Electrochemistry and Spectroelectrochemistry of Metallocenyl β-Diketonato Dicarbonyl Complexes of Rhodium(I)—Cytotoxicity of [Rh(FcCOCHCOCF3)(CO)2]. Inorganics 2024, 12, 321. [Google Scholar] [CrossRef]
- Nafady, A.; Chin, T.T.; Geiger, W.E. Manipulating the Electrolyte Medium to Favor Either One-Electron or Two-Electron Oxidation Pathways for (Fulvalendiyl)dirhodium Complexes. Organometallics 2006, 25, 1654–1663. [Google Scholar] [CrossRef]
- Ramollo, G.K.; López-Gómez, M.J.; Liles, D.C.; Matsinha, L.C.; Smith, G.S.; Bezuidenhout, D.I. Rhodium(I) Ferrocenylcarbene Complexes: Synthesis, Structural Determination, Electrochemistry, and Application as Hydroformylation Catalyst Precursors. Organometallics 2015, 34, 5745–5753. [Google Scholar] [CrossRef]
- Conradie, J.; Cameron, T.S.; Aquino, M.A.S.; Lamprecht, G.J.; Swarts, J.C. Synthetic, electrochemical and structural aspects of a series of ferrocene-containing dicarbonyl β-diketonato rhodium(I) complexes. Inorganica Chim. Acta 2005, 358, 2530–2542. [Google Scholar] [CrossRef]
- U. COSMO, version 1.48; Bruker AXS Inc.: Madison, WI, USA, 2003.
- Bruker. SAINT-PLUS, version 7.12; including XPREP; Bruker AXS Inc.: Madison, WI, USA, 2004. [Google Scholar]
- SADABS, version 2016/2; Bruker AXS Inc.: Madison, WI, USA, 2016.
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Gritzner, G.; Kuta, J. Recommendations on reporting electrode potentials in nonaqueous solvents (Recommendations 1983). Pure Appl. Chem. 1984, 56, 461–466. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ligand (2) | Complex (6) |
|---|---|---|
| Crystal data | ||
| Chemical formula | C23H15F3O2 | C31H26F3O2Rh |
| Mr | 380.35 | 590.43 |
| Crystal system, space group | Orthorhombic, Pbca | Monoclinic, P21/c |
| Temperature (K) | 101 | 100 |
| a, b, c (Å) | 14.7394(9), 6.0976(4), 37.668(3) | 23.920(3), 10.749(2), 9.539(1) |
| α, β, γ (°) | 90, 90, 90 | 90, 91.001(8), 90 |
| V (Å3) | 3385.4(4) | 2452.6(6) |
| Z | 8 | 4 |
| µ (mm−1) | 0.988 | 6.068 |
| Crystal size (mm) | 0.239 × 0.048 × 0.001 | 0.190 × 0.032 × 0.011 |
| Data collection | ||
| Diffractometer | Bruker D8 Venture 4K Kappa Photon III C28 diffractometer | Bruker D8 Venture 4K Kappa Photon III C28 diffractometer |
| Wavelength (Å) | 1.54178 (CuKα) | 1.54178 (CuKα) |
| Absorption correction | Multi-scan SADABS2016/2 [3] | Multi-scan SADABS2016/2 [3] |
| Tmin, Tmax | 0.666, 0.754 | 0.569, 0.754 |
| No. of measured, independent, and observed [I > 2σ(I)] reflections | 54,753, 3662, 2887 | 27,666, 5125, 3489 |
| Rint | 0.0663 | 0.886 |
| 2Θ range for data collection/° | 4.692 to 158.948 | 3.694 to 163.776 |
| Completeness (%) | 99.50 | 99.10 |
| Refinement | ||
| R [F2 > 2σ(F2)], wR(F2), S | 0.0505, 0.1323, 1.043 | 0.0806, 0.2016, 1.093 |
| No. of reflections | 3662 | 5125 |
| No. of parameters | 258 | 334 |
| Δρmax, Δρmin (e Å−3) | 0.30, −0.32 | 2.48, −2.01 |
| CCDC No. | 2,466,425 | 2,466,393 |
| Compound | Distances (Å) | Angles (°) | Torsion Angles (°) | Reference | |||
|---|---|---|---|---|---|---|---|
| O1–O2 | O1–C1 | O2–C3 | C20–C23 | C1–C2–C3 | O1–C1–C17–C22 | ||
| 2 | 2.434(2) | 1.287(2) | 1.301(2) | 1.499(3) | 119.8(2) | 159.2(2) | This work |
| 6 | 2.945(7) | 1.28(1) | 1.27(1) | 1.49(1) | 127.7(8) | 149.7(9) | This work |
| HUPPOZ | 2.529(4) | 1.264(3) | 1.310(3) | 1.494(5) | 122.4(3) | −170.5(3) | [28] |
| FAXWAD01 | 2.436(1) | 1.295(2) | 1.295(2) | 1.505(2) | 119.8(2) | −172.0(2) | [29] |
| ACUVUS | 2.463(1) | 1.278(1) | 1.310(1) | N/A | 120.39(8) | 172.12(8) | [30] |
| BICQEM | 2.52(6) | 1.32(1) | 1.32(1) | N/A | 121.2(4) | −154(1) | [27] |
| PPDOCR20 | 2.849(6) | 1.283(6) | 1.283(6) | N/A | 125.6(3) | 153.9(5) | [31] |
| CUNZEQ | 2.86(1) | 1.30(1) | 1.28(1) | N/A | 124.9(9) | 161.2(9) | [32] |
| WICFUN | 2.910(3) | 1.282(3) | 1.282(3) | N/A | 127.1(3) | 153.5(2) | [33] |
| D—H···A | Type of Interaction | D—H (Å) | H···A (Å) | D···A (Å) | D—H···A (°) |
|---|---|---|---|---|---|
| (1) Ligand | |||||
| O2—H2···O1 | H-bond | 0.97(2) | 1.52(2) | 2.434(2) | 151(3) |
| C14—H14A···C9 i | π–CH | 0.99 | 2.68 | 3.639(3) | 162(1) |
| C14—H14B···Cg1 ii | π–CH | 0.99 | 3.21 | 3.875(2) | 125.7(1) |
| (2) Complex | |||||
| C21—H21···O1 iii | H-bond | 0.95 | 2.50 | 3.24(1) | 134 |
| C22—H22···Rh1 iv | Metal-H | 0.95 | 3.07 | 3.897(9) | 146 |
| Planes | Atoms Used to Generate the Plane | Ligand 2 (°) | Complex 6 (°) |
|---|---|---|---|
| Red—Green | C4, C5, C6, C7, C15, and C16 | 18.6(1) | 24.7(3) |
| Green—Blue | O1, C1, C2, C3, and O2 | 21.5(1) | 30.7(3) |
| Red—Blue | C17, C18, C19, C20, C21 and C22 | 17.7(3) | 30.8(3) |
| Compound | λmax (nm) | ε (M−1cm−1) | λmax (nm) | Compound | λmax (nm) | ε (M−1cm−1) | λmax (nm) |
|---|---|---|---|---|---|---|---|
| 1 | 349 | 16,383 | 458, 516 | 5 | 357 | 24,151 | 460, 517 |
| 2 | 370 | 31,030 | 457, 518 | 6 | 366 | 16,643 | 457, 514 |
| 3 | 366 | 39,448 | 455, 515 | 7 | 362 | 21,319 | 454, 514 |
| 4 | 369 | 32,462 | 456, 514 | 8 | 366 | 17,047 | 455, 513 |
| Compound | Wave | Epa (mV) | ipa (μA) | ∆Ep (mV) | E°′ (mV) | ipc/ipa |
|---|---|---|---|---|---|---|
| 1 | A | −1480 * | 4.6 * | 140 | −1410 | 0.80 * |
| 2 | A | −1835 * | 7.3 * | 104 | −1783 | 0.51 * |
| 3 | A | −2019 * | 12.8 * | 132 | −1953 | 0.49 * |
| 4 | A | −2058 * | 12.5 * | 137 | −1989 | 0.47 * |
| 5 | A | −1886 * | 2.8 * | 82 | −1845 | 0.82 * |
| B | 361 | 3.1 | 122 | 300 | 0.29 | |
| 6 | A | −2073 * | 3.1 * | 103 | −2022 | 0.49 * |
| B | 536 | 1.3 | 105 | 483 | 0.61 | |
| 7 | A | −2129 * | 2.3 * | 89 | −2084 | 0.78 * |
| B | 286 | 1.7 | 142 | 215 | 0.47 | |
| 8 | A | −2000 * | 3.2 * | 88 | −1956 | 0.28 * |
| B | 326 | 1.2 | 130 | 315 | 0.42 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacobs, F.J.F.; Khoza, S.; Fourie, E. Fluorene-Containing β-Diketonato Ligands and Their Rhodium(I) Complexes—A Characterization and Crystallographic Study. Inorganics 2025, 13, 255. https://doi.org/10.3390/inorganics13080255
Jacobs FJF, Khoza S, Fourie E. Fluorene-Containing β-Diketonato Ligands and Their Rhodium(I) Complexes—A Characterization and Crystallographic Study. Inorganics. 2025; 13(8):255. https://doi.org/10.3390/inorganics13080255
Chicago/Turabian StyleJacobs, Frederick Jacobus Francois, Siyanda Khoza, and Eleanor Fourie. 2025. "Fluorene-Containing β-Diketonato Ligands and Their Rhodium(I) Complexes—A Characterization and Crystallographic Study" Inorganics 13, no. 8: 255. https://doi.org/10.3390/inorganics13080255
APA StyleJacobs, F. J. F., Khoza, S., & Fourie, E. (2025). Fluorene-Containing β-Diketonato Ligands and Their Rhodium(I) Complexes—A Characterization and Crystallographic Study. Inorganics, 13(8), 255. https://doi.org/10.3390/inorganics13080255