

Synthesis of B-Substituted Anthracenyl and Pyrenyl Derivatives of ortho-Carborane

 , , ,
, , ,  and
and 
Abstract

1. Introduction
2. Results and Discussion
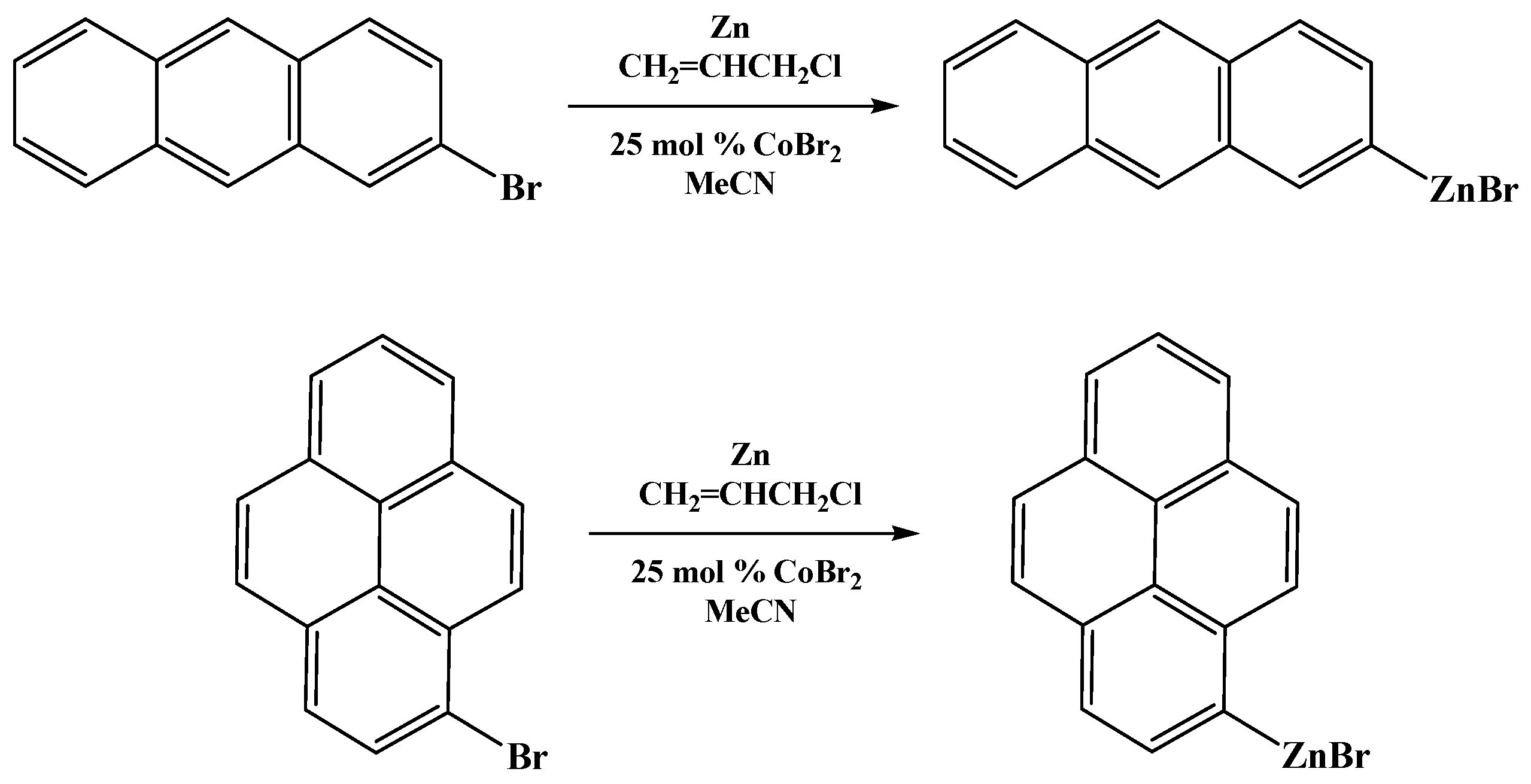
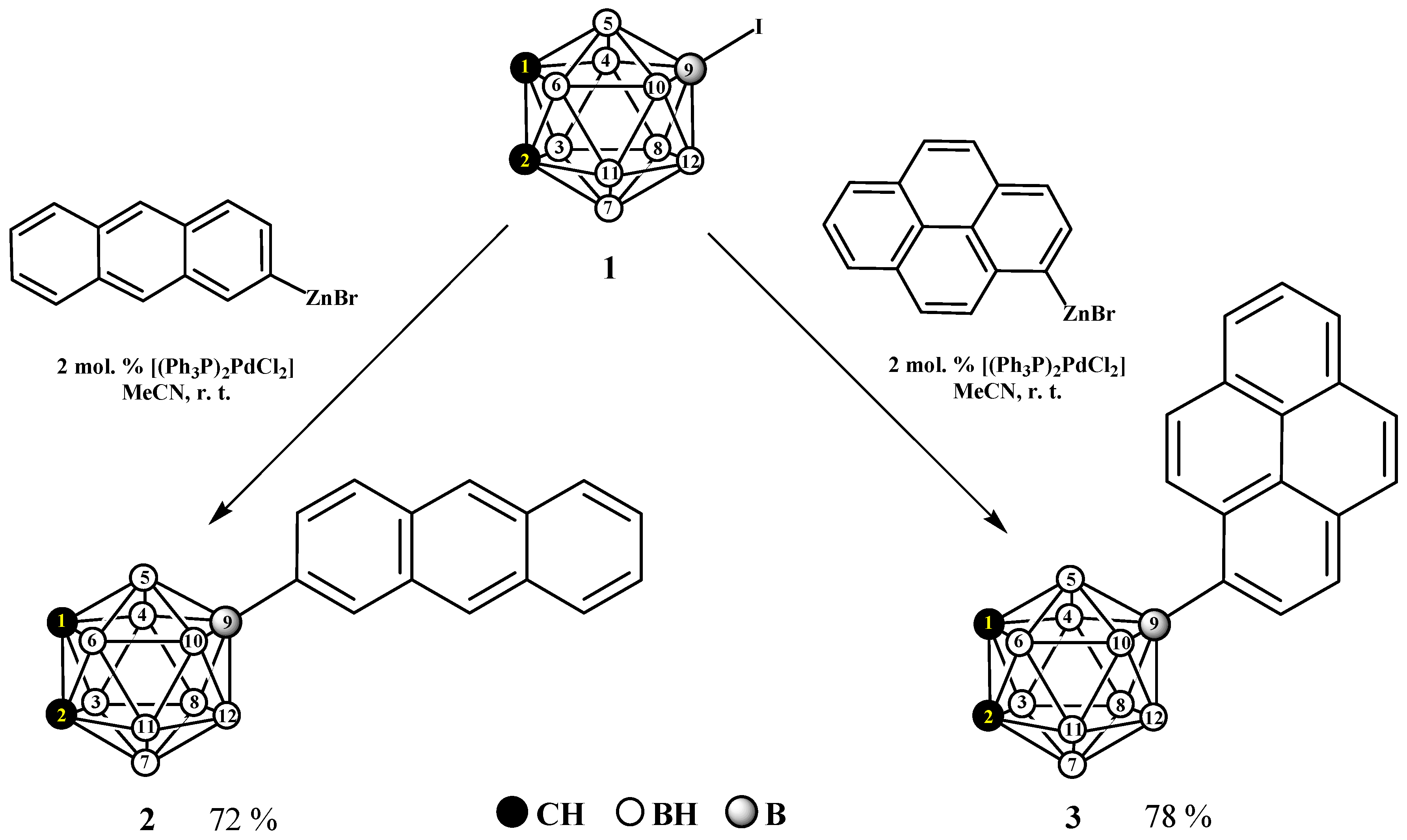
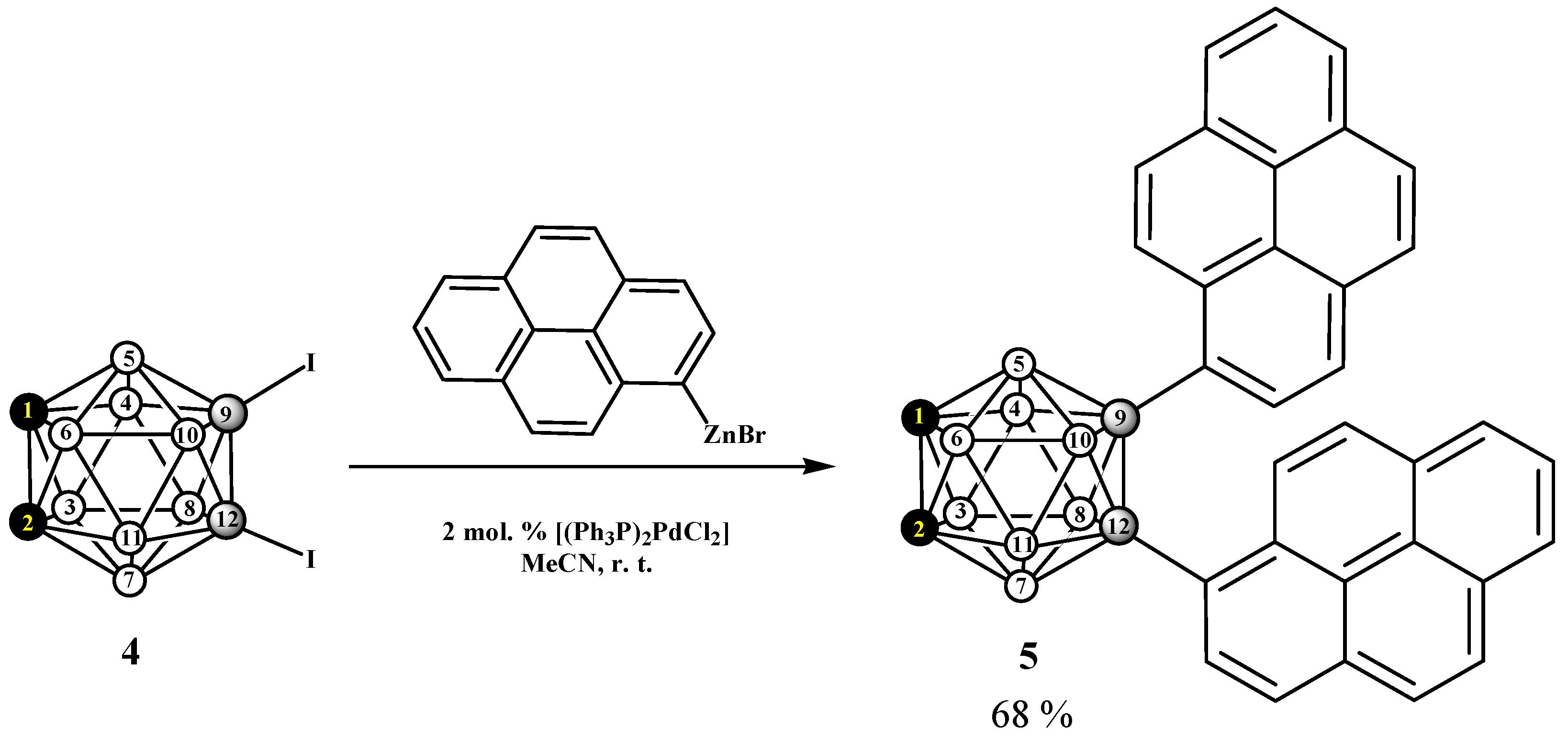
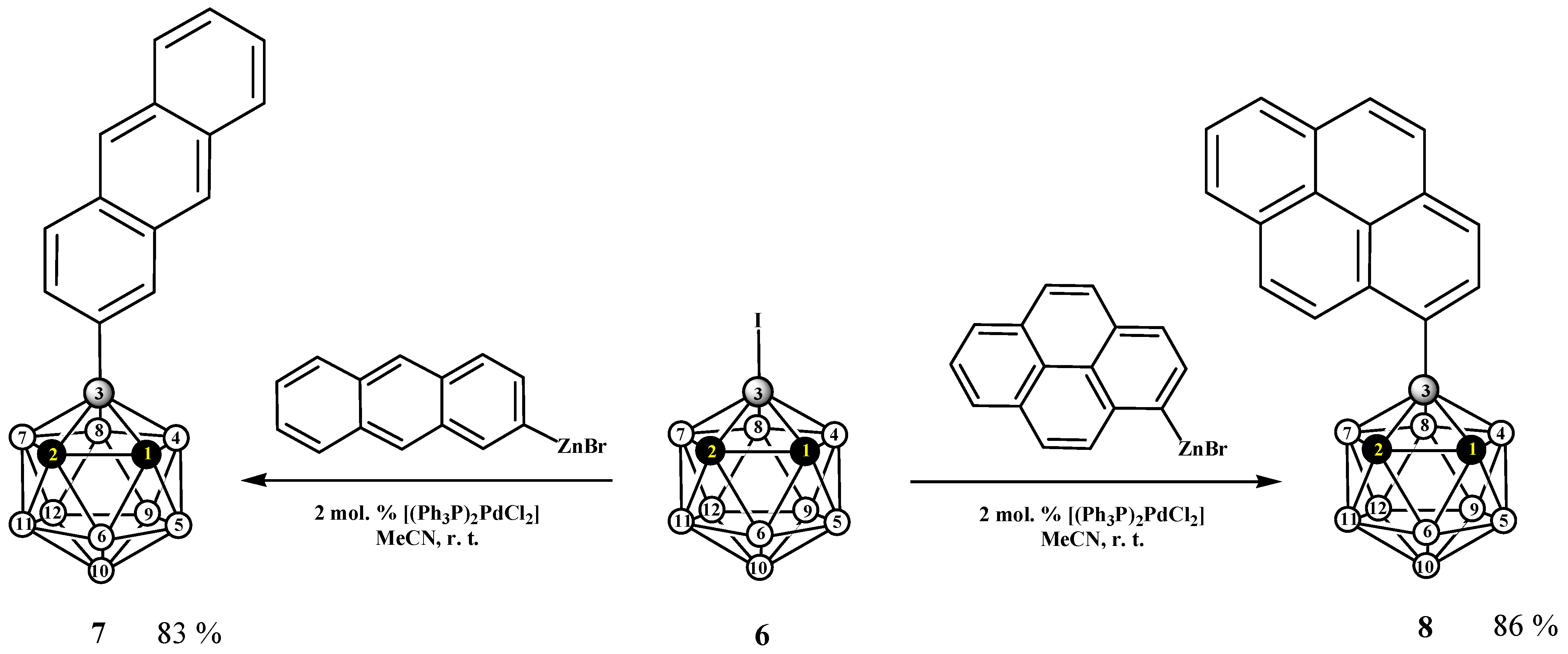
2.1. Synthesis and Characterization
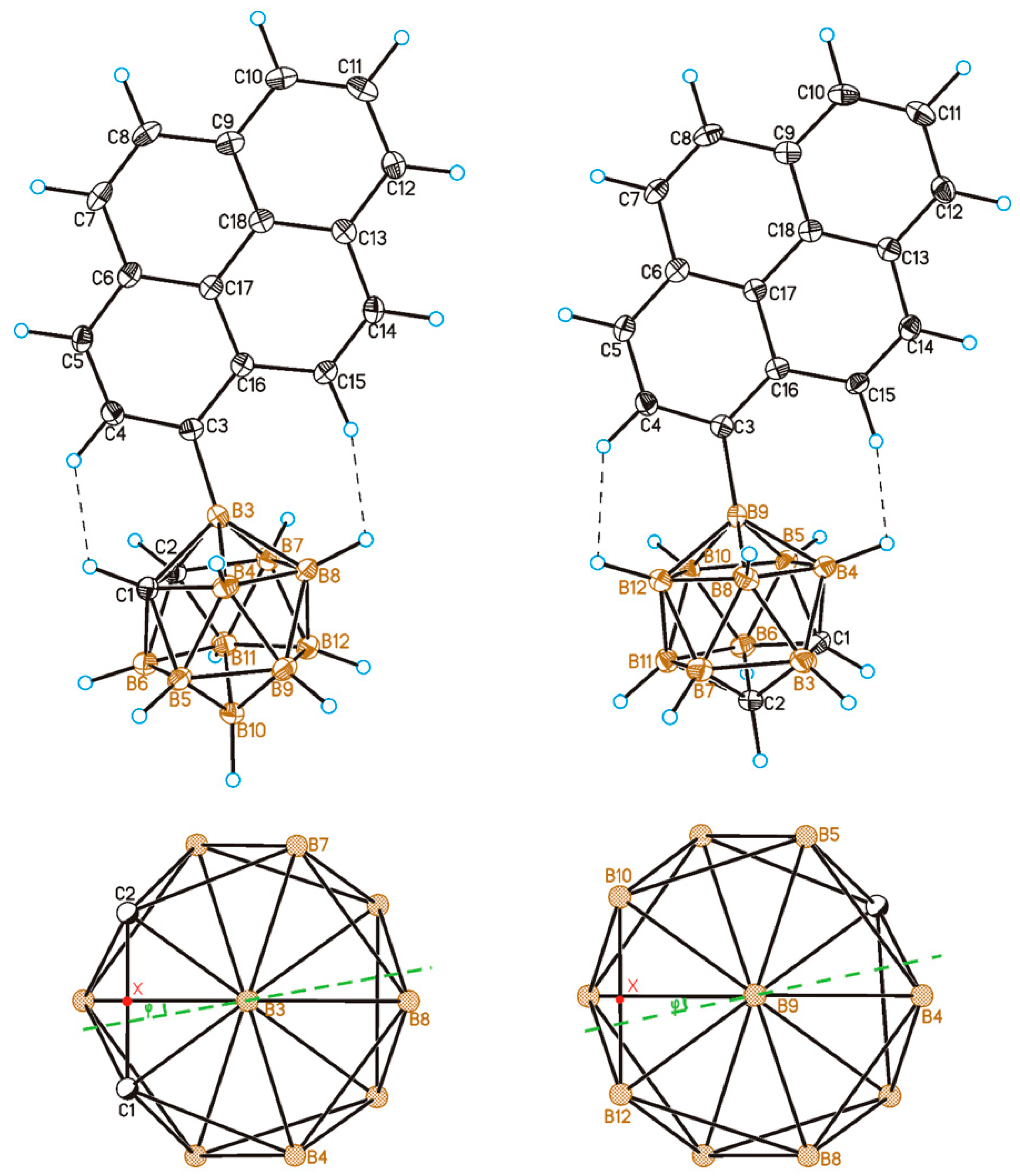
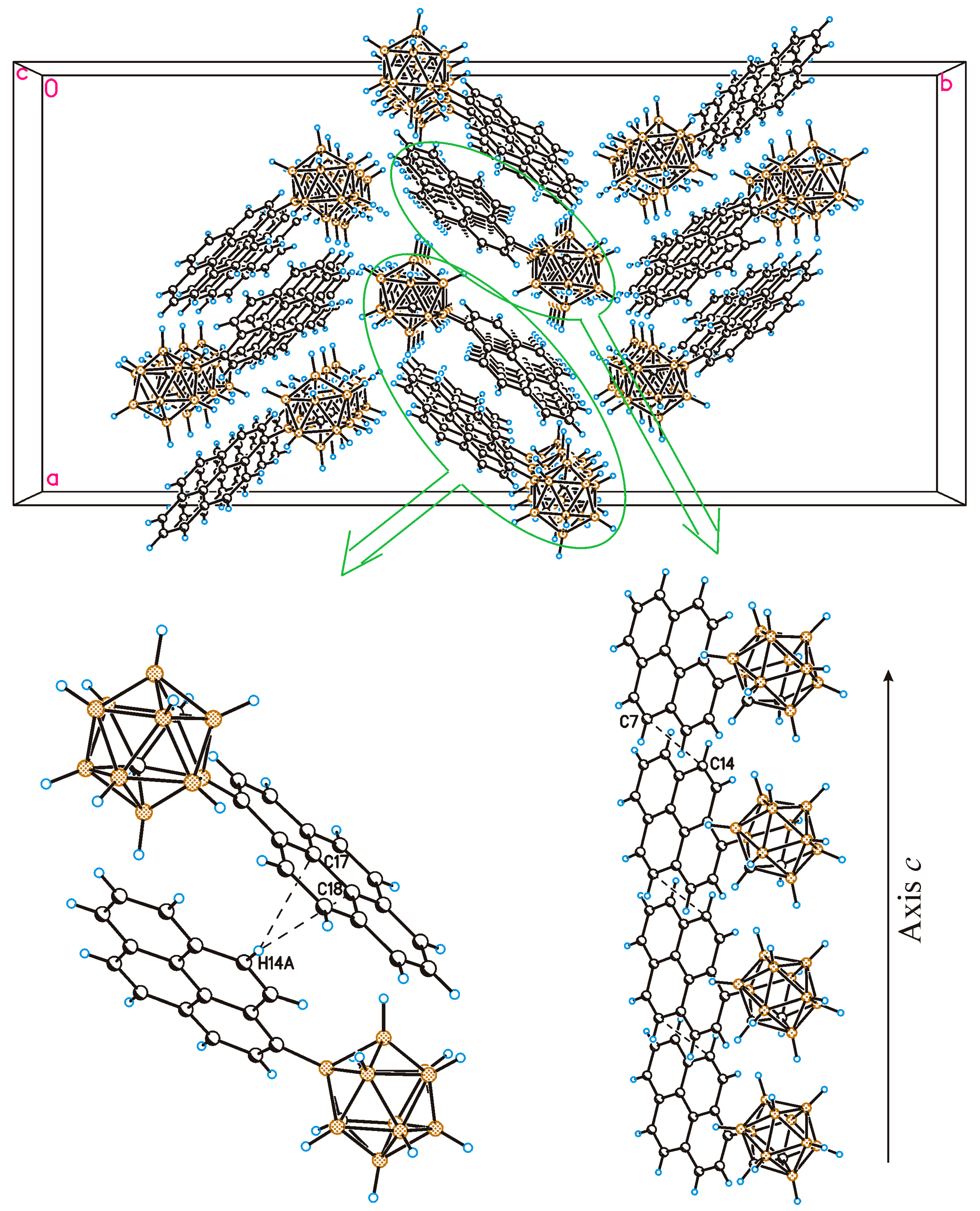
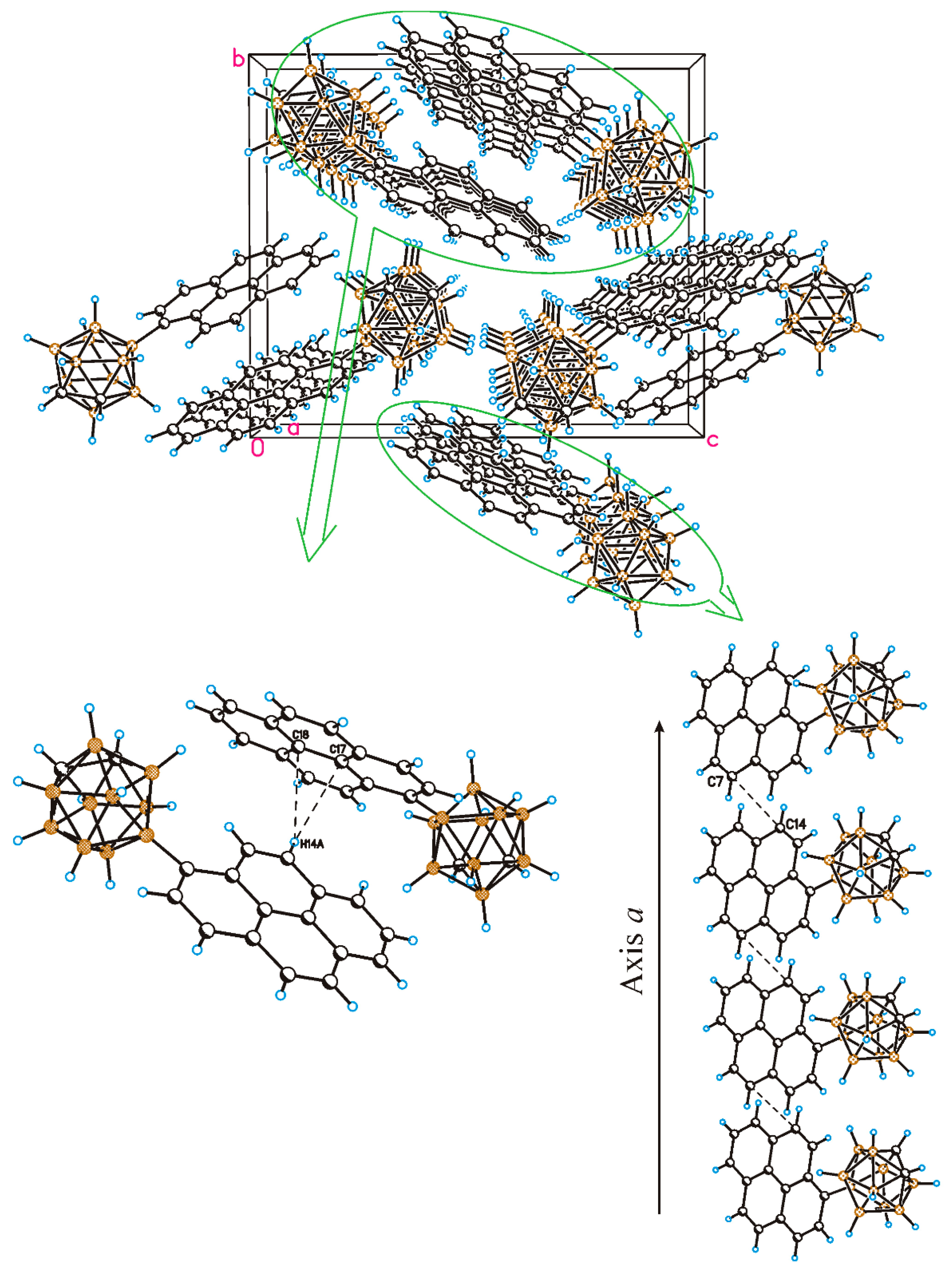
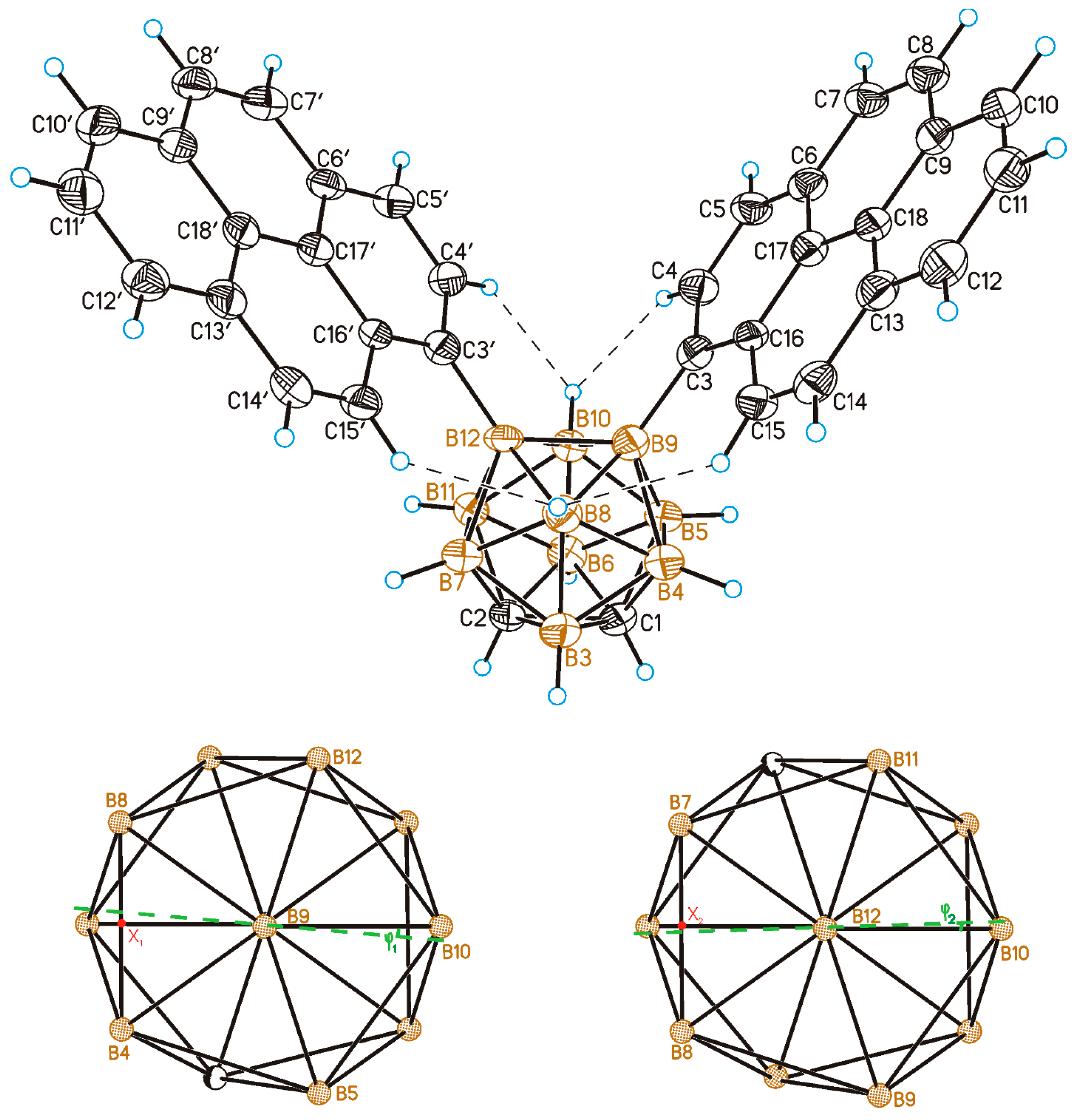
2.2. Solid-State Structure and Crystal Packing
3. Materials and Methods
3.1. General Methods
3.2. General Procedure for Synthesis of 9-Polycyclicaryl-ortho-Carboranes
3.2.1. 9-(Anthracen-2′-yl)-ortho-carborane (2)
3.2.2. 9-(Pyren-1′-yl)-ortho-carborane (3)
3.3. Synthesis of 9,12-Bis(pyren-1′-yl)-ortho-Carborane (5)
3.4. General Procedure for Synthesis of 3-Polycyclicaryl-ortho-Carboranes
3.4.1. 3-(Anthracen-2′-yl)-ortho-carborane (7)
3.4.2. 3-(Pyren-1′-yl)-ortho-carborane (8)
3.5. Single Crystal X-Ray Diffraction Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yamamoto, K.; Endo, Y. Utility of boron clusters for drug design. Hansch-Fujita hydrophobic parameters of dicarba-closo-dodecaboranyl groups. Bioorg. Med. Chem. Lett. 2001, 11, 2389–2392. [Google Scholar] [CrossRef] [PubMed]
- Issa, F.; Kassiou, M.; Rendina, L.M. Boron in drug discovery: Carboranes as unique pharmacophores in biologically active compounds. Chem. Rev. 2011, 111, 5701–5722. [Google Scholar] [CrossRef] [PubMed]
- Scholz, M.; Hey-Hawkins, E. Carbaboranes as pharmacophores: Properties, synthesis, and application strategies. Chem. Rev. 2011, 111, 7035–7062. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y. Carboranes as hydrophobic pharmacophores: Applications for design of nuclear receptor ligands. In Boron-Based Compounds: Potential and Emerging Applications in Medicine; Hey-Hawkins, E., Viñas Teixidor, C., Eds.; John Wiley & Sons Ltd.: Oxford, UK, 2018; pp. 3–19. [Google Scholar] [CrossRef]
- Vinogradova, S.V.; Valetskii, P.M.; Kabachii, Y.A. Polymers with arylenedicarbadodecaborane fragments in the chain. Russ. Chem. Rev. 1995, 64, 365–388. [Google Scholar] [CrossRef]
- Zhang, X.; Rendina, L.M.; Müllne, M. Carborane-containing polymers: Synthesis, properties, and applications. ACS Polym. Au 2024, 4, 7–33. [Google Scholar] [CrossRef]
- Ochi, J.; Tanaka, K.; Chujo, Y. Recent progress in the development of solid-state luminescent o-carboranes with stimuli responsivity. Angew. Chem. Int. Ed. 2020, 132, 9925–9939. [Google Scholar] [CrossRef]
- Tanaka, K.; Gon, M.; Ito, S.; Ochi, J.; Chujo, Y. Recent progresses in the mechanistic studies of aggregation-induced emission-active boron complexes and clusters. Coord. Chem. Rev. 2022, 472, 214779. [Google Scholar] [CrossRef]
- Ran, Z.; Zhao, M.; Shi, J.; Ji, L. Luminescent o-carboranyl-containing molecules: A promising platform for stimuli-responsive smart materials. Dye. Pigment. 2025, 232, 112484. [Google Scholar] [CrossRef]
- Qu, Q.; Fu, M.; Lin, C.; Geng, Y.; Li, Y.; Yuan, Y. Synthesis, properties and application of o-carborane-based π-conjugated macrocycles. Org. Chem. Front. 2023, 10, 3293–3299. [Google Scholar] [CrossRef]
- Zhu, M.; Zhou, Q.; Cheng, H.; Sha, Y.; Bregadze, V.I.; Yan, H.; Sun, Z.; Li, X. Boron-cluster embedded necklace-shaped nanohoops. Angew. Chem. Int. Ed. 2023, 62, e202213470. [Google Scholar] [CrossRef]
- Heying, T.L.; Ager, J.W.; Clark, S.L.; Mangold, D.J.; Goldstein, H.L.; Hillman, M.; Polak, R.J.; Szymanski, J.W. A new series of organoboranes. I. Carboranes from the reaction of decaborane with acetylenic compounds. Inorg. Chem. 1963, 2, 1089–1092. [Google Scholar] [CrossRef]
- You, D.K.; So, H.; Ryu, C.H.; Kim, M.; Lee, K.M. Strategic molecular design of closo-ortho-carboranyl luminophores to manifest thermally activated delayed fluorescence. Chem. Sci. 2021, 12, 8411–8423. [Google Scholar] [CrossRef] [PubMed]
- Ohta, K.; Goto, A.T.; Endo, Y. 1,2-Dicarba-closo-dodecaboran-1-yl naphthalene derivatives. Inorg. Chem. 2005, 44, 8569–8573. [Google Scholar] [CrossRef] [PubMed]
- Naito, H.; Morisaki, Y.; Chujo, Y. o-Carborane-based anthracene: A variety of emission behaviors. Angew. Chem. Int. Ed. 2015, 54, 5084–5087. [Google Scholar] [CrossRef]
- Kim, S.; So, H.; Lee, J.H.; Hwang, H.; Kwon, H.; Park, M.H.; Lee, K.M. Photophysical properties of spirobifluorene-based o-carboranyl compounds altered by structurally rotating the carborane cages. Molecules 2019, 24, 4135. [Google Scholar] [CrossRef]
- Nishino, K.; Tanaka, K.; Chujo, Y. Tuning of sensitivity in thermochromic luminescence by regulating molecular rotation based on triphenylamine-substituted o-carboranes. Asian J. Org. Chem. 2019, 8, 2228–2232. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.H.; So, H.; Kim, M.; Mun, M.S.; Hwang, H.; Park, M.H.; Lee, K.M. Insights into the effects of substitution position on the photophysics of mono-o-carborane-substituted pyrenes. Inorg. Chem. Front. 2020, 7, 2949–2959. [Google Scholar] [CrossRef]
- Wada, K.; Hashimoto, K.; Ochi, J.; Tanaka, K.; Chujo, Y. Rational design for thermochromic luminescence in amorphous polystyrene films with bis-o-carborane-substituted enhanced conjugated molecule having aggregation-induced luminochromism. Aggregate 2021, 2, e93. [Google Scholar] [CrossRef]
- Yuhara, K.; Tanaka, K.; Chujo, Y. Regulation of solid-state dual-emission properties by switching luminescence processes based on a bis-o-carborane-modified anthracene triad. Mater. Chem. Front. 2022, 6, 1414–1420. [Google Scholar] [CrossRef]
- Coult, R.; Fox, M.A.; Gill, W.R.; Herbertson, P.L.; MacBride, J.A.H.; Wade, K. C-arylation and C-heteroarylation of icosahedralcarboranes via their copper(I) derivatives. J. Organomet. Chem. 1993, 462, 19–29. [Google Scholar] [CrossRef]
- Fujii, S.; Yamada, A.; Nakano, E.; Takeuchi, Y.; Mori, S.; Masuno, H.; Kagechika, H. Design and synthesis of nonsteroidal progesterone receptor antagonists based on C,C’-diphenylcarborane scaffold as a hydrophobic pharmacophore. Eur. J. Med. Chem. 2014, 84, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Naito, H.; Nishino, K.; Morisaki, Y.; Tanaka, K.; Chujo, Y. Solid-state emission of the anthracene-o-carborane dyad from the twisted-intramolecular charge transfer in the crystalline state. Angew. Chem. Int. Ed. 2017, 56, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.; Lu, Z.; Xie, Z. Light-promoted copper-catalyzed cage C-arylation of o-carboranes: Facile synthesis of 1-aryl-o-carboranes and o-carborane-fused cyclics. New J. Chem. 2021, 45, 14944–14948. [Google Scholar] [CrossRef]
- Tang, C.; Xie, Z. Nickel-catalyzed cross-coupling reactions of o-carboranyl with aryl iodides: Facile synthesis of 1-aryl-o-carboranes and 1,2-diaryl-o-carboranes. Angew. Chem. Int. Ed. 2015, 54, 7662–7665. [Google Scholar] [CrossRef]
- Wu, X.; Guo, J.; Cao, Y.; Zhao, J.; Jia, W.; Chen, Y.; Jia, D. Mechanically triggered reversible stepwise tricolor switching and thermochromism of anthracene-o-carborane dyad. Chem. Sci. 2018, 9, 5270–5277. [Google Scholar] [CrossRef]
- Wu, X.; Guo, J.; Lv, Y.; Jia, D.; Zhao, J.; Shan, H.; Jin, X.; Ma, Y. Aggregation-induced emission characteristics of o-carborane-functionalized fluorene and its heteroanalogs: The influence of heteroatoms on photoluminescence. Mater. Chem. Front. 2020, 4, 257–267. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, C.; Li, N.; Lv, Y.; Chia, J.; Guo, J. ESIPT-modulated HBT-o-carborane dyads with efficient aggregation-induced emission and acid-induced discolouration properties. J. Mater. Chem. C 2024, 12, 16809–16816. [Google Scholar] [CrossRef]
- Kalinin, V.N.; Kobel’kova, N.I.; Astakhin, A.V.; Gusev, A.I.; Zakharkin, L.I. Synthesis and crystal structure of B-aryl-o-carboranes and electronic effects of B-o-carboranyl groups. J. Organomet. Chem. 1978, 149, 9–12. [Google Scholar] [CrossRef]
- Zakharkin, L.I.; Kovredov, A.I.; Ol’shevskaya, V.A.; Shaugumbekova, Z.S. Synthesis of B-organo-substituted 1,2-, 1,7-, and 1,12-dicarbaclosododecarboranes(12). J. Organomet. Chem. 1982, 226, 217–222. [Google Scholar] [CrossRef]
- Lee, H.; Knobler, C.B.; Hawthorne, M.F. Supramolecular self-assembly directed by carborane C–H. F interactions. Chem. Commun. 2000, 2485–2486. [Google Scholar] [CrossRef]
- Fox, M.A.; Wade, K. Model compounds and monomers for phenylene ether carboranylene ketone (PECK) polymer synthesis: Preparation and characterization of boron-arylated ortho-carboranes bearing carboxyphenyl, phenoxyphenyl or benzoylphenyl substituents. J. Mater. Chem. 2002, 12, 1301–1306. [Google Scholar] [CrossRef]
- Bayer, M.J.; Herzog, A.; Diaz, M.; Harakas, G.A.; Lee, H.; Knobler, C.B.; Hawthorne, M.F. The synthesis of carboracycles derived from B,B’-bis(aryl) derivatives of icosahedral ortho-carborane. Chem. Eur. J. 2003, 9, 2732–2744. [Google Scholar] [CrossRef] [PubMed]
- Spokoyny, A.M.; Li, T.C.; Farha, O.K.; Machan, C.W.; She, C.; Stern, C.L.; Marks, T.J.; Hupp, J.T.; Mirkin, C.A. Electronic tuning of nickel-based bis(dicarbollide) redox shuttles in dye-sensitized solar cells. Angew. Chem. Int. Ed. 2010, 49, 5339–5343. [Google Scholar] [CrossRef]
- Tominaga, M.; Morisaki, Y.; Chujo, Y. Luminescent polymer consisting of 9,12-linked o-carborane. Macromol. Rapid Commun. 2013, 34, 1357–1562. [Google Scholar] [CrossRef]
- Anufriev, S.A.; Sivaev, I.B.; Bregadze, V.I. Synthesis of 9,9’,12,12’-substituted cobalt bis(dicarbollide) derivatives. Russ. Chem. Bull. 2015, 64, 712–717. [Google Scholar] [CrossRef]
- Anderson, K.P.; Mills, H.A.; Mao, C.; Kirlikovali, K.O.; Axtell, J.C.; Rheingold, A.L.; Spokoyny, A.M. Improved synthesis of icosahedral carboranes containing exopolyhedral B-C and C-C bonds. Tetrahedron 2018, 75, 187–191. [Google Scholar] [CrossRef]
- Kelemen, Z.; Pepiol, A.; Lupu, M.; Sillanpää, R.; Hänninen, M.M.; Teixidor, F.; Viñas, C. Icosahedral carboranes as scaffolds for congested regioselective polyaryl compounds: The distinct distance tuning of C–C and its antipodal B–B. Chem. Commun. 2019, 55, 8927–8930. [Google Scholar] [CrossRef]
- Milewski, M.; Caminade, A.-M.; Hey-Hawkins, E.; Lönnecke, P. Pitfalls of a structure determination: The structure of closo-9-[4-(dibenzylamino)phenyl]-1,2-dicarbadodecaborane(12). Acta Cryst. E 2022, 78, 1145–1150. [Google Scholar] [CrossRef]
- Anufriev, S.A.; Shmal’ko, A.V.; Suponitsky, K.Y.; Sivaev, I.B. One-pot synthesis of B-aryl carboranes with sensitive functional groups using sequential cobalt- and palladium-catalyzed reactions. Catalysts 2020, 10, 1348. [Google Scholar] [CrossRef]
- Viñas, C.; Barberà, G.; Oliva, J.M.; Teixidor, F.; Welch, A.J.; Rosair, G.M. Are halocarboranes suitable for substitution reactions? The case for 3-I-1,2-closo-C2B10H11: Molecular orbital calculations, aryldehalogenation reactions, 11B NMR Interpretation of closo-carboranes, and molecular structures of 1-Ph-3-Br-1,2-closo-C2B10H10 and 3-Ph-1,2-closo-C2B10H11. Inorg. Chem. 2001, 40, 6555–6562. [Google Scholar] [CrossRef]
- Ramachandran, B.M.; Knobler, C.B.; Hawthorne, M.F. Example of an intramolecular, non-classical C–H⋯π hydrogen bonding interaction in an arylcarborane derivative. J. Mol. Struct. 2006, 785, 167–169. [Google Scholar] [CrossRef]
- Qiu, Z.; Xie, Z. Palladium/Nickel-cocatalyzed cycloaddition of 1,3-dehydro-o-carborane with alkynes. Facile synthesis of C,B-substituted carboranes. J. Am. Chem. Soc. 2010, 132, 16085–16093. [Google Scholar] [CrossRef] [PubMed]
- Anufriev, S.A.; Shmal’ko, A.V.; Suponitsky, K.Y.; Sivaev, I.B. Synthesis of 3-aryl-ortho-carboranes with sensitive functional groups. Molecules 2021, 26, 7297. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, K.; Ohta, K.; Endo, Y. Synthesis of 3-aryl-1,2-dicarba-closo-dodecaboranes by Suzuki-Miyaura coupling reaction. Heterocycles 2010, 80, 369. [Google Scholar] [CrossRef]
- Cheng, R.; Qiu, Z.; Xie, Z. Iridium-catalysed regioselective borylation of carboranes via direct B–H activation. Nat. Commun. 2017, 8, 14827. [Google Scholar] [CrossRef]
- Zhao, D.; Xie, Z. Visible-light-promoted photocatalytic B−C coupling via a boron-centered carboranyl radical: Facile synthesis of B(3)-arylated o-carboranes. Angew. Chem. Int. Ed. 2016, 55, 3166–3170. [Google Scholar] [CrossRef]
- Ochi, J.; Tanaka, K.; Chujo, Y. Investigation of the substitution site effect on o-carborane-based chromophores by anthracene introduction at the B(3) position. Bull. Chem. Soc. Jpn. 2023, 96, 98–102. [Google Scholar] [CrossRef]
- Mattejata, M.; Ménard, G. Selective heterogeneous capture and release of actinides using carborane-functionalized electrodes. Chem. Commun. 2023, 59, 9710–9713. [Google Scholar] [CrossRef]
- Gosmini, C.; Moncomble, A. Cobalt-catalyzed cross-coupling reactions of aryl halides. Isr. J. Chem. 2010, 50, 568–576. [Google Scholar] [CrossRef]
- Rykowski, S.; Gurda-Woźna, D.; Orlicka-Płocka, M.; Fedoruk-Wyszomirska, A.; Giel-Pietraszuk, M.; Wyszko, E.; Kowalczyk, A.; Stączek, P.; Bak, A.; Kiliszek, A.; et al. Design, synthesis, and evaluation of novel 3-carboranyl-1,8-naphthalimide derivatives as potential anticancer agents. Int. J. Mol. Sci. 2021, 22, 2772. [Google Scholar] [CrossRef]
- Flieger, S.; Takagaki, M.; Kondo, N.; Lutz, M.R.; Gupta, Y.; Ueda, H.; Sakurai, Y.; Moran, G.; Kempaiah, P.; Hosmane, N.; et al. Carborane-containing hydroxamate MMP ligands for the treatment of tumors using boron neutron capture therapy (BNCT): Efficacy without tumor cell entry. Int. J. Mol. Sci. 2023, 24, 6973. [Google Scholar] [CrossRef] [PubMed]
- Ueberham, L.; Gündel, D.; Kellert, M.; Deuther-Conrad, W.; Ludwig, F.-A.; Lönnecke, P.; Kazimir, A.; Kopka, K.; Brust, P.; Moldovan, R.-P.; et al. Development of the high-affinity carborane-based cannabinoid receptor type 2 PET ligand [18F]LUZ5-d8. J. Med. Chem. 2023, 66, 5242–5260. [Google Scholar] [CrossRef] [PubMed]
- Ueberham, L.; Schädlich, J.; Schramke, K.; Braun, S.; Selg, C.; Laube, M.; Lönnecke, P.; Pietzsch, J.; Hey-Hawkins, E. Carborane-based analogues of celecoxib and flurbiprofen, their COX inhibition potential and COX selectivity index. ChemMedChem 2025, 20, e202500166. [Google Scholar] [CrossRef]
- Choi, J.; Kim, S.; Ahn, M.; Kim, J.; Cho, D.W.; Kim, D.; Eom, S.; Im, D.; Kim, Y.; Kim, S.H.; et al. Singlet fission dynamics modulated by molecular configuration in covalently linked pyrene dimers, Anti- and Syn-1,2-di(pyrenyl)benzene. Commun. Chem. 2023, 6, 16. [Google Scholar] [CrossRef]
- Wu, X.; Guo, J.; Zhao, J.; Che, Y.; Jia, D.; Chen, Y. Multifunctional luminescent molecules of o-carborane-pyrene dyad/triad: Flexible synthesis and study of the photophysical properties. Dye. Pigment. 2018, 154, 44–51. [Google Scholar] [CrossRef]
- Berger, S.; Diehl, B.W.K. 13C and 1H NMR chemical shifts of 2-substituted anthracenes. Magn. Reson. Chem. 1989, 27, 201–203. [Google Scholar] [CrossRef]
- Kalinin, V.N.; Ol’shevskaya, V.A. Some aspects of the chemical behavior of icosahedral carboranes. Russ. Chem. Bull. 2009, 57, 815–836. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Anufriev, S.A.; Shmalko, A.V. How substituents at boron atoms affect the CH-acidity and the electron-withdrawing effect of the ortho-carborane cage: A close look on the 1H NMR spectra. Inorg. Chim. Acta 2023, 547, 121339. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Anufriev, S.A.; Shmalko, A.V. Electron-withdrawing effect and CH-acidity of carboranes: Effect of nature and position of substituents. Phosphorus Sulfur Silicon Relat. Elem. 2024, 199, 299–302. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Anufriev, S.A.; Sivaev, I.B.; Suponitsky, K.Y.; Bregadze, V.I. Practical synthesis of 9-methylthio-7,8-nido-carborane [9-MeS-7,8-C2B9H11]−. Some evidences of BH···X hydride-halogen bonds in 9-XCH2(Me)S-7,8-C2B9H11 (X = Cl, Br, I). J. Organomet. Chem. 2017, 849–850, 315–323. [Google Scholar] [CrossRef]
- Anufriev, S.A.; Erokhina, S.A.; Suponitsky, K.Y.; Godovikov, I.A.; Filippov, O.A.; Fabrizi de Biani, F.; Corsini, M.; Chizhov, A.O.; Sivaev, I.B. Methylsulfanyl-stabilized rotamers of cobalt bis(dicarbollide). Eur. J. Inorg. Chem. 2017, 2017, 4444–4451. [Google Scholar] [CrossRef]
- Suponitsky, K.Y.; Anisimov, A.A.; Anufriev, S.A.; Sivaev, I.B.; Bregadze, V.I. 1,12-Diiodo-ortho-carborane: A classic textbook example of the dihalogen bond. Crystals 2021, 11, 396. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Rozas, I.; Elguero, J.; Molins, E. About the evaluation of the local kinetic, potential and total energy densities in closed-shell interactions. Chem. Phys. Lett. 2001, 336, 457–461. [Google Scholar] [CrossRef]
- Andrews, J.S.; Zayas, J.; Jones, M. 9-Iodo-o-carborane. Inorg. Chem. 1985, 24, 3715–3716. [Google Scholar] [CrossRef]
- Zheng, Z.; Jiang, W.; Zinn, A.A.; Knobler, C.B.; Hawthorne, M.F. Facile electrophilic iodination of icosahedral carboranes. Synthesis of carborane derivatives with boron-carbon bonds via the palladium-catalyzed reaction of diiodocarboranes with Grignard reagents. Inorg. Chem. 1995, 34, 2095–2100. [Google Scholar] [CrossRef]
- Zhao, D.; Xie, Z. [3-N2-o-C2B10H11][BF4]: A useful synthon for multiple cage boron functionalizations of o-carborane. Chem. Sci. 2016, 7, 5635–5639. [Google Scholar] [CrossRef]
- Itatani, H.; Bailar, J.C. Homogenous catalysis in the reactions of olefinic substances. V. Hydrogenation of soybean oil methyl ester with triphenylphosphine and triphenylarsine palladium catalysts. J. Am. Oil Chem. Soc. 1967, 44, 147–151. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 7th ed.; Butterworth-Heinemann: Oxford, UK, 2013. [Google Scholar] [CrossRef]
- APEX2 and SAINT; Bruker AXS Inc.: Madison, WI, USA, 2014.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Kudin, K.N., Jr.; Burant, J.C.; Millam, J.M.; et al. Gaussian 03, Revision E.01; Gaussian, Inc.: Wallingford, UK, 2004. [Google Scholar]
- Keith, T.A. AIMAll, Version 15.05.18; TK Gristmill Software: Overland Park, KS, USA, 2015.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound 8 | Compound 3 | |||
|---|---|---|---|---|
| X-Ray | Calcd. | X-Ray | Calcd. | |
| Angle φ ** | 10.2(4) | 34.3 | 11.0(2) | 36.3 |
| C(4)-H(4A)⋯H(1)-C(1) | 2.05 | 1.93 [−2.6] | - | - |
| C(15)-H(15A)⋯H(8)-B(8) | 1.94 | 2.18 [−1.9] | - | - |
| C(4)-H(4A)⋯H(12)-B(12) | - | - | 2.25 | 2.06 [−2.2] |
| C(15)-H(15A)⋯H(4)-B(4) | - | - | 1.89 | 2.23 [−1.4] |
| X-Ray | Calcd. | |
|---|---|---|
| Angle φ1 ** | 4.6(4) | 1.6 |
| Angle φ2 ** | 0.5(4) | 1.5 |
| C(4)-H(4A)⋯H(10)-B(10) | 2.02 | 2.08 [−2.2] |
| C(15)-H(15A)⋯H(8)-B(8) | 2.18 | 2.14 [−2.0] |
| C(4′)-H(4′A)⋯H(10)-B(10) | 2.09 | 2.08 [2.2] |
| C(15′)-H(15B)⋯H(8)-B(8) | 2.11 | 2.14 [−2.0] |
| 3 | 5 | 8 | |
|---|---|---|---|
| Formula | C18H20B10 | C34H28B10 | C18H20B10 |
| FW | 344.44 | 544.66 | 344.44 |
| Crystal system | Orthorhombic | Monoclinic | Orthorhombic |
| Space group | P212121 | P21/n | Fdd2 |
| a, Å | 7.2182(2) | 8.6601(8) | 21.9384(16) |
| b, Å | 14.4891(5) | 35.244(3) | 47.186(4) |
| c, Å | 17.2004(6) | 9.7598(9) | 6.9542(5) |
| α, deg. | 90 | 90 | 90 |
| β, deg. | 90 | 109.323(4) | 90 |
| γ, deg. | 90 | 90 | 90 |
| V, Å3 | 1798.91(10) | 2811.1(4) | 7198.9(9) |
| Z | 4 | 4 | 16 |
| ρcryst, g·cm−3 | 1.272 | 1.287 | 1.271 |
| F(000) | 712 | 1128 | 2848 |
| μ, mm−1 | 0.064 | 0.068 | 0.063 |
| θ range, deg. | 1.84–27.03 | 2.29–26.08 | 2.05–27.35 |
| Rflns. collected | 23,984 | 29,493 | 18,983 |
| Indep. rflns./Rint | 3937/0.0522 | 5557/0.1121 | 3927/0.0660 |
| Completeness to theta θ, % | 100 | 99.8 | 99.6 |
| Ref. parameters | 297 | 437 | 297 |
| GOF (F2) | 1.043 | 1.030 | 1.031 |
| Rflns. with I > 2σ(I) | 3374 | 3124 | 3488 |
| R1(F) (I > 2σ(I)) a | 0.0397 | 0.0760 | 0.0438 |
| wR2(F2) (all data) b | 0.0912 | 0.1897 | 0.1028 |
| Largest diff. peak/hole, e·Å−3 | 0.164/−0.237 | 0.390/−0.287 | 0.197/−0.203 |
| CCDC Number | 2,435,230 | 2,435,231 | 2,435,229 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shmal’ko, A.V.; Anufriev, S.A.; Suponitsky, K.Y.; Antoshkina, E.P.; Sivaev, I.B.; Bregadze, V.I. Synthesis of B-Substituted Anthracenyl and Pyrenyl Derivatives of ortho-Carborane. Inorganics 2025, 13, 138. https://doi.org/10.3390/inorganics13050138
Shmal’ko AV, Anufriev SA, Suponitsky KY, Antoshkina EP, Sivaev IB, Bregadze VI. Synthesis of B-Substituted Anthracenyl and Pyrenyl Derivatives of ortho-Carborane. Inorganics. 2025; 13(5):138. https://doi.org/10.3390/inorganics13050138
Chicago/Turabian StyleShmal’ko, Akim V., Sergey A. Anufriev, Kyrill Yu. Suponitsky, Evgeniia P. Antoshkina, Igor B. Sivaev, and Vladimir I. Bregadze. 2025. "Synthesis of B-Substituted Anthracenyl and Pyrenyl Derivatives of ortho-Carborane" Inorganics 13, no. 5: 138. https://doi.org/10.3390/inorganics13050138
APA StyleShmal’ko, A. V., Anufriev, S. A., Suponitsky, K. Y., Antoshkina, E. P., Sivaev, I. B., & Bregadze, V. I. (2025). Synthesis of B-Substituted Anthracenyl and Pyrenyl Derivatives of ortho-Carborane. Inorganics, 13(5), 138. https://doi.org/10.3390/inorganics13050138