Abstract
The reactivity of the benzenedithiolate (bdt)-bridged complex [Fe2(CO)6(µ-bdt)] with arsine, stibine and phosphine ligands has been studied. The new mono- and disubstituted complexes [Fe2(CO)5(EPh3)(µ-bdt)] (E = As, 1; E = Sb 3) and [Fe2(CO)4(EPh3)2(µ-bdt)] (E = As, 2; E = Sb, 4) and the previously reported [Fe2(CO)4(PPh2H)2(µ-bdt)] (5) have been prepared by Me3NO-initiated carbonyl substitution reactions of [Fe2(CO)6(µ-bdt)] with appropriate ligands at 80 °C. Spectroscopic and single-crystal X-ray diffraction studies reveal that in all cases the introduced ligands occupy apical coordination site(s) lying trans to the iron–iron bond. Their electrochemistry has been probed by cyclic voltammetry and selected complexes have been tested as proton reduction catalysts. Monosubstituted complexes 1 and 3 show two irreversible reductions at ca. −1.7 V and −2.0 V, respectively, relative to Fc+/Fc, while the disubstituted complexes 2 and 5 show a single irreversible reduction at ca. −2.2 V and −1.84 V, respectively. Complexes 1, 3 and 5 can catalyse electrocatalytic proton reduction in the presence of either p-toluene sulfonic acid (TsOH) or trifluoroacetic acid (CF3CO2H).
1. Introduction
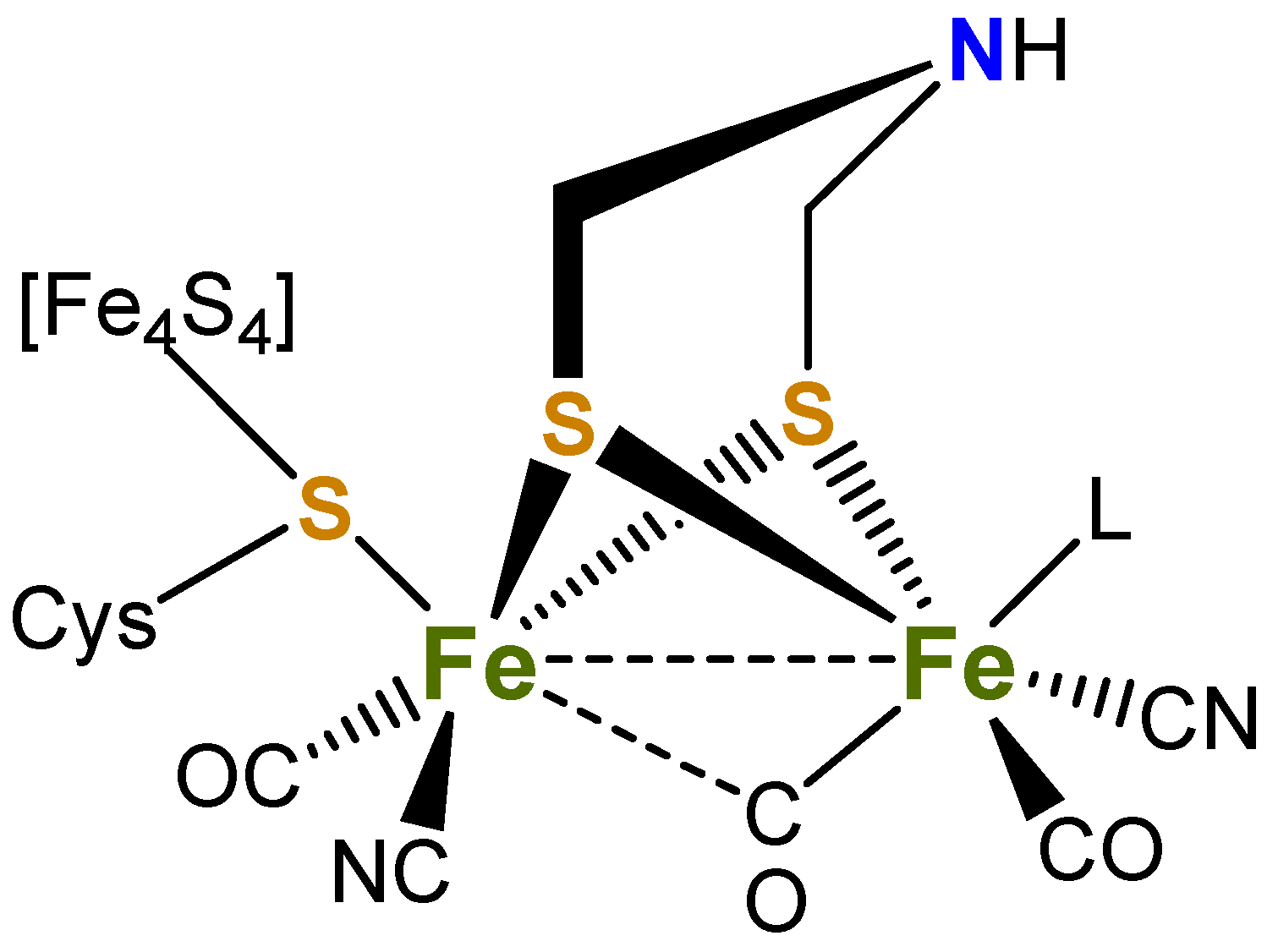
Over the past two decades, there has been a concerted effort to understand the structure and electrochemical properties of a wide range of dithiolate-supported diiron carbonyl complexes [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19], focusing on their role in the reduction of protons to dihydrogen [9,20,21,22,23,24,25], as they closely resemble the active H-cluster site of [FeFe]-hydrogenases (Figure 1) [26,27,28,29,30]. To mimic the role of the cyanide groups, numerous studies have been performed on the substitution of carbonyl ligands in [Fe2(CO)6(µ-dithiolate)] for tertiary phosphines, typically yielding the corresponding mono-and di-substituted derivatives [9,20,21,22,23,24]. Most of these studies focus on the biologically relevant [Fe2(CO)6(µ-SCH2XCH2S)] [X = CH2, pdt = propanedithiolate; X = NR, adt = aminodithiolate) with less attention being paid to related models with rigid two-atom linking groups such as ethanedithiolate (edt) and benzenedithiolate (bdt) bridging ligands [31,32,33,34,35,36,37,38,39,40,41,42].
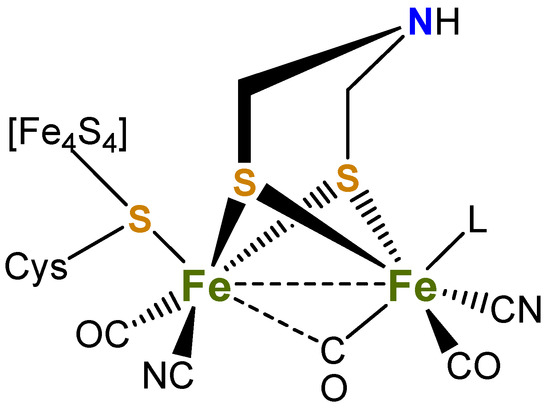
Figure 1.
Active site of [FeFe]-hydrogenase (L = H2O or vacant site).
Varying the bridgehead moieties of the diiron dithiolate complexes influences the electron density of the diiron centre and thus reduction potentials, and, based on IR data, the electron density at the metal core varies in the order pdt > edt > bdt [1]. The bridgehead group can also serve to stabilise generated anions, for example in [Fe2(CO)6(µ-bdt)] via an interaction between sulfur pπ and benzene pπ orbitals. Further, it is known that [Fe2(CO)6(µ-bdt)] undergoes a two-electron reduction process during the catalytic activity to produce H2, being the result of potential inversion [34,35]. While the singly reduced complex [Fe2(CO)6(µ-bdt)]− is unstable, the dianionic species is sufficiently stable to be identified and partially characterised by X-ray absorption spectroscopy (EXAFS, XANES) [35]. Computational modelling of the dianion suggests that the two iron ions are reduced, and subsequent structural rearrangement leads to the formation of [Fe2(µ-CO)(CO)5(µ-bdt)]2− with one ruptured Fe–S bond and a bridging carbonyl ligand [35]. The relative stabilities of [Fe2(CO)6(µ-bdt)] and its corresponding reduced species makes it a good candidate for investigations as a proton reduction catalyst. Kaur-Ghumaan and co-workers have detailed the synthesis of mono- and disubstituted triarylphosphine complexes, [Fe2(CO)5L(μ-bdt)] and [Fe2(CO)4L2(μ-bdt)] (L = PPh3, PPh2Me, PPh2H), showing that mono-substituted derivatives were active proton-reduction catalysts in the presence of CF3CO2H and HClO4 in CH2Cl2 and MeCN [33] and that the current was larger than related complexes with pdt and odt (odt = oxadithiolate) ligands [43,44,45].
While there has been much focus on the use of phosphines as cyanide surrogates, the heavier group 15 homologues have, in comparison, been neglected. In order to address this, we recently reported a comparative study on the synthesis, structural characterization and electrochemical properties of a series of edt-bridged complexes, viz. [Fe2(CO)5(EPh3)(μ-edt)] and [Fe2(CO)4(EPh3)2(μ-edt)] (E = P, As, Sb) [46], all being catalytically active for proton reduction in acetonitrile. To further evaluate the potential role of AsPh3 and SbPh3 in [FeFe]-ase biomimics, we have turned our attention to the related chemistry of arsine and stibine derivatives of [Fe2(CO)6(µ-bdt)]. Herein, we report the synthesis and structural characterization of the monosubstituted [Fe2(CO)5(EPh3)(µ-bdt)] (E = As, 1; E = Sb 3) and disubstituted [Fe2(CO)4(EPh3)2(µ-bdt)] (E = As, 2; E = Sb, 4) derivatives, together with the secondary phosphine complex [Fe2(CO)4(PPh2H)2(µ-bdt)] (5), and a preliminary investigation of their electrochemical and proton reduction behaviour.
2. Results and Discussion
2.1. Synthesis and Spectroscopic Characterisation of 1–5
Reaction of [Fe2(CO)6(µ-bdt)] with two equivalents of AsPh3 or SbPh3 in the presence of Me3NO.H2O in MeCN at 80 °C afforded the mono-substituted complexes, [Fe2(CO)5(µ-bdt)(EPh3)] (1, E = As, 39%; 3, E = Sb, 42%) as the major products, together with smaller amounts of disubstituted products [Fe2(CO)4(µ-bdt)(EPh3)2] (2, E = As, 17%; 4, E = Sb, 22%) (Scheme 1). Monosubstituted complexes alone were isolated from room temperature reactions in CH2Cl2. In contrast, a similar reaction between [Fe2(CO)6(µ-bdt)] and Ph2PH led to the isolation of only disubstituted [Fe2(CO)4(PPh2H)2(μ-bdt)] (5) in moderate yield (51%). The PPh2H analogue of 1 and 2, i.e., [Fe2(CO)5(PPh2H)(μ-bdt)], and 5 were previously synthesised by Kaur-Ghumaan and co-workers from a room temperature reaction between [Fe2(CO)6(µ-bdt)] and PPh2H in toluene [33]. These authors also noted that when the reaction was carried out under refluxing conditions it afforded mainly 5, which is consistent with the isolation of only 5 in our case. Since [Fe2(CO)5(PPh2H)(μ-bdt)] was structurally characterised and its electrochemical and electrocatalytic properties were also investigated properly [33], we deliberately chose the method which affords only 5 in order to maximise its yield. Complexes 1–5 are air-stable in the solid state and soluble in polar organic solvents, but insoluble or sparingly soluble in non-polar organic solvents. They have been characterised by IR and 1H NMR spectroscopy, elemental analysis, mass spectrometry, electrochemical studies, and single-crystal X-ray diffraction analyses. Full details of the synthetic procedures and spectroscopic data can be found in the experimental section.
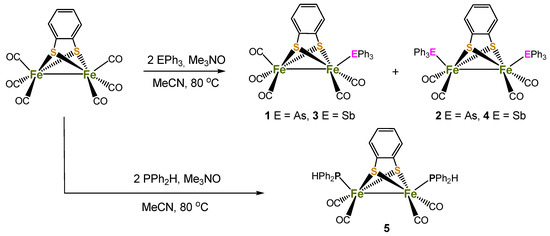
Scheme 1.
Schematic depiction of synthetic routes to compounds 1–5.
IR spectra in the carbonyl region are characteristic and informative with patterns and positions of bands being very similar to those for phosphine-substituted edt-and pdt-bridged complexes [33,42,46]. Mass spectra are also in good agreement with the proposed compositions, showing molecular ion peaks at m/z 698.20, 976.42, 745.04, 1070.10 and 736.31 for 1–5, respectively, in addition to peaks corresponding to the sequential loss of all COs. 1H NMR spectra are not very informative but show resonances attributed to EPh3 (E = As, Sb) and C6H4 ligands in the region 7.42–5.61 ppm. The molecular structures of 1–5 were determined by X-ray diffraction analysis of single crystals that were obtained by slow diffusion of n-hexane into CH2Cl2 solutions.
2.2. Structural Studies of Complexes 1–5
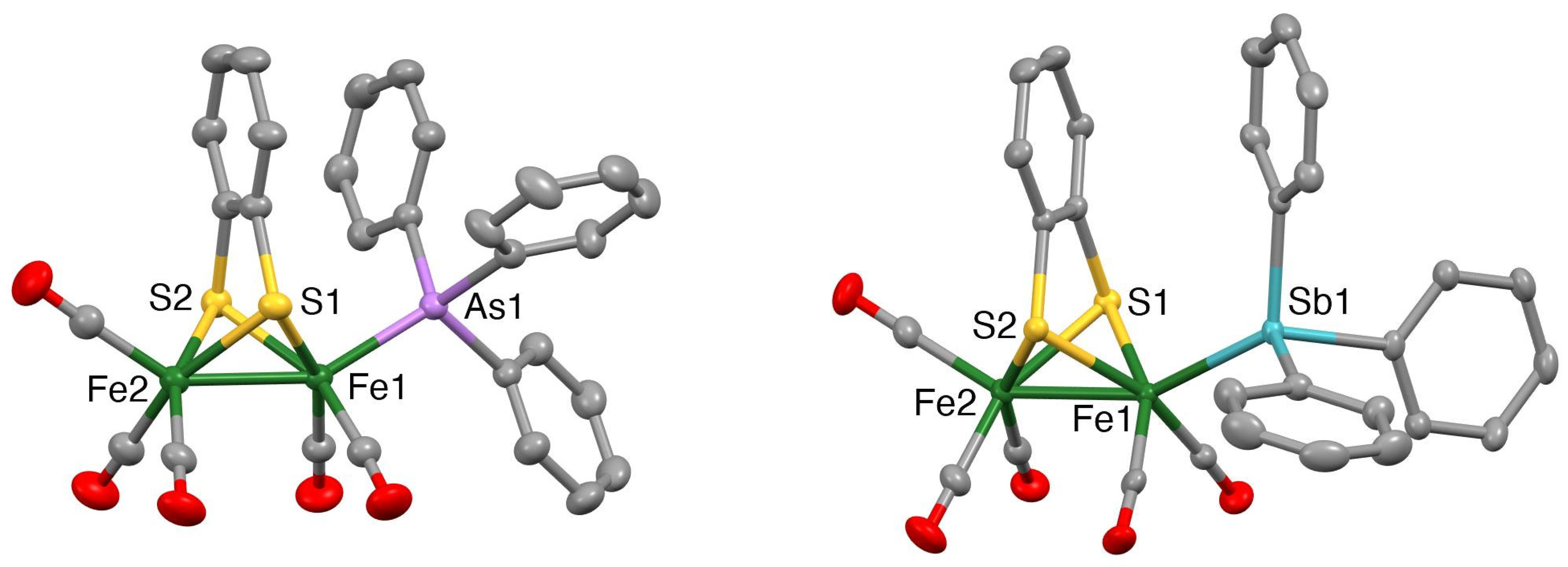
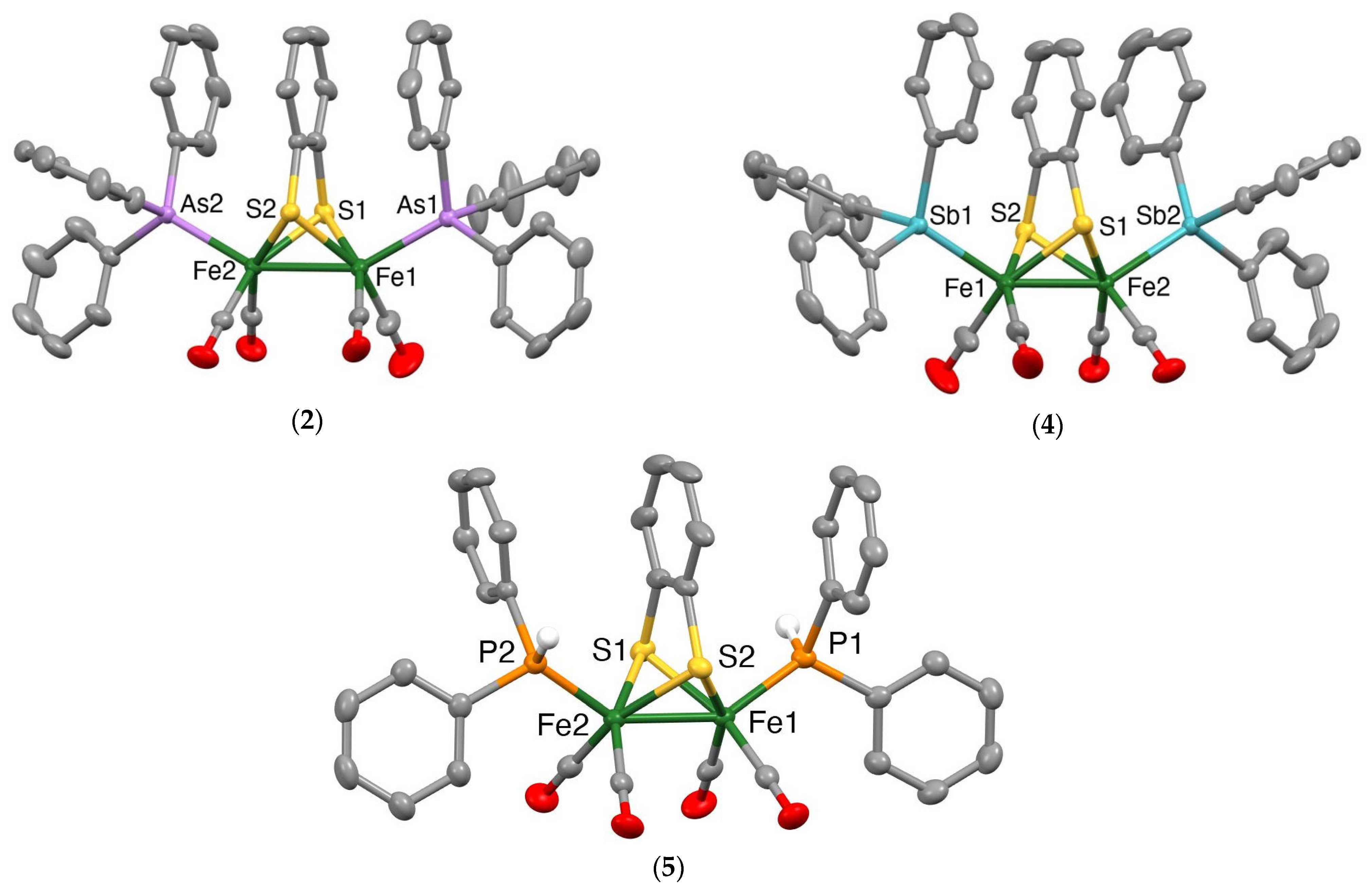
Mercury drawings of the molecular structures of the monosubstituted clusters 1 and 3 are depicted in Figure 2, while those of the disubstituted clusters 2, 4 and 5 are shown in Figure 3. Selected bond lengths and angles for 1–5 together with those for [Fe2(CO)6(µ-bdt)] [45], [Fe2(CO)5(µ-bdt)(PPh3)] [31] and [Fe2(CO)4(µ-bdt)(PPh3)2] [31] are summarised in Table 1 and Tables S1–S5. In all structures, the group 15 ligands are coordinated at an apical site, lying trans to the Fe−Fe bond. The Fe−Fe bond distance in 1 [2.4850(3) Å] is similar to that in [Fe2(CO)6(µ-bdt)] [2.480(2) Å] [47] and slightly longer than that found in 3 [2.4664(4) Å]. The Fe−As and Fe−Sb bond distances in 1–4 are comparable to those of the corresponding diiron-edt complexes [46]. Likewise, the Fe−Fe distances in disubstituted 2 (2.4840(3) Å), 4 (2.4716(3) Å) and 5 (2.4755(5) Å) are close to those observed in [Fe2(CO)4)(PPh3)2(µ-bdt)] (2.4989(6) Å) [33], [Fe2(CO)4(AsPh3)2(µ-edt)] (2.4989(6) Å) [46] and [Fe2(CO)4(SbPh3)2(µ-edt)] (2.4741(4) Å) [46]. As expected, the Fe−As bond distances in 2 (2.3418(2) and 2.3437(2) Å) are significantly shorter than the Fe−Sb bond distances in 4 (2.4788(5)−2.4815(4) Å), and the Fe−P bond distances in 5 (2.2163(6)−2.2194(7) Å) are shorter than the Fe−As/Sb bond distances for 2 and 4. The molecular structures of [Fe2(CO)4(µ-bdt)(AsPh3)2] (2), [Fe2(CO)4(µ-bdt)(SbPh3)2] (4) and [Fe2(CO)4(µ-bdt)(SbPh3)2] (5) reveal close alignment and proximities of arene rings in the benzenedithiolate and phenyl rings of the arsine/stibine substituents (Figure 3). The distances between the centres of the substituent phenyl rings and the centre of the arene ring of the benzenedithiolate are ca 3.62 and 3.55 Å for 2, 3.68 and 3.73 Å for 4, and 3.95 and 3.70 Å for 5, all suggestive of intramolecular p-p interactions. In [Fe2(CO)5(µ-bdt)(AsPh3)] (1) and [Fe2(CO)5(µ-bdt)(AsPh3)] (2), arene rings in close proximity are not well aligned (i.e., the arene planes are far from parallel) and the substituent arene to benzenedithiolate distances are larger than 4.1 Å for both 1 and 2. There are no significant intermolecular interactions in the crystal structures of 1–5.
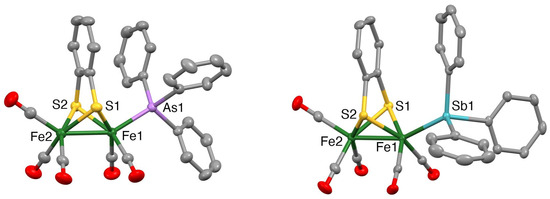
Figure 2.
Mercury plots of the molecular structures of [Fe2(CO)5(AsPh3)(µ-bdt)] (1) (left) and [Fe2(CO)5(SbPh3)(µ-bdt)] (3) (right). Hydrogen atoms are omitted for clarity. Thermal ellipsoids are plotted at the 50% probability level.
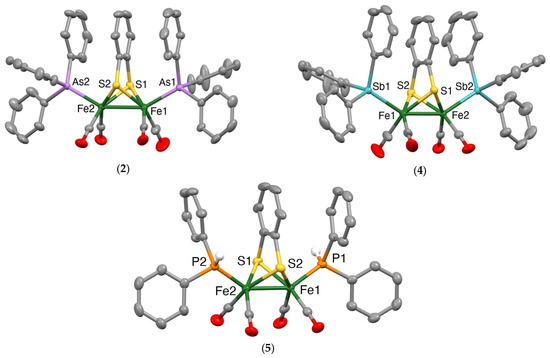
Figure 3.
Mercury plots of the molecular structures of [Fe2(CO)4(AsPh3)2(µ-bdt)] (2) (top left), Fe2(CO)4(SbPh3)2(µ-bdt)] (4) (top right) and Fe2(CO)4(PPh2H)2(µ-bdt)] (5) (bottom). Phenyl ring hydrogen atoms are omitted for clarity. Thermal ellipsoids are plotted at the 50% probability level.

Table 1.
Selected structural parameters for [Fe2(CO)6(µ-SC6H4S)] [47], 1−5, [Fe2(CO)5(µ-SC6H4S)(PPh3)] [33] and [Fe2(CO)4(µ-SC6H4S)(PPh3)2] [33].
2.3. Protonation Studies
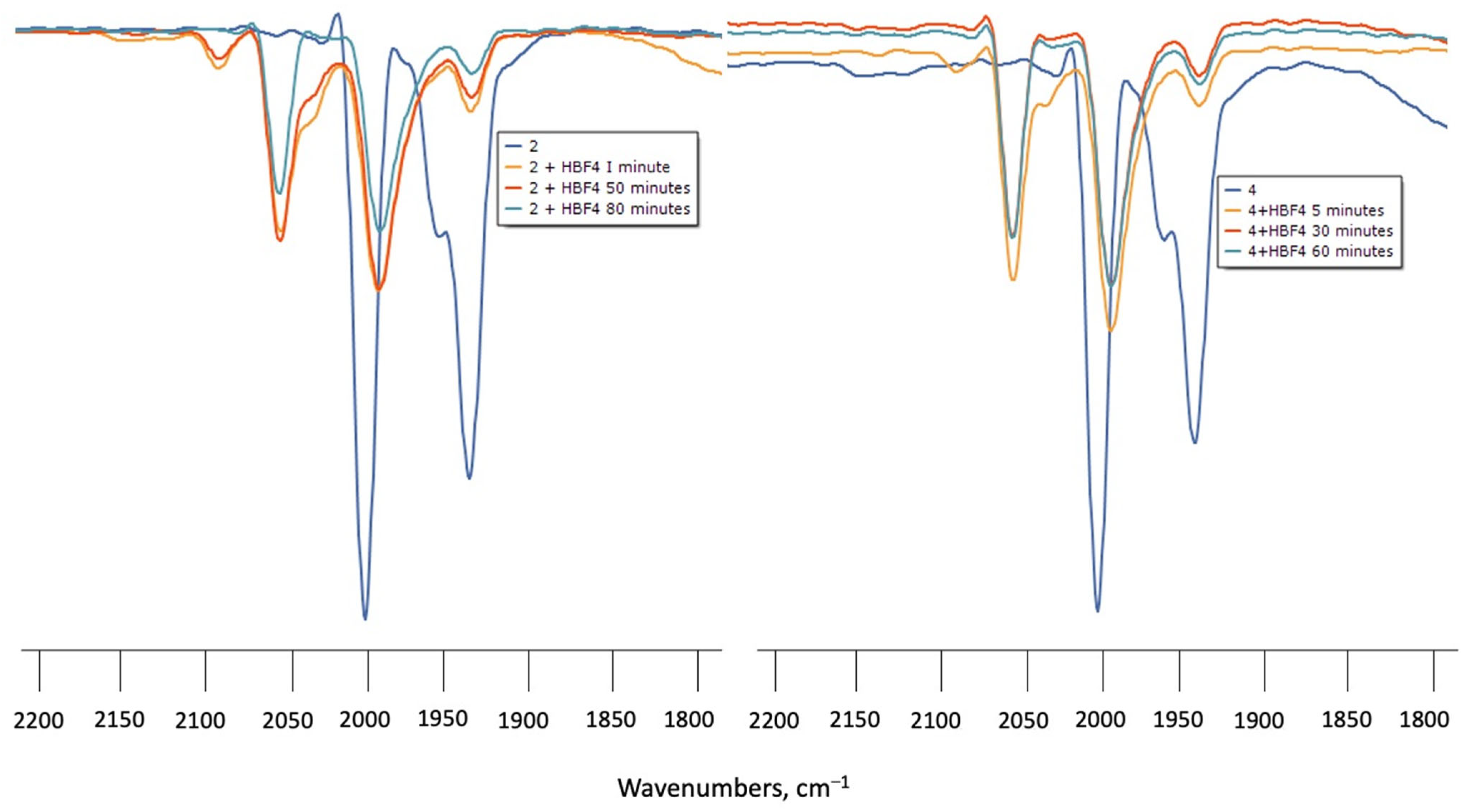
Protonation of 1–4 in CH2Cl2 was monitored by IR spectroscopy under N2 at room temperature using HBF4.Et2O as the proton source. Upon addition of excess HBF4.Et2O to the solutions of 1 or 3, the ν(CO) absorptions did not change even after standing for 6 h, indicating that the diiron centre is not basic enough to undergo protonation. Treatment of CH2Cl2 solutions of 2 or 4 with HBF4.Et2O in air resulted in a rapid (1 min) colour change from yellow to light yellow. IR spectra showed new ν(CO) absorption bands at ca. 50–60 cm−1 higher in energy with respect to the neutral complexes (Figure 4). However, attempts to see the hydride resonances in NMR spectra were unsuccessful in both cases which implies that, instead of protonation, 2 and 4 underwent oxidation when treated with HBF4.Et2O in the presence of air, and led to the formation of the oxidised species [Fe2(CO)4(µ-bdt)(AsPh3)2]+ (2+) and [Fe2(CO)4(µ-bdt)(SbPh3)2]+ (4+). The 50–60 cm−1 blue shift of the highest energy ν(CO) absorption can be compared with analogous phosphine-substituted cations [48,49,50]
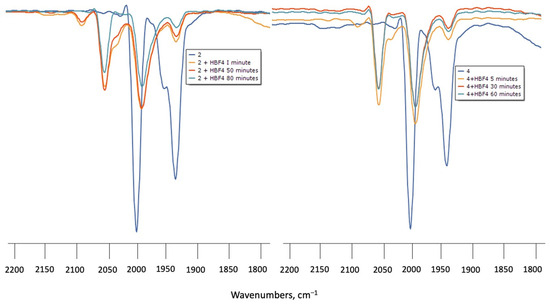
Figure 4.
(left) FTIR spectra of 2 in CH2Cl2 turquoise (complex 2), orange (complex 2 + HBF4 after 1 min), red (complex 2 + HBF4 after 50 min) and green (complex 2 + HBF4 after 80 min) (right) FTIR of 4 in CH2Cl2 turquoise (complex 4), orange (complex 4 + HBF4 after 1 min), red (complex 4 + HBF4 after 30 min) and (complex 4 + HBF4 after 60 min).
2.4. Electrochemical Studies
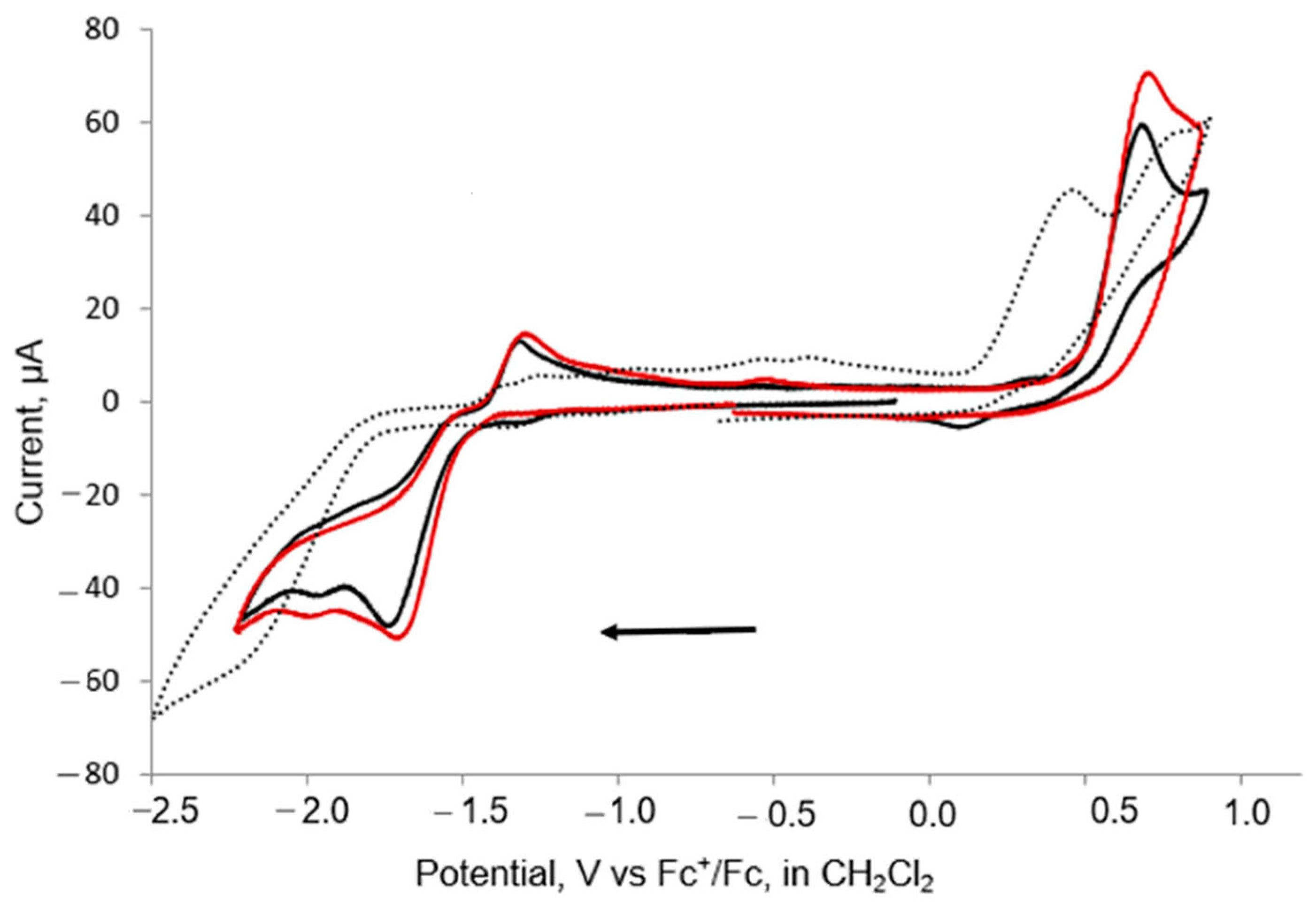
The electrochemical properties of 1–3 were investigated by cyclic voltammetry (CV) in CH2Cl2 under a nitrogen atmosphere. Recently, we observed that the electrochemical properties of [Fe2(CO)5(L)(μ-edt)] depend on the nature of the substituents. Potential inversion leading to a two-electron reduction was observed for the parent complex [Fe2(CO)6(μ-edt)] and [Fe2(CO)5 (μ-edt)(SbPh3)], while for L = PPh3 or AsPh3 a quasi-reversible one-electron reduction was detected [46]. The mono-substituted complexes 1 and 3 show similar oxidation and reduction behaviour, and they show an irreversible reduction peak at ca. −1.7 V vs. Fc+/Fc, followed by another small reduction at ca. −2.0 V for both at 0.1 V/s−1 scan rate (Figure 5 and Table 2). At the return scan, three small anodic peaks were observed at ca. −1.2, −0.8 and −0.4 V, and upon increasing the scan rate, these anodic peaks became broader. Both 1 and 3 also exhibit irreversible oxidation at ca. 0.60 V at a scan rate of 0.1 V/s−1 (Figure 5). The reduction and oxidation potentials were shifted by ca. 0.32 and 0.25 V toward more negative potential with respect to the redox peaks observed for the parent complex [40] due to the increasing electron density at the iron centre. For disubstituted 2, an irreversible reduction was observed at ca.−2.2 V together with an irreversible oxidation at 0.4 V that was followed by a small quasi-reversible oxidation at ca. 0.8 V. The disubstituted complex 2 reduces at ca. 0.5 V more negative potential than the monosubstituted complexes 1 and 3, as expected due to the increased electron density in the diiron centre as compared to 1 and 3. Similar electrochemical behaviour was reported for [Fe2(CO)5(µ-bdt)(PPh3)] and [Fe2(CO)4(µ-bdt)(PPh3)2] [33].
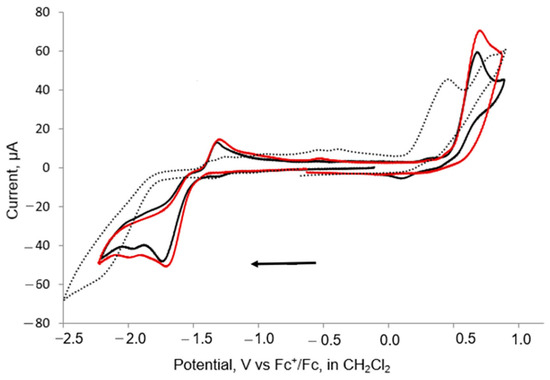
Figure 5.
CVs of 1 (black), 3 (red) and 2 (dots) in CH2Cl2 at 0.1 V/s scan rate (1 mM solution, supporting electrolyte [NBu4] [PF6], glassy carbon electrode, potential vs. Fc+/Fc; initial negative sweep—see arrow).
Table 2.
Summary of electrochemical behaviour of 1–5 and related complexes in CH2Cl2. Potentials are referenced to Fc+/Fc.
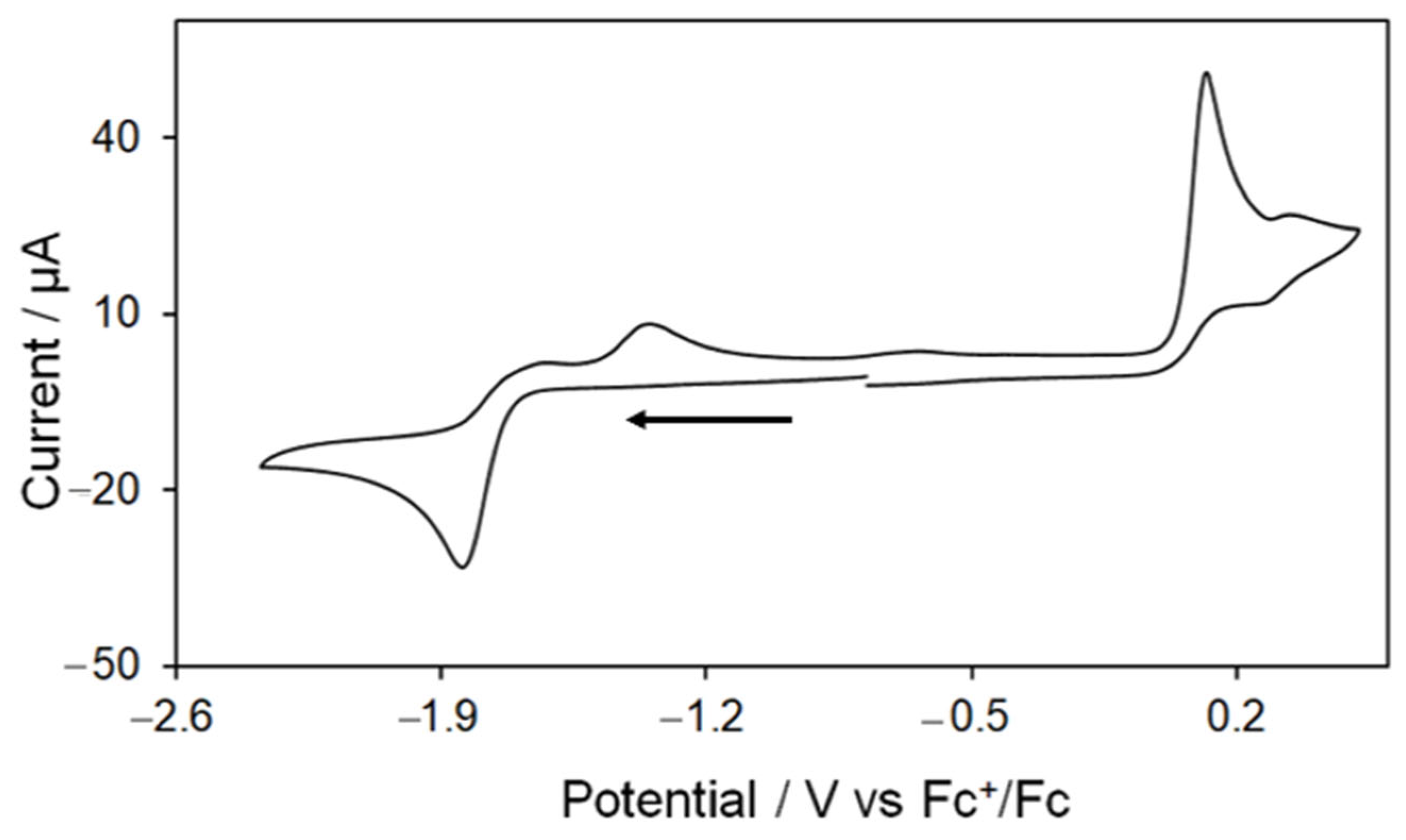
As previously mentioned, Kaur-Ghumaan and co-workers have studied the redox behaviour of 5 in MeCN [33], but did not employ this complex for electrocatalytic proton reduction. An initial cyclovoltammetric study showed an electrochemical behaviour for 5 that is in full agreement with the results obtained by Kaur-Ghumaan and coworkers [33]. Thus, the CV of 5 shows an irreversible oxidation at 0.12 V followed by a small quasi-reversible oxidation at 0.32 V and an irreversible reduction at −1.84 V at scan rate 0.1 V/s (Figure 6). The reversibility of the first oxidative process does not improve when the potential is cycled below 0.25 V. Two small oxidative features are also seen on the return scan at −1.34 V and −0.63 V due to the oxidation of product(s) formed in the reductive process. The CV does not show any remarkable change when the scan rate is varied (scanning the negative potential window first), but a series of small overlapping reductive features have been observed between −1.0 to −1.6 V at higher scan rates (≥0.25 V/s) when the positive potential window is scanned first as a result of the reduction of products formed during the oxidative processes (Figure S8).
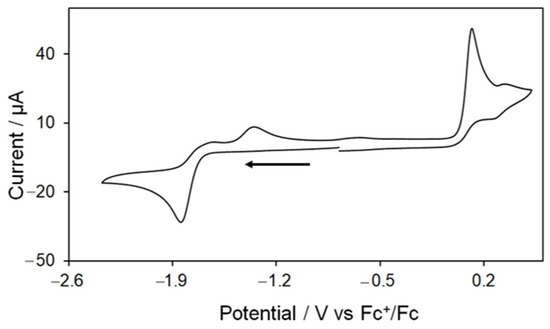
Figure 6.
Cyclic voltammogram of [Fe2(CO)4(PPh2H)2(μ-bdt)] (5) in MeCN (1 mM solution, supporting electrolyte [NBu4] [PF6], glassy carbon electrode, potential vs. Fc+/Fc scan rate = 0.1 V/s; initial negative sweep—see arrow).
2.5. Electrocatalytic Studies
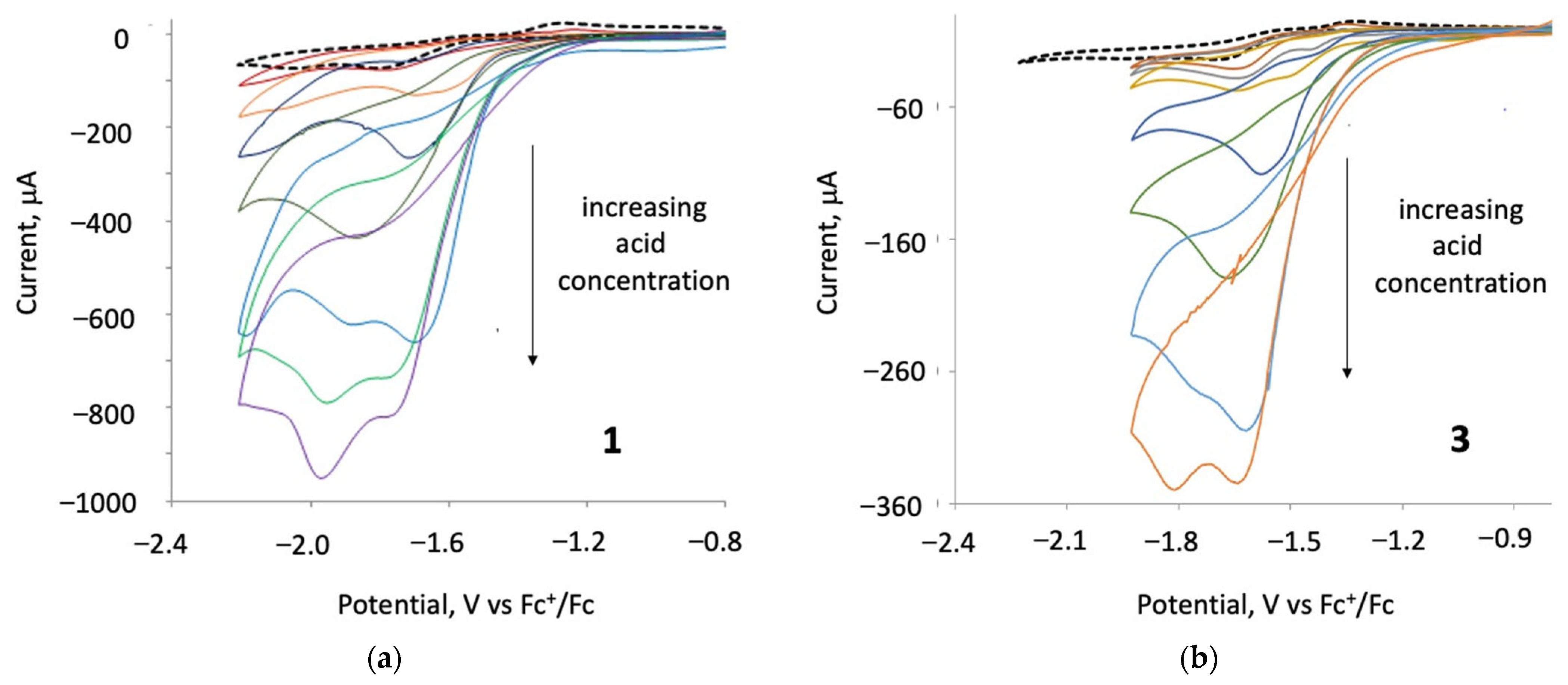
Catalytic studies were carried out on 1–3 in the presence of para-toluene sulfonic acid (p-TsOH) and, in each case, a catalytic response was observed. The protonation experiment of 1–4 in the presence of 2–3 equivalents of p-TsOH or HBF4.Et2O in CH2Cl2 did not show any evidence of protonation (Figures S1 and S4). After the addition of three equivalents of p-TsOH to 1 and 3, a new catalytic reduction peak appeared at ca. −1.6 V for 1 and at ca. −1.4 V for 3 (Figure 7). Upon increasing acid concentration, these new first reduction peaks show catalytic behaviour—they increase and are shifted to more negative potentials, merging with the second reduction waves for each of the two complexes [39], a feature which may correspond to chemically irreversible reductions of 1 and 3. Upon the addition of 18 equivalents of acid, new catalytic peaks occur at ca. −1.9 and −1.8 V for 1 and 3, respectively. The height of the reduction wave increases with the addition of successive molar equivalents of p-TsOH, indicating that 1 and 3 catalyse proton reduction at both their first and second reduction potentials. In the presence of 3–18 equivalents of p-TsOH, complex 2 showed two new (non-catalytic) reduction waves at ca. −1.2 and −1.5 V vs. Fc+/Fc and a catalytic wave at −2.2 V (Figures S9 and S12).
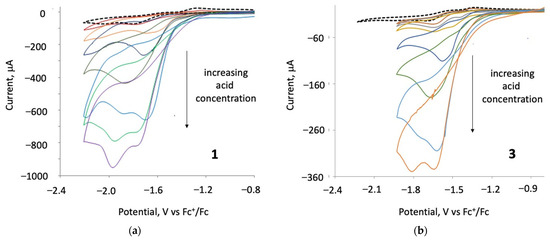
Figure 7.
(a) CVs of 1 in the absence and presence of 1–36 molar equivalents of TsOH and (b) CVs of 3 in the absence and presence of 1–27 molar equivalents of TsOH (1 mM solution in CH2Cl2, supporting electrolyte [NBu4] [PF6], scan rate 0.2 Vs−1 for 1, and 0.05 Vs−1 for 3, glassy carbon electrode, potential vs. Fc+/Fc).
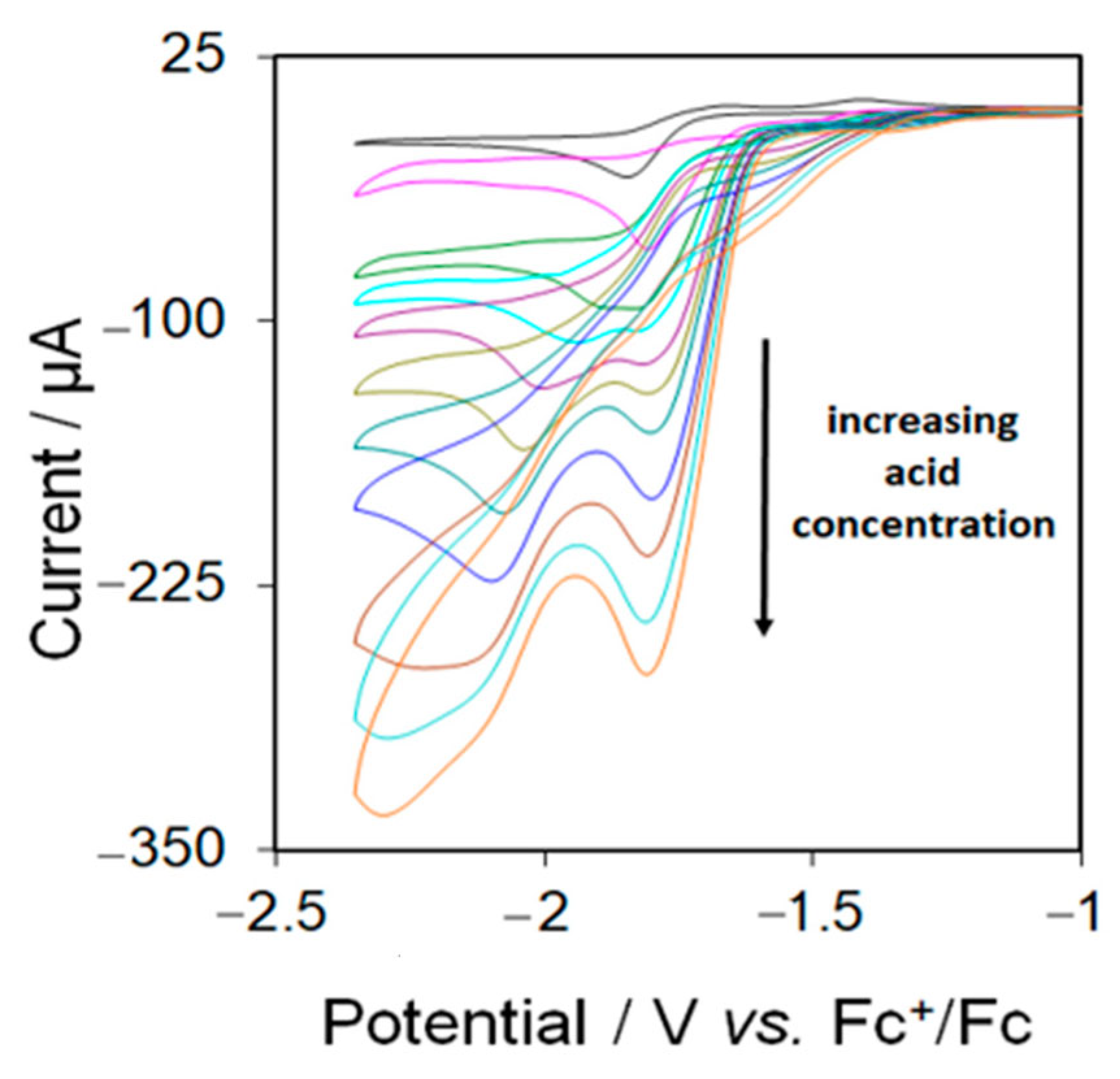
Electrocatalytic testing of 5 was carried out in MeCN using CF3CO2H as the proton source. Complex 5 displays catalytic waves at its first reduction potential in the presence of acids. Figure 8 shows the CVs upon addition of up to 10 equivalents of acid to an MeCN solution of 5. The height of the first reduction peak increases gradually with the concentration of acid which is characteristic of electrocatalytic proton reduction. Since 5 triggers a catalytic wave at its first reduction potential, we can suggest that an EC mechanism is operating at this potential for proton reduction. A new catalytic peak is also observed at −1.94 after addition of 3 molar equivalents of acid which gradually shifts to more negative potential as the concentration of acid is increased.
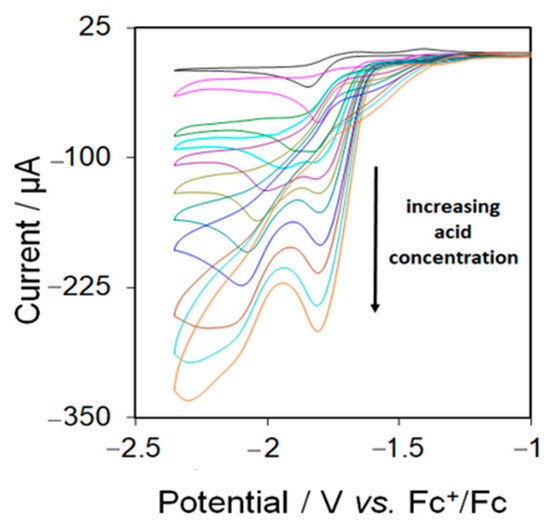
Figure 8.
Cyclic voltammograms of 5 in the absence and in the presence of 1–10 molar equivalents of CF3CO2H (1 mM solution in MeCN, supporting electrolyte [NBu4] [PF6], scan rate 0.1 Vs−1, glassy carbon electrode, potential vs. Fc+/Fc).
3. Summary
In this paper, we have described the synthesis of two monosubstituted complexes [Fe2(CO)5(EPh3)(μ-bdt)] (E = As, 1; and 3, Sb), and three disubstituted complexes [Fe2(CO)4(EPh3)2(μ-bdt)] (E = As, 2; and 4, Sb) and [Fe2(CO)4(PPh2H)2(μ-bdt)] (5) by carbonyl substitution reactions of the precursor benzene dithiolate complex [Fe2(CO)6(μ-bdt)]. All have been unambiguously characterised by spectroscopic techniques and single-crystal X-ray diffraction studies. The molecular structures reveal that the introduced ligands lie in the apical sites trans to the Fe−Fe bond, which is characteristic of these types of complexes. Complexes 1–3 have been examined as proton-reduction catalysts in the presence of p-TsOH. Cyclic voltammetry of 1 and 3 show chemically irreversible reduction at ca. −1.7 and −2.0 V vs. Fc+/Fc. Complex 2 shows an irreversible reduction at ca. −2.2 V. The first reduction peak of 2 is ca. 0.5 V more negative than the first reduction of 1 and 3, which is assigned to the increase of electron density on the metal centre when more electron-donating arsine ligands replace two CO ligands. The proton reduction ability of 1–3 and 5 have also been tested using p-TsOH or CF3CO2H as the proton source. All these complexes can catalyse reduction of protons to dihydrogen under electrocatalytic conditions in the presence of an acid.
4. Experimental
4.1. General Procedures
Unless otherwise stated, all reactions and manipulations were carried out under a nitrogen atmosphere using standard Schlenk techniques. Reagent-grade solvents were dried by standard procedures and were freshly distilled prior to use. All chromatographic separations and ensuing work-up were carried out in air. Triphenylarsine, triphenylstibine, diphenylphosphine, p-toluenesulfonic acid, trifluoroacetic acid, tetrabutylammonium hexafluorophosphate, benzene1−1,2-dithiol were used as supplied by Sigma-Aldrich (St. Louis, MO, USA) and [Fe2(CO)6(µ-bdt)] was prepared according to the literature procedure [40]. IR spectra were recorded on Nicolet 6700 FT-IR or Nicolet Avatar 360 FT-IR-spectrophotometers (Thermo Fisher Nicolet, Madison, WI, USA). NMR spectra were recorded on a Varian Unity 500 MHz spectrometer (Varian Inc., Palo Alto, CA, USA). Fast atom bombardment (FAB) mass spectra were obtained on a JEOL SX-102 spectrometer (JEOL, Tokyo, Japan) using 3-nitrobenzyl alcohol as the matrix and CsI as the calibrant. Products were separated in air by TLC plates coated with 0.25 mm of silica gel (HF254-type 60, E. Merck, Germany). Elemental analyses were performed by the Microanalytical Laboratories of Wazed Miah Science Research Centre at Jahangirnagar University.
4.2. Synthesis of [Fe2(CO)5(AsPh3)(µ-bdt)] (1) and [Fe2(CO)4(AsPh3)2(µ-bdt)] (2)
An MeCN solution (30 mL) of [Fe2(CO)6(µ-bdt)] (100 mg, 0.238 mmol) and AsPh3 (74 mg, 0.476 mmol) was added dropwise to an MeCN solution (5 mL) of Me3NO (18 mg, 0.24 mmol). The reaction mixture was heated at 80 °C for 6 h. The solvent was removed under reduced pressure and the residue chromatographed by TLC on silica gel. Elution with n-hexane/CH2Cl2 (3:1 v/v) developed two bands. The faster-moving orange band afforded [Fe2(CO)5(AsPh3)(µ-bdt)] (1) (65 mg, 39%) as orange crystals, while the slower-moving red band afforded [Fe2(CO)4(AsPh3)2(µ-bdt)] (2) (40 mg, 17%) as deep red crystals after recrystallization from hexane/CH2Cl2 at 4 °C. In a separate experiment, a CH2Cl2 solution (7 mL) of Me3NO (5.4 mg, 0.072 mmol) was added dropwise to a CH2Cl2 solution (20 mL) of 1 (50 mg, 0.072 mmol) and AsPh3 (22 mg, 0.072 mmol), and the reaction mixture was stirred at room temperature for 24 h. The solvent was removed under reduced pressure and the residue was chromatographed by TLC on silica gel. Elution with n-hexane/CH2Cl2 (1:1 v/v) developed two bands. The faster moving band was unreacted 1, while the second band afforded 2 (30 mg, 42%).
Analytical and spectroscopic data for 1: Anal. Calcd for C29H19Fe2O5S2As: C, 49.89; H, 2.74. Found: C, 49.80; H, 2.89. IR (ν(CO), CH2Cl2): 2052 vs, 1992 s, 1935 w cm−1. 1H NMR (CDCl3): δ 7.43 (m, 6H, phenyl), 7.34 (m, 9H, phenyl), 6.35 (s, 2H, C6H4), 6.18 (s, 2H, C6H4). ESI-MS: m/z 698.20.
Analytical and spectroscopic data for 2: Anal. Calcd for C46H34Fe2O4S2As2: C, 56.58; H, 3.51. Found: C, 56.69; H, 3.67. IR (ν(CO), CH2Cl2): 2000 vs, 1955 s, 1936 m cm−1. 1H NMR (CDCl3): δ 7.42 (m, 12H, phenyl), 7.36 (m, 18H, phenyl), 7.13 (s, 2H, C6H4), 7.03 (s, 2H, C6H4). ESI-MS: m/z 976.42.
4.3. Synthesis of [Fe2(CO)5(SbPh3)(µ-SC6H4S)] (3) and [Fe2(CO)4(SbPh3)2(µ-SC6H4S)] (4)
An MeCN solution (25 mL) of [Fe2(CO)6(µ-bdt)] (100 mg, 0.238 mmol) and SbPh3 (84 mg, 0.476 mmol) was added dropwise to a solution of Me3NO (18 mg, 0.24 mmol) in MeCN (5 mL). The reaction mixture was heated at 80 °C for 5 h. The solvent was removed under reduced pressure and the residue chromatographed by TLC on silica gel. Elution with n-hexane/CH2Cl2 (3:1 v/v) developed three bands which afforded the following compounds, in order of elution: unreacted [Fe2(CO)6(µ-bdt)] (3 mg), [Fe2(CO)5(SbPh3)(µ-SC6H4S)] (3) (75 mg, 42%) and [Fe2(CO)4(SbPh3)2(µ-SC6H4S)] (4) (55 mg, 22%) as orange crystals after recrystallization from hexane/CH2Cl2 at 4 °C. In a separate experiment, a CH2Cl2 solution (20 mL) of 3 (50 mg, 0.067 mmol) and SbPh3 (24 mg, 0.067 mmol) was added to a solution of Me3NO (5 mg, 0.067 mmol) in the same solvent (7 mL) and the resulting solution was stirred at room temperature for 24 h. The solvent was removed by rotary evaporation and the residue chromatographed by TLC on silica gel. Elution with n-hexane/CH2Cl2 (1:1, v/v) developed two bands. The faster moving band was unreacted 3 (trace) and the second band afforded 4 (46 mg, 64%).
Analytical and spectroscopic data for 3: Anal. Calcd. for C29H19Fe2O5S2Sb: C, 46.75; H, 2.57. Found: C, 46.88; H, 2.69. IR (ν(CO), CH2Cl2): 2053 s, 1992 s, 1935 m cm−1. 1H NMR (CDCl3): δ 7.47 (m, 10H, phenyl), 7.36 (m, 6H, phenyl), 6.57 (s, 2H, C6H4), 6.13 (s, 2H, C6H4). ESI-MS: m/z 745.04.
Analytical and spectroscopic data for 4: Anal. Calcd for C46H34Fe2O4S2Sb2: C, 51.63; H, 3.20. Found: C, 51.58; H, 3.23. Data for 4: IR (ν(CO), CH2Cl2): 2000 vs, 1958 m, 1938 s cm−1. 1H NMR (CDCl3): δ 7.46 (m, 10H, phenyl), 7.34 (m, 20H, phenyl), 6.11 (m, 2H, C6H4), 5.70 (m, 2H, C6H4). ESI-MS: m/z 1070.10.
4.4. Synthesis of [Fe2(CO)4(PPh2H)2(µ-SC6H4S)] (5) [34]
To an MeCN solution (25 mL) of [Fe2(CO)6(µ-bdt)] (100 mg, 0.238 mmol) and PPh2H (89 mg, 0.478 mmol) was added dropwise a solution of Me3NO (18 mg, 0.24 mmol) in MeCN (5 mL). The reaction mixture was heated at 80 °C for 4 h. The solvent was removed under reduced pressure and the residue chromatographed by TLC on silica gel. Elution with n-hexane/CH2Cl2 (3:1 v/v) developed three bands which afforded only [Fe2(CO)4(PPh2H)2(µ-SC6H4S)] (5) [33] (67 mg, 51%) as red crystals after recrystallization from n-hexane/CH2Cl2 at 4 °C.
4.5. X-Ray Crystallography
Single crystals of 1–5 suitable for X-ray diffraction studies were obtained by recrystallization from hexane/CH2Cl2 at 4 °C. The X-ray diffraction data were collected on an Agilent technologies Oxford Diffraction Supernova diffractometer (1,4), Bruker Kappa Apex II diffractometer (2,3), or Bruker Smart 1K CCD diffractometer (5) using Mo Kα radiation (λ = 0.71073 Å). The CrysAlisPro [51] version 1.171.36.28 (1,4) Denzo/Scalepack [52] (2,3) or SAINT [53] version 7.68A (5) software packages were used for cell refinements and data reductions. A Multi-scan absorption correction (1,3,5) or numerical absorption correction (2, 4) was applied to the intensities before the structure solutions (SADABS) [54]. The structures were solved by the charge-flipping method using the Superflip [55] program (1–4) or direct methods (5), SHELXS [56]. Structural refinements were carried out using the SHELXL-2014 [57] program with the Olex2 [58] and SHELXLE [59] graphical user interfaces. Hydrogen atoms in 1–4 were positioned geometrically and constrained to ride on their parent atoms, with C-H = 0.95 Å and Uiso = 1.2·Ueq (parent atom). In 5, hydrogen atoms were also located geometrically but refined isotropically. The details of data collection and structure refinement are summarised in Table 3.
Table 3.
Crystal data and structure refinement details for compounds 1–5.
4.6. Electrochemistry
Electrochemical measurements were carried out in deoxygenated CH2Cl2 and acetonitrile solutions with 0.1 M TBAPF6 as the supporting electrolyte. The working electrode was a 3 mm diameter glassy carbon electrode that was polished with 0.3 μm alumina slurry prior to each scan. The counter electrode was a platinum wire and the quasi-ᵟreference electrode was a silver wire. All CVs were referenced to the Fc+/Fc redox couple. A Pine WaveNow potentiostat (Pine Research Instrumentation Inc., Durham, NC, USA) was used for all electrochemical measurements. Catalysis studies were carried out by adding equivalents of HOTs (Sigma-Aldrich, St. Louis, MO, USA).
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/inorganics13020063/s1, Figure S1: FTIR of 1 in CH2Cl2 blue (complex 1), orange (complex 1 + HBF4 after 1 min) and red (complex 1 + HBF4 after 60 min); Figure S2: FTIR of 2 in CH2Cl2 blue (complex 2), orange (complex 2 + HBF4 after 1 min), (complex 2 + HBF4 after 60 min) and green (complex 2 + HBF4 after 80 min); Figure S3: FTIR of 3 in CH2Cl2 blue (complex 3), orange (complex 3 + HBF4 after 1 min), (complex 3 + HBF4 after 60 min) and green (complex 3 + HBF4 after 80 min); Figure 4: FTIR of 4 in CH2Cl2 blue (complex 4), orange (complex 4 + HBF4 after 1 min) and (complex 4 + HBF4 after 60 min); Figure S5: CVs of 1 in CH2Cl2 (1 mM solution, supporting electrolyte [NBu4][PF6] at 10–2000 mV/sec scan rate, glassy carbon electrode, potential vs. Fc+/Fc); Figure S6: CVs of 3 in CH2Cl2 (1 mM solution, supporting electrolyte [NBu4][PF6] at 10–2000 mV/sec scan rate, glassy carbon electrode, potential vs. Fc+/Fc); Figure S7: CVs of 2 in CH2Cl2 (1 mM solution, supporting electrolyte [NBu4][PF6] at 25, 50, 100 and 500 mV/sec scan rate, glassy carbon electrode, potential vs. Fc+/Fc); Figure S8: CVs of [Fe2(CO)4(PHPh2)2(μ-bdt)] (5) at various scan rates in acetonitrile (1 mM solution, supporting electrolyte [NBu4][PF6], glassy carbon electrode, potential vs. Fc+/Fc); Figure S9: CVs of 2 (black dots) in the absence and presence of 1, 3, 6, 13, 27 molar equivalents of TsOH (1 mM solution in CH2Cl2, supporting electrolyte [NBu4][PF6], scan rate 200 mVs−1, glassy carbon electrode, potential vs. Fc+/Fc); Figure S10: CVs of 1 (black) in the absence of acid, 16 molar equivalents of TsOH itself (red dots) and complex 1 in the presence of 18 molar equivalents of TsOH (blue) (1 mM solution in CH2Cl2, supporting electrolyte [NBu4][PF6], scan rate 0.05 Vs−1, glassy carbon electrode, potential vs. Fc+/Fc); Figure S11: CVs of 3 (black dots) in the absence acid, 16 molar equivalents of TsOH itself (red) and complex 3 in the presence of 18 molar equivalents of TsOH (blue) (1 mM solution in CH2Cl2, supporting electrolyte [NBu4][PF6], scan rate 0.05 Vs−1, glassy carbon electrode, potential vs. Fc+/Fc); Figure S12: CVs of 2 (black dots) in the absence of acid, 16 molar equivalents of TsOH itself (red dots) and complex 2 in the presence of 18 molar equivalents of TsOH (blue) (1 mM solution in CH2Cl2, supporting electrolyte [NBu4][PF6], scan rate 200 mVs−1, glassy carbon electrode, potential vs. Fc+/Fc); Figure S13: CVs of 5 in the absence and in the presence of 1–10 molar equivalents of CF3CO2H (1 mM solution in MeCN, supporting electrolyte [NBu4][PF6], scan rate 0.1 Vs−1, glassy carbon electrode, potential vs. Fc+/Fc); Table S1: Selected structural parameters for bond lengths [Å] and angles [°] for 1; Table S2: Selected structural parameters for bond lengths [Å] and angles [°] for 2; Table S3: Selected structural parameters for bond lengths [Å] and angles [°] for 3; Table S4: Selected structural parameters for bond lengths [Å] and angles [°] for 4; Table S5: Selected structural parameters for bond lengths [Å] and angles [°] for 5; Table S6: FTIR for [Fe2(CO)6(μ-bdt)][47] (C), 1-4, [Fe2(CO)5(μ-bdt)(PPh3)][33] and [Fe2(CO)4(μ-bdt)(PPh3)2][33] in CH2Cl2; Table S7: Atomic coordinates (×104) and equivalent isotropic displacement parameters (Å2 × 103) for [Fe2(CO)5(μ-bdt)(AsPh3)] (1). U(eq) is defined as one third of the trace of the orthogonalized Uij tensor; Table S8. Atomic coordinates (×104) and equivalent isotropic displacement parameters (Å2 × 103) for [Fe2(CO)5(μ-bdt)(SbPh3)] (2). U(eq) is defined as one third of the trace of the orthogonalized Uij tensor; Table S9: Atomic coordinates (×104) and equivalent isotropic displacement parameters (Å2 × 103) for [Fe2(CO)4(μ-bdt)(AsPh3)2] (3). U(eq) is defined as one third of the trace of the orthogonalized Uij tensor; Table S10: Atomic coordinates (×104) and equivalent isotropic displacement parameters (Å2 × 103) for [Fe2(CO)4(μ-bdt)(SbPh3)2] (4). U(eq) is defined as one third of the trace of the orthogonalized Uij tensor; Table S11: Cartesian coordinates for [Fe2(CO)4(μ-bdt)(PPh2H)2] (5).
Author Contributions
Synthetic work was performed by A.R. and U.K. with input from F.R.A. and S.E.K.; Single crystal X-ray diffraction studies were performed by S.G., G.H. and M.H.; Protonation, electrochemistry and catalytic studies were carried out by A.R. and S.G. with input from G.H. and E.N.; Initial drafting of the manuscript was done by A.R. and S.E.K. with input from S.G., G.H. and E.N.; The manuscript was finally revised jointly by G.H. and E.N. All authors have read and agreed to the published version of the manuscript.
Funding
We gratefully acknowledge the Ministry of Science and Technology, Bangladesh, for financial support.
Data Availability Statement
The data presented in this study are openly available in [Cambridge Structural Database] at [ccdc.cam.ac.uk], reference number [CCDC 2339448, 2339449, 2339450, 2339451, 2339452]. All other data are available from the authors upon request.
Acknowledgments
We are grateful to (the late) Manzurul Karim for providing X-ray quality crystals of 5 for diffraction studies.
Conflicts of Interest
The authors declare no competing conflicts of interest.
References
- Hogarth, G. An unexpected leading role for [Fe2(CO)6(μ-pdt)] in our understanding of [FeFe]-H2ases and the search for clean hydrogen production. Coord. Chem. Rev. 2023, 490, 215174. [Google Scholar] [CrossRef]
- Evans, D.J.; Pickett, C.J. Chemistry and the hydrogenases. Chem. Soc. Rev. 2003, 32, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Georgakaki, I.P.; Thomson, L.M.; Lyon, E.J.; Hall, M.B.; Darensbourg, M.Y. Fundamental properties of small molecule models of Fe-only hydrogenase: Computations relative to the definition of an entatic state in the active site. Coord. Chem. Rev. 2003, 238–239, 255–266. [Google Scholar] [CrossRef]
- Rauchfuss, T.B. Research on soluble metal sulfides: From polysulfido complexes to functional models for the hydrogenases. Inorg. Chem. 2004, 43, 14–26. [Google Scholar] [CrossRef]
- Liu, X.; Ibrahim, S.K.; Tard, C.; Pickett, C.J. Iron-only hydrogenase: Synthetic, structural and reactivity studies of model compounds. Coord. Chem. Rev. 2005, 249, 1641–1652. [Google Scholar] [CrossRef]
- Sun, L.; Åkermark, B.; Ott, S. Iron hydrogenase active site mimics in supramolecular systems aiming for light-driven hydrogen production. Coord. Chem. Rev. 2005, 249, 1653–1663. [Google Scholar] [CrossRef]
- Capon, J.-F.; Gloaguen, F.; Schollhammer, P.; Talarmin, J. Catalysis of the electrochemical H2 evolution by di-iron sub-site models. Coord. Chem. Rev. 2005, 249, 1664–1676. [Google Scholar] [CrossRef]
- Tard, C.; Pickett, C.J. Structural and functional analogues of the active sites of the [Fe]-, [NiFe]-, and [FeFe]-hydrogenases. Chem. Rev. 2009, 109, 2245–2274. [Google Scholar] [CrossRef]
- Felton, G.A.N.; Mebi, C.A.; Petro, B.J.; Vannucci, A.K.; Evans, D.H.; Glass, R.S.; Lichtenberger, D.L. Review of electrochemical studies of complexes containing the Fe2S2 core characteristic of [FeFe]-hydrogenases including catalysis by these complexes of the reduction of acids to form dihydrogen. J. Organomet. Chem. 2009, 694, 2681–2699. [Google Scholar] [CrossRef]
- Lubitz, W.; Ogata, H.; Rudiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef]
- Li, Y.; Rauchfuss, T.B. Synthesis of diiron(I) dithiolato carbonyl complexes. Chem. Rev. 2016, 116, 7043–7077. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, M.; He, C.; Li, G.; Liu, X.; Chen, C.; Åkermark, B.; Sun, L. Influence of tertiary phosphanes on the coordination configurations and electrochemical properties of iron hydrogenase model complexes: Crystal structures of [(μ-S2C3H6)Fe2(CO)6–nLn] (L = PMe2Ph,n = 1, 2; PPh3, P(OEt)3, n = 1). Eur. J. Inorg. Chem. 2005, 2506–2513. [Google Scholar] [CrossRef]
- Adam, F.I.; Hogarth, G.; Richards, I.; Sanchez, B.E. Models of the iron-only hydrogenase: Structural studies of chelating diphosphine complexes [Fe2(CO)4(µ-pdt)(κ2P,P′-diphosphine)]. Dalton Trans. 2007, 24, 2495–2498. [Google Scholar] [CrossRef] [PubMed]
- Adam, F.I.; Hogarth, G.; Richards, I. Models of the iron-only hydrogenase: Reactions of [Fe2(CO)6(µ-pdt)] with small bite-angle diphosphines yielding bridge and chelate diphosphine complexes [Fe2(CO)4(diphosphine)(µ-pdt)]. J. Organomet. Chem. 2007, 692, 3957–3968. [Google Scholar] [CrossRef]
- Li, J.-R.; Hu, M.-Y.; Lü, S.; Gu, X.-L.; Jing, X.-B.; Zhao, P.-H. Amine-containing tertiary phosphine-substituted diiron ethanedithioate (edt) complexes Fe2(μ-edt)(CO)6-nLn (n= 1, 2): Synthesis, protonation, and electrochemical properties. Appl. Organomet. Chem. 2020, 34, e5929. [Google Scholar] [CrossRef]
- Natarajan, M.; Kumar, N.; Joshi, M.; Stein, M.; Ghumaan, S.K. Mechanism of Diiron Hydrogenase Complexes Controlled by Nature of Bridging Dithiolate Ligand. Chem. Open 2022, 11, e202100238. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, X.; Zhang, T.; Li, B.; Jiang, S.; Zhang, G.; Hai, L.; Wanga, J.; Shao, X. The influence of phosphine ligand substituted [2Fe2S] model complexes as electro-catalyst on proton reduction. RSC Adv. 2018, 8, 42262–42268. [Google Scholar] [CrossRef]
- Peralta-Arriaga, S.L.; Fernandez-Teran, R.J.; Shipp, J.D.; Royle, C.E.; Chekulaev, D.; Morris, M.J.; Weinstein, J.A. Photophysics of Fe-Fe hydrogenase mimic complexes for hydrogen evolution. J. Organomet. Chem. 2024, 1004, 122940. [Google Scholar] [CrossRef]
- Liu, X.-F.; Wang, S.-J.; Zhao, P.-H. Di-iron dithiolato complexes with 3-bromothiophene moiety: Preparation, structures, and electrochemistry. J. Mol. Struct. 2023, 1294, 136454. [Google Scholar] [CrossRef]
- Wang, N.; Wang, M.; Chen, L.; Sun, L. Reactions of [FeFe]-hydrogenase models involving the formation of hydrides related to proton reduction and hydrogen oxidation. Dalton Trans. 2013, 42, 12059–12071. [Google Scholar] [CrossRef]
- Heinekey, D.M. Hydrogenase enzymes: Recent structural studies and active site models. J. Organomet. Chem. 2009, 694, 2671–2680. [Google Scholar] [CrossRef]
- Lian, M.; He, J.; Yu, X.-Y.; Mu, C.; Liu, X.-F.; Li, Y.-L.; Jiang, Z.-Q. Diiron ethanedithiolate complexes with acetate ester: Synthesis, characterization and electrochemical properties. J. Organomet. Chem. 2018, 870, 90–96. [Google Scholar] [CrossRef]
- Capon, J.-F.; Gloaguen, F.; Petillon, F.Y.; Schollhammer, P.; Talarmin, J. On the electrochemistry of diiron dithiolate complexes related to the active site of the [FeFe]H2ase. C. R. Chim. 2008, 11, 842–851. [Google Scholar] [CrossRef]
- Capon, J.-F.; Gloaguen, F.; Petillon, F.Y.; Schollhammer, P.; Talarmin, J. Electron and proton transfers at diiron dithiolate sites relevant to the catalysis of proton reduction by the [FeFe]-hydrogenases. Coord. Chem. Rev. 2009, 253, 1476–1494. [Google Scholar] [CrossRef]
- Tschierlei, S.; Ott, S.; Lomoth, R. Spectroscopically characterized intermediates of catalytic H2 formation by [FeFe] hydrogenase models. Energy Environ. Sci. 2011, 4, 2340–2352. [Google Scholar] [CrossRef]
- Peters, J.W.; Lanzilotta, W.N.; Lemon, B.J.; Seefeldt, L.C. X-ray Crystal structure of the Fe-only hydrogenase (CpI) from clostridium pasteurianum to 1.8 angstrom resolution. Science 1998, 282, 1853–1858. [Google Scholar] [CrossRef]
- Nicolet, Y.; Piras, C.; Legrand, P.; Hatchikian, C.E.; Fontecilla-Camps, J.C. Desulfovibrio desulfuricans iron hydrogenase: The structure shows unusual coordination to an active site Fe binuclear center. Structure 1999, 13, 13–23. [Google Scholar] [CrossRef]
- Nicolet, Y.; De Lacey, A.L.; Vernede, X.; Fernandez, V.M.; Hatchikian, E.C.; Fontecilla-Camps, J.C. Crystallographic and FTIR spectroscopic evidence of changes in Fe coordination upon reduction of the active site of the Fe-only hydrogenase from Desulfo- vibrio desulfuricans. J. Am. Chem. Soc. 2001, 123, 1596–1601. [Google Scholar] [CrossRef]
- Nicolet, Y.; Cavazza, C.; Fontecilla-Camps, J.C. Fe-only hydrogenases: Structure, function and evolution. J. Inorg. Biochem. 2002, 91, 1–8. [Google Scholar] [CrossRef]
- Armstrong, F.A. Hydrogenases: Active site puzzles and progress. Curr. Opin. Chem. Biol. 2004, 8, 133–140. [Google Scholar] [CrossRef]
- Gloaguen, F.; Morvan, D.; Capon, J.-F.; Schollhammer, P.; Talarmin, J.-F. Electrochemical proton reduction at mild potentials by monosubstituted diiron organometallic complexes bearing a benzenedithiolate bridge. J. Electroanal. Chem. 2007, 603, 15–20. [Google Scholar] [CrossRef]
- Schwartz, L.; Singh, P.S.; Eriksson, L.; Lomoth, R.; Ott, S. Tuning the electronic properties of Fe2(µ-arenedithiolate) (CO)6-n(PMe3)n (n = 0, 2) complexes related to the [Fe-Fe]-hydrogenase active site. C. R. Chim. 2008, 11, 875–889. [Google Scholar] [CrossRef]
- Pandey, I.K.; Mobin, S.M.; Deibel, N.; Sarkar, B.; Kaur-Ghumaan, S. Diiron benzenedithiolate complexes relevant to the [FeFe] Hydrogenase active site. Eur. J. Inorg. Chem. 2015, 17, 2875–2882. [Google Scholar] [CrossRef]
- Wright, R.J.; Zhang, W.; Yang, X.; Fasulo, M.; Tilley, T.D. Isolation, observation, and computational modelling of proposed intermediates in catalytic proton reductions with the hydrogenase mimic Fe2(CO)6S2C6H4. Dalton Trans. 2012, 41, 73–82. [Google Scholar] [CrossRef]
- Oudsen, J.P.H.; Venderbosch, B.; Martin, D.J.; Korstanje, T.J.; Reek, J.N.H.; Tromp, M. Spectroscopic and theoretical investigation of the [Fe2(bdt)(CO)6] hydrogenase mimic and some catalyst intermediates. Phys. Chem. Chem. Phys. 2019, 21, 14638. [Google Scholar] [CrossRef]
- Quentel, F.; Passard, G.; Gloaguen, F. Electrochemical hydrogen production in aqueous micellar solution by a diiron benzenedithiolate complex relevant to [FeFe] hydrogenases. Energy Environ. Sci. 2012, 5, 7757–7761. [Google Scholar] [CrossRef]
- Felton, G.A.N.; Vannucci, A.K.; Chen, J.; Lockett, L.T.; Okumura, N.; Petro, B.J.; Zakai, U.I.; Evans, D.H.; Glass, R.S.; Lichtenberger, D.L. Hydrogen generation from weak acids: Electrochemical and computational studies of a diiron hydrogenase mimic. J. Am. Chem. Soc. 2007, 129, 12521–12530. [Google Scholar] [CrossRef]
- McKennis, S.J.; Kyba, P.E. Linked Bis(µ-phosphido) and Related ligand for metallic cluster. Application to the hexacarbonyldiironmoiety. Organometallics 1983, 2, 1249–1251. [Google Scholar] [CrossRef]
- Capon, J.-F.; Gloaguen, F.; Schollhammer, P.; Talarmin, J. Activation of proton by the two-electron reduction of a di-iron organometallic complex. J. Electroanal. Chem. 2006, 595, 47–52. [Google Scholar] [CrossRef]
- Capon, J.-F.; Gloaguen, F.; Schollhammer, P.; Talarmin, J. Electrochemical proton reduction by thiolate-bridged hexacarbonyl diiron clusters. J. Electroanal. Chem. 2004, 566, 241–247. [Google Scholar] [CrossRef]
- Li, P.; Wang, M.; Pan, J.; Chen, L.; Wang, N.; Sun, L. [FeFe]-Hydrogenase active site models with relatively low reduction potentials: Diiron dithiolate complexes containing rigid bridges. J. Inorg. Biochem. 2008, 102, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Hizbullah, L.; Rahaman, A.; Safavi, S.; Haukka, M.; Tocher, D.A.; Lisensky, G.C.; Nordlander, E. Synthesis of phosphine derivatives of [Fe2(CO)6(μ-sdt)](sdt = SCH2SCH2S) and investigation of their proton reduction capabilities. J. Inorg. Biochem. 2023, 246, 112272. [Google Scholar] [CrossRef]
- Chong, D.; Georgakaki, I.P.; Mejia-Rodriguez, R.; Chinchilla, J.S.; Soriaga, M.P.; Darensbourg, M.Y. Electrocatalysis of hydrogen production by active site analogues of the iron hydrogenase enzyme: Structure/function relationships. Dalton Trans. 2003, 4158–4163. [Google Scholar] [CrossRef]
- Song, L.-C.; Yang, Z.-Y.; Bian, H.-Z.; Liu, Y.; Wang, H.-T.; Liu, X.-F.; Hu, Q.-M. Diiron oxadithiolate type models for the active site of iron-only hydrogenases and biomimetic: Hydrogen evolution catalyzed by Fe2(μ-SCH2OCH2S-μ)(CO)6. Organometallics 2005, 24, 6126–6135. [Google Scholar] [CrossRef]
- Song, L.-C.; Li, C.-G.; Ge, J.-H.; Yang, Z.-Y.; Wang, H.-T.; Zhang, J.; Hu, Q.-M. Synthesis and structural characterization of the mono- and diphosphine-containing diiron propanedithiolate complexes related to [FeFe]-hydrogenases. Biomimetic H2 evolution catalyzed by (µ-pdt)Fe2(CO)4[(Ph2P)2N(n-Pr)]. J. Inorg. Biochem. 2008, 102, 1973–1979. [Google Scholar] [CrossRef]
- Ghosh, S.; Rahaman, A.; Orton, G.; Gregori, G.; Bernat, M.; Kulsume, U.; Hollingsworth, N.; Holt, K.B.; Kabir, S.E.; Hogarth, G. Synthesis, molecular structures and electrochemical investigations of [FeFe]-hydrogenase biomimics [Fe2(CO)6-n(EPh3)n(μ-edt)] (E = P, As, Sb; n = 1, 2). Eur. J. Inorg. Chem. 2019, 42, 4506–4515. [Google Scholar] [CrossRef]
- Cabeza, J.A.; Martinez-García, M.A.; Riera, V.; Ardura, D.; García-Granda, S. Binuclear iron(I), ruthenium(I), and osmium(I) hexacarbonyl complexes containing a bridging benzene-1,2-dithiolate ligand. Synthesis, X-ray structures, protonation reactions, and EHMO calculations. Organometallics 1998, 17, 1471–1477. [Google Scholar] [CrossRef]
- Orton, G.R.F.; Belazregue, S.; Cockcroft, J.K.; Hartl, F.; Hogarth, G. Biomimics of [FeFe]-hydrogenases with a pendant amine: Diphosphine complexes [Fe2(CO)4{μ-S(CH2)nS}{κ2-(Ph2PCH2)2NR}] (n = 2,3; R = Me, Bn) towards H2 oxidation catalysts. J. Organomet. Chem. 2023, 991, 122673. [Google Scholar] [CrossRef]
- Cheng, M.; Wang, M.; Zheng, D.; Sun, L. Effect of the S-to-S bridge on the redox properties and H2 activation performance of diiron complexes related to the [FeFe]-hydrogenase active site. Dalton Trans. 2016, 45, 17687–17696. [Google Scholar] [CrossRef]
- Wang, N.; Wang, M.; Wang, Y.; Zheng, D.; Han, H.; Ahlquist, M.S.G.; Sun, L. Catalytic activation of H2 under mild conditions by an [FeFe]- hydrogenase model via an active μ-hydride species. J. Am. Chem. Soc. 2013, 135, 13688–13691. [Google Scholar] [CrossRef]
- Agilent. CrysAlisPro, Agilent Technologies Inc.: Yarnton, UK, 2014.
- Otwinowski, Z.; Minor, W. Volume 276: Macromolecular Crystallography, Part A. In Methods in Enzymology; Carter, C.W., Sweet, J., Eds.; Academic Press: New York, NY, USA, 1997; pp. 307–326. [Google Scholar]
- Bruker AXS. SAINT, Bruker AXS, Inc.: Madison, WI, USA, 2009.
- Sheldrick, G.M. SADABS—Bruker AXS Scaling and Absorption Correction; Bruker AXS, Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Palatinus, L.; Chapuis, G. SUPERFLIP—A Computer Program for the Solution of Crystal Structures by Charge Flipping in Arbitrary Dimensions. J. Appl. Cryst. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H.J. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt Graphical User Interface for SHELXL. J. Appl. Cryst. 2011, 44, 1281–1284. [Google Scholar] [CrossRef]
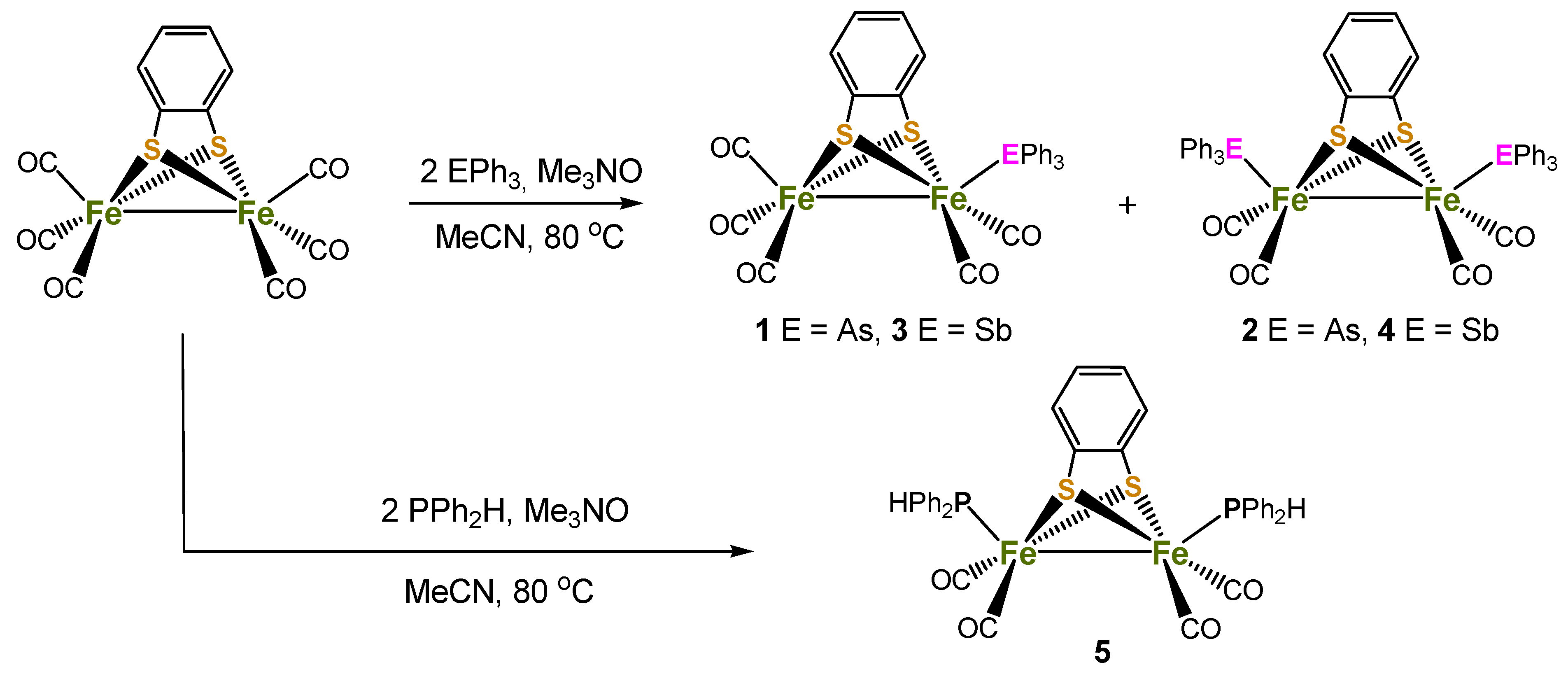
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).