Abstract
A ferrocenium-catalyzed synthesis of trisubstituted tetrahydrofurans by dehydrative cyclization of diols is reported. Treatment of γ-phenyl-γ-butyrolactone or valerolactone with 2–3 equivalents of MeLi, n-BuLi, or PhLi yielded the corresponding substituted 1,4-butanediols in 41–86% yields. Subsequent dehydrative cyclization of the diols under non-inert conditions using catalytic ferrocenium tetrafluoroborate (10 mol%) produced trisubstituted tetrahydrofurans in 72–83% yields after 48–72 h at 45–70 °C in CH2Cl2. This study demonstrates ferrocenium-catalyzed dehydrative cyclization for the first time, offering a convenient route to substituted tetrahydrofurans in two steps from commercial or easily accessible starting materials.
1. Introduction
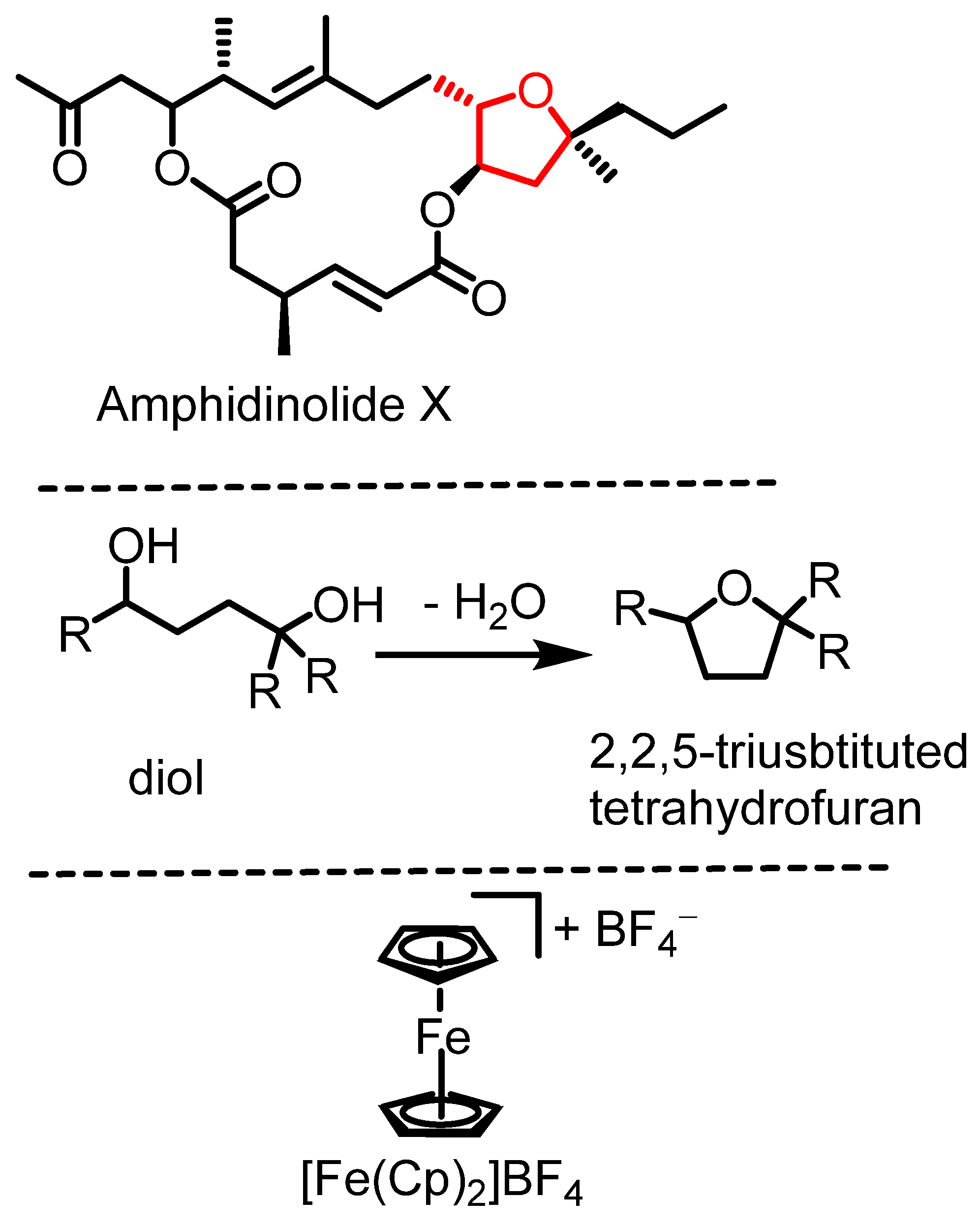
Highly substituted tetrahydrofurans are key structural motifs found in numerous natural products with bioactive properties, including antibiotics, antivirals, and anticancer agents [1,2]. Their architecture contributes to molecular stability, making them valuable scaffolds in drug design and medicinal chemistry [3,4]. Many tetrahydrofurans or polycyclic ethers [5] possess biological activities [6,7], such as amphidinolide X (Scheme 1, top) [8]. Synthetic access to these structures has been described through metal-catalyzed carboetherification reactions [1,9], intramolecular epoxide-opening reactions [10], or hydroalkoxidations [1,11], among other methods [8].
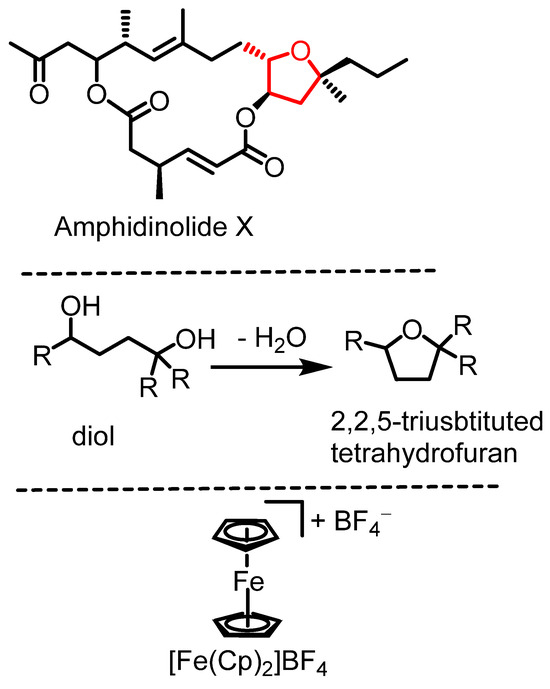
Scheme 1.
Amphidinolide X, dehydrative diol cyclization, and ferrocenium tetrafluoroborate.
The intramolecular version of the substitution of OH units by another one in the same molecule constitutes the dehydrative cyclization of diols to the corresponding tetrahydrofuran species (Scheme 1, middle). This type of cyclization reaction is underdeveloped [12] and allows access to highly substituted tetrahydrofuran species. Brønsted acids are generally effective in catalyzing diol cyclization reactions and related processes [13], but they often require elevated reaction temperatures [14]. For more complex synthetic targets, Brønsted acid catalysis can pose challenges such as limited functional group tolerance, the need for elevated temperatures, and the risk of carbocation rearrangements. Lewis acids can cyclize diols to give cyclic ethers as well, but also tend to require high reaction temperatures [15,16]. Alternative methods for diol cyclization typically involve using excess reagents, such as carbonate activators [17] or establishing leaving groups [1,18,19,20]. More complex diol cyclization strategies exist, e.g., employing selenium reagents [21] or cerium(IV) ammonium nitrate, which has been employed in the synthesis of a tertiary, 2,2-vinyl-disubstituted tetrahydrofuran through a radical cyclization mechanism [22].
Access to 1,4-disubstituted and 1,2,3,4-tetrasubstituted tetrahydrofurans by diastereoselective, dehydrative cyclizations of diols has been described to be catalyzed by FeCl3 (20% catalyst load) [23]. To the best of our knowledge, other iron-catalyzed dehydrative cyclization reactions to produce highly substituted tetrahydrofuran derivatives have not been reported. Catalytic, dehydrative cyclization offers a significant advantage by eliminating the need for complex reagents or auxiliaries (such as leaving groups), generating only water as a byproduct in accordance with green chemistry principles [24]. Iron catalysis [25] plays a crucial role in developing new catalytic methods due to its abundance, low cost, relative non-toxicity, and environmental friendliness [26,27]. Ferrocenium cations have emerged as effective and versatile catalysts in the past two decades [28,29], valued for their tunability, benchtop stability, and ease of handling. For example, ferrocenium salts are air-stable, whereas FeCl3 is water-sensitive. Herein, we present dehydrative cyclization reactions catalyzed by [FeCp2]BF4 (Scheme 1, bottom) without the exclusion of air or water for the synthesis of trisubstituted tetrahydrofurans. The approach allows for access to 2,2,5-trisubstituted tetrahydrofuran derivatives in two steps from commercial or easily accessible starting materials.
2. Results and Discussion
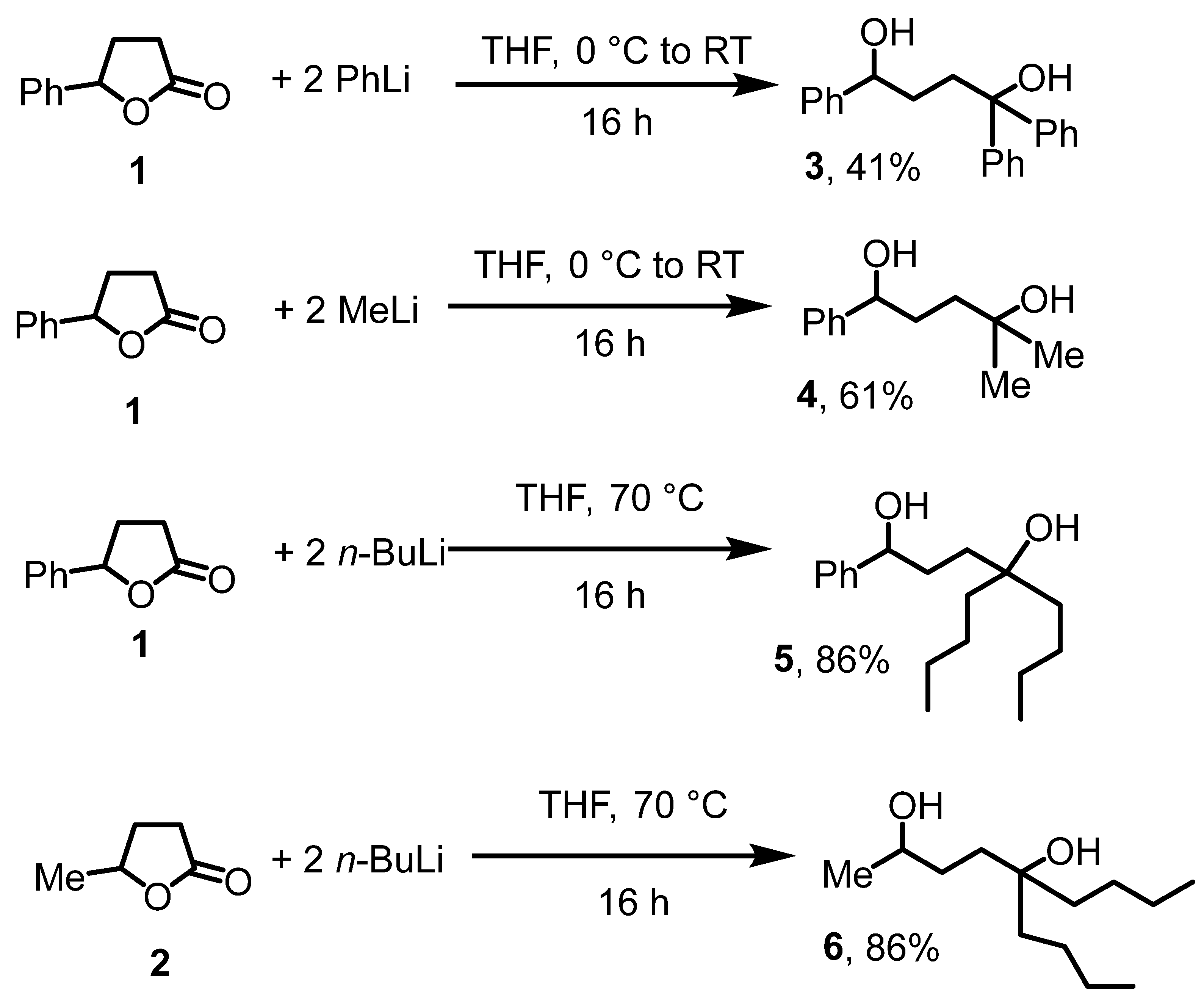
First, we sought access to substituted 1,4-diol precursor compounds. The nucleophilic ring opening of cyclic butyrolactones has previously been employed for that purpose [30]. Accordingly, we treated commercial γ-phenyl-γ-butyrolactone (1) with three equivalent lithium organyls, followed by quenching with water and extraction with diethyl ether (Scheme 2). That way, reaction of 1 with PhLi, MeLi, and n-BuLi gave the trisubstituted diols 3, 4, and 5 in 86 to 41% isolated yields. The cyclic, commercial valerolactone 2 gave under similar conditions the purely aliphatic diol 6. The structure of the diols was unequivocally established by NMR and IR. The C=O stretch for the ester functionality in 1 disappeared. Instead, a broad band around 3400 cm−1 was observed for the OH groups in the diols 3–6, establishing the presence of the alcohols. The OH groups were also observed as broad resonances in the 1H NMR spectra between 2 and 3 ppm. Furthermore, a diagnostic resonance for the HCOH proton was observed for all three diols around 4.5 ppm. Diols 3 [31] and 4 [32] are documented in the literature and were obtained through alternative synthetic routes; the NMR data are consistent with published values.
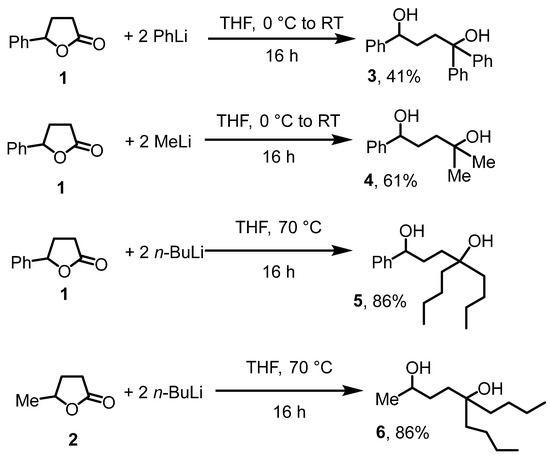
Scheme 2.
Diol syntheses.
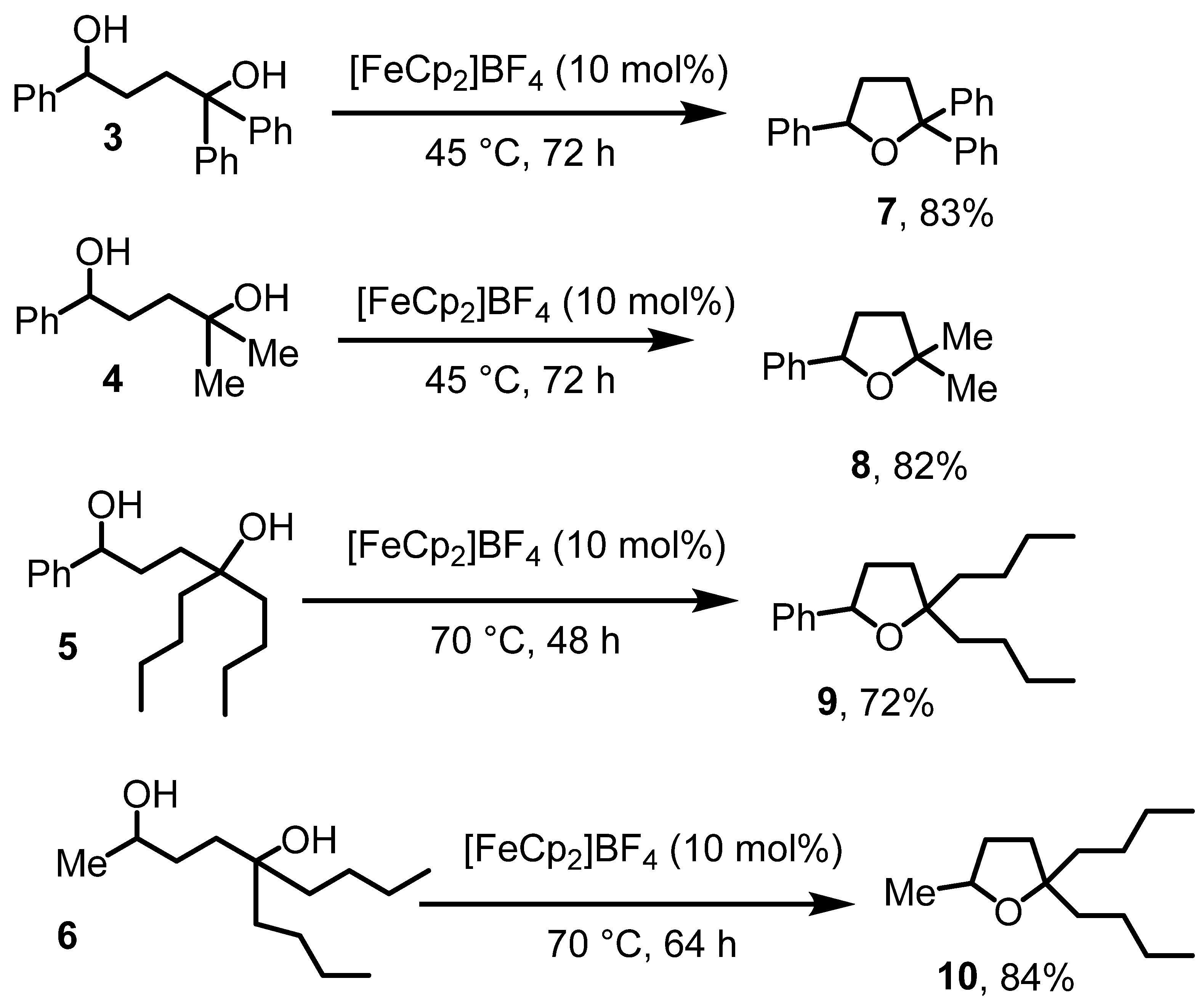
We next sought cyclization of the diols to the tetrahdyrofurans, drawing knowledge from the literature referenced above about Lewis acid-catalyzed dehydrative diol cyclization reactions [12]. After brief screening efforts, we determined that ferrocenium tetrafluoroborate, [FeCp2]BF4, is catalytic active for the title reaction (Scheme 3). Accordingly, when the diols 3–6 were treated with [FeCp2]BF4 in CH2Cl2 at 45–70 °C for two to three days, the corresponding trisubstituted tetrahydrofurans 7–10 were isolated by filtration through Al2O3 in 83–72% yields. The isolation by filtration was easy, because the catalyst and potential residual amounts of the diols were much more polar than the substituted tetrahydrofuran products 7–10. The identity of the products was again established by IR and NMR. The OH stretch of the diol starting materials in the IR spectra disappeared for the products. The diagnostic resonance for the HCOH proton shifted by about 0.4 ppm upfield. Most importantly, the formation of the five-membered ring system rendered some of the ring protons and side chain carbons diasterotopic, resulting in characteristic peak splitting in the NMR spectra that were not observed in the starting materials. The tetrahydrofuran derivatives 7 and 8 were known. For 7, no spectroscopic details were published [33] and for 8, NMR data matched the literature values [34]. Especially products 9 and 10 contained small amounts of ferrocene, derived from the ferrocenium salt employed in the reactions. Although ferrocene could be removed by column chromatography, the prolonged reaction time for products 9 and 10 appeared to cause the decomposition of the ferrocenium salts, facilitating their removal.
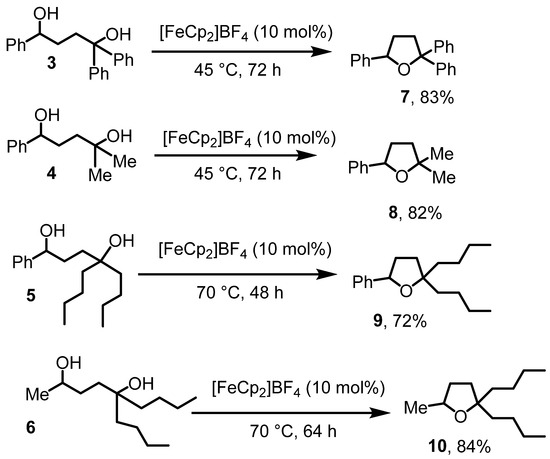
Scheme 3.
Diol cyclizations to afford substituted tetrahydrofurans.
The chemistry in Scheme 3 establishes for the first time ferrocenium cations as catalysts for dehydrative etherification reactions of diols. While the reaction does not work at room temperature at appreciable rates, the reaction temperature of 45 °C is similar or lower than other Lewis acid-catalyzed dehydrative cyclization reactions to afford tetrahydrofuran derivatives [14]. The reactions to afford 8 and 9 can also be performed at lower temperatures with comparable yields. Consequently, the reaction temperature can be adjusted to the desired time frame.
We conducted control experiments to evaluate the efficiency of the catalytic system. The cyclization of 6 failed in the absence of a catalyst, confirming that the dehydrative diol cyclization reaction requires a Lewis acid, as previously demonstrated by others [15,16]. Using KPF6 as a Lewis acid catalyst (which is not very well soluble in CH2Cl2), the cyclization of 6 yielded 53% of product 10 under otherwise identical reaction conditions and workup methods. Similarly, catalytic amounts of FeCl3 facilitated the cyclization of 5, producing 9 in a 56% yield under the same conditions. However, the NMR spectra (see Supplementary Data) of products 9 and 10 from both reactions were less clean compared to those obtained using [FeCp2]BF4 as the catalyst and the yields were also lower. Nevertheless, diols 3–6 can generally be cyclized with a Lewis acid catalyst. Ferrocenium salts offer an advantage over purely inorganic Lewis acids, as their catalytic properties can be fine-tuned through modifications to their cyclopentadienyl ring systems and they tend to be well soluble in CH2Cl2.
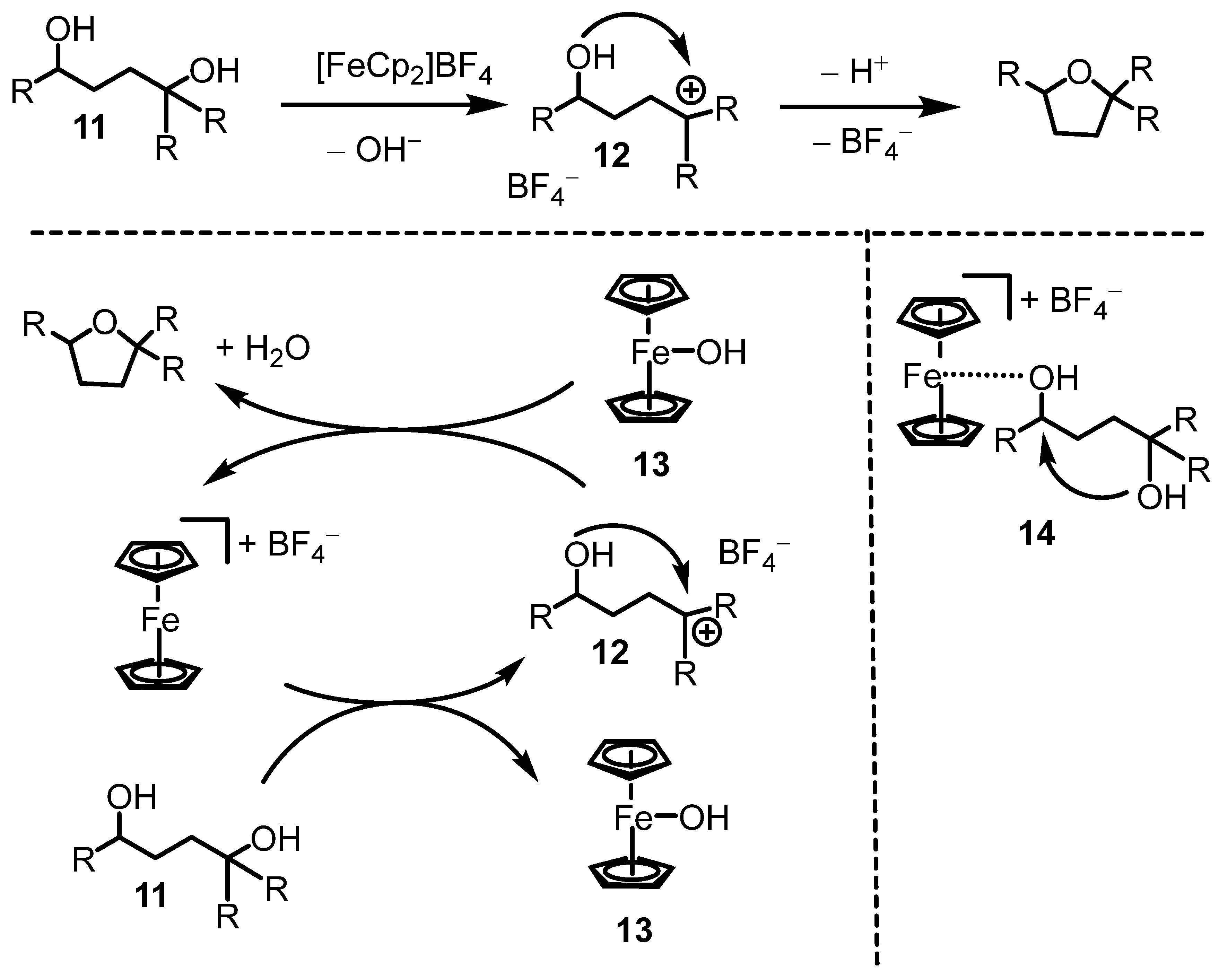
Mechanistically, we assumed the reaction proceeds through a tertiary carbocation intermediate (12, Scheme 4, top). Ferrocenium ions are known to generate carbocations from alcohols. Ferroceniumboronic acid hexafluoroantimonate has been reported to generate benzylic carbocations, which persist in solution as solvated ion pairs with the hexafluoroantimonate counterion [35]. Within the carbocation 12 (persisting as an ion pair with the BF4− counterion), an intramolecular attack of the other OH group on the carbocation would afford the tetrahydrofuran product. As shown in the detailed cycle in Scheme 4 (bottom left), carbocation formation would generate the Fe-OH species 13, though its presence in our reaction was not confirmed. While species 13 has been reported in the literature [36], it remains relatively unexplored [37]. Cyclization of 12 would release a proton, regenerating the ferrocenium cation and yielding the tetrahydrofuran product along with a molecule of water. Alternatively, the ferrocenium cation could polarize one of the C–OH bonds in the diol starting material through species 14 (Scheme 4, bottom right). In this pathway, bond polarization may occur at the sterically less hindered secondary carbon, facilitating intramolecular attack. As indicated above, the products contained varying amounts of ferrocene. This may be an indication that the ferrocenium cation does not completely decompose during catalysis, and 13 or 14 are transient species throughout the catalytic cycle that are finally reduced to ferrocene.
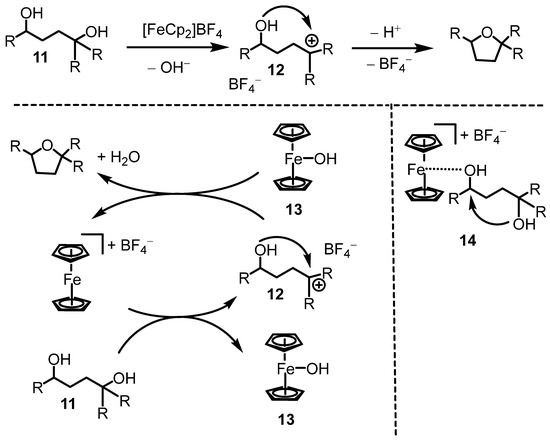
Scheme 4.
Mechanistic picture.
We did not perform mechanistic experiments to verify the mechanism. However, cyclization of 1,4-butanediol, a primary diol, was not possible under the conditions in Scheme 4, presumably due to the fact that it does not form stable carbocations. We are currently investigating if a ferrocenium cation with a chiral counterion can generate enantiomeric excesses through asymmetric counterion-directed catalysis [38,39,40].
3. Experimental Section
General. Commercial γ-phenyl-γ-butyrolactone (1, TCI America, Portland, OR, USA), valorolactone (2), ferrocenium tetrafluoroborate ([FeCp2]BF4), and magnesium sulfate (Sigma-Aldrich, St. Louis, MO, USA) were used as received. PhLi in cyclohexane ether (7:3), MeLi in diethyl ether, and BuLi in hexanes (all Sigma-Aldrich, St. Louis, MO, USA) were used as received and titrated prior to use [41]. THF and diethyl ether were distilled from Na/benzophenone, and CH2Cl2 was distilled from P2O5 prior to use. Column chromatography was performed with silica gel 60 (70–230 mesh, SiliCycle, Québec City, QC, Canada) or Al2O3 (neutral, Brockmann I, 50–100 μm, Acros Organics, Newark, NJ, USA). The diol syntheses were performed under inert conditions utilizing Schlenk techniques. The cyclization reactions were performed without exclusion of air and water with dried CH2Cl2. NMR spectra were obtained at room temperature on a Bruker Avance 300 MHz instrument (Bruker, Billerica, MA, USA) and referenced a residual solvent signal; all assignments are tentative. EI mass spectrometry was performed on a Bruker Maxis Plus (maXis HD) quadrupole time-of-flight mass spectrometer. IR spectra were recorded on Thermo Nicolet 670 FTIR (Thermo Fisher Scientific, Waltham, MA, USA).
1,1,4-triphenylbutane-1,4-diol (3):
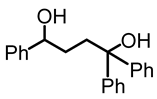
To a Schlenk flask, γ-phenyl-γ-butyrolactone (1, 0.10 g, 0.61 mmol) and diethyl ether (3 mL) were added and cooled to 0 °C. After that, phenyl lithium (0.77 M in cyclohexane-ether, 2 mL, 1.54 mmol) was added dropwise, using a needle and a syringe, and the mixture was stirred for 6 h at 0 °C. After 6 h, the reaction mixture was allowed to warm to room temperature and quenched with water (5 mL). The aqueous and the organic layers were separated. The aqueous layer was extracted twice with diethyl ether. The combined ether fractions were dried with MgSO4 and filtered, and the solvent was removed to obtain the product as a white solid (0.08 g, 0.251 mol, 41%). NMR (δ, CDCl3) 1H: 7.27–7.23 (m, 4H, aromatic), 7.14–7.09 (m, 11H, aromatic), 4.50 (t, 1H, JHH = 6 Hz, OCH), 3.01 (br s, 2H, OH), 2.31–2.23 (m, 2H, CH2), 1.67–1.61 (m, 2H, CH2).13C{1H}: 146.9 (s, aromatic), 144.5 (s, aromatic), 128.5 (s, aromatic), 128.2 (s, aromatic), 127.5 (s, aromatic), 126.84 (s, aromatic), 126.76 (s, aromatic), 126.22 (s, aromatic), 126.16 (s, aromatic), 125.9 (s, aromatic), 78.0 (s, COH), 74.6 (s, COH), 38.0 (s, CH2), 33.4 (s, CH2). IR (ATR, neat): ν 3293 (br), 3056 (w), 3026 (w), 2927 (m), 2861 (m) cm−1.
1,1,4-trimethylbutane-1,4-diol (4):
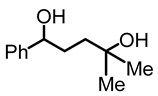
To a Schlenk flask, γ-phenyl-γ-butyrolactone (1, 0.501 g, 3.09 mmol) and diethyl ether (3 mL) were added and cooled to 0 °C. After that, methyl lithium (1.3 M in diethyl ether, 6 mL, 7.8 mmol) was added dropwise, using a needle and a syringe, and the mixture was stirred for 6 h at 0 °C. After 6 h, the reaction mixture was allowed to warm to room temperature and quenched with water (5 mL). The aqueous and the organic layers were separated. The aqueous layer was extracted twice with diethyl ether. The combined ether fractions were dried with MgSO4 and filtered, and the solvent was removed to obtain the product as a yellow oil (0.368 g, 1.89 mmol, 61%). NMR (δ, CDCl3) 1H: 7.26–7.18 (m, 5H, aromatic), 4.57 (dd, 1H, JHH = 8 Hz, JHH = 5 Hz, OCH), 2.83 (br s, 2H, OH), 1.78–1.72 (m, 2H, CH2), 1.57–1.54 (m, 1 H, CHH), 1.45–1.40 (m, 1 H, CHH), 1.11 (br s, 6H, CH3).13C{1H}: 145.0 (s, aromatic), 128.5 (s, aromatic), 128.4 (s, aromatic), 127.3 (s, aromatic), 125.9 (s, aromatic), 74.6 (s, COH), 70.6 (s, COH), 39.8 (s, CH2), 34.0 (s, CH2), 29.7 (s, CH3), 28.9 (s, CH3′). IR (ATR, neat): ν 3398 (br), 3062 (w), 2962 (m), 2933 (m) cm−1.
1,1,4-tri(n-butyl)butane-1,4-diol (5):
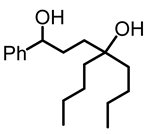
To a pressure vial with a Teflon screw cap, γ-phenyl-γ-butyrolactone (1, 0.250 g, 1.54 mmol) and THF (5 mL) were added. n-Butyl lithium (1.3 M in hexanes, 6 mL, 7.8 mmol) was added dropwise, using a needle and a syringe. The pressure vial was closed, and the yellow mixture was kept for 16 h at 65 °C, resulting in a dark red solution. The reaction mixture was allowed to cool to room temperature and quenched with water (10 mL). The aqueous and the organic layers were separated. The aqueous layer was extracted three times with diethyl ether. The combined ether fractions were dried with MgSO4 and filtered, and the solvent was removed to obtain the product as a yellow oil (0.369 g, 1.33 mmol, 86%). NMR (δ, CDCl3) 1H: 7.24–7.16 (m, 5H, aromatic), 4.54 (dd, 1H, JHH = 8 Hz, JHH = 5 Hz, OCH), 2.78 (br s, 2H, OH), 1.74–1.63 (m, 2H, CH2), 1.54–1.37 (m, 2H, CH2), 1.34–1.29 (m, 3H, CH2), 1.20–1.05 (m, 9H, CH2), 1.11 (t, JHH = 7 Hz, 6H, 2CH3).13C{1H}: 145.0 (s, aromatic), 128.4 (s, aromatic), 127.4 (s, aromatic), 125.9 (s, aromatic), 74.8 (s, COH), 74.3 (s, COH), 39.1 (s, CH2), 38.6 (s, CH2), 35.4 (s, CH2), 33.2 (s, CH2), 25.8 (s, CH2), 25.7 (s, CH2), 23.3 (s, CH2), 14.2 (s, CH3). IR (ATR, neat): ν 3400 (br), 3026 (vw), 2952 (s), 2928 (s), 2858 (s) cm−1. HRMS ESI [M + Na]+ calcd for C18H30O2Na+ 301.2143; found 301.2248.
5-butylnonane-2,5-diol (6):
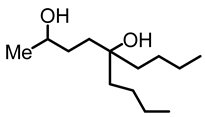
To a pressure vial with a Teflon screw cap, valerolactone (2, 0.300 g, 3.00 mmol) and THF (3 mL) were added. n-Butyl lithium (1.4 M in hexanes, 6 mL, 8.4 mmol) was added dropwise, using a needle and a syringe. The pressure vial was closed, and the yellow mixture was kept for 16 h at 65 °C, resulting in a dark red solution. The reaction mixture was allowed to cool to room temperature and quenched with water (10 mL). The aqueous and the organic layers were separated. The aqueous layer was extracted three times with diethyl ether. The combined ether fractions were dried with MgSO4 and filtered, and the solvent was removed to obtain the product as a light yellow oil (0.560 g, 2.59 mmol, 86%). NMR (δ, CDCl3) 1H: NMR (δ, CDCl3) 1H: 3.74–3.69 (m, 1H, OCH), 2.72 (br s, 2H, OH), 1.54–1.36 (m, 8H, CH2), 1.30–1.20 (m, 8H, CH2), 1.13 (t, 3H, JHH = 6 Hz, CH3), 0.85 (t, 6H, JHH = 6 Hz, 2CH3).13C{1H}: 74.2 (s, COH), 68.4 (s, COH), 39.1 (s, CH2), 38.5 (s, CH2), 35.4 (s, CH2), 32.9 (s, CH2), 25.8 (s, CH2), 25.7 (s, CH2), 23.5 (s, CH2), 23.3 (s, CH3), 14.1. (s, CH3). IR (ATR, neat): ν 3380 (br), 2956 (s), 2927 (s), 2859 (s) cm−1.
2,2,5-triphenyltetrahydrofuran (7):
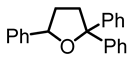
In a 7 mL vial with Teflon screw cup, the diol 3 (0.050 g, 0.157 mmol) and [FeCp2]BF4 (0.005 g, 0.018 mmol) were combined and the mixture was stirred at 45 °C for 3 days. The reaction mixture was filtered through a short pad of silica gel, and the solvent was removed in high vacuum to obtain the product (7) as a yellow, viscous oil (0.039 g, 0.130 mmol, 83%). NMR (δ, CDCl3) 1H: 7.50–7.44 (m, 4H, aromatic), 7.30–7.13 (m, 11H, aromatic), 5.08 (t, 1H, JHH = 7 Hz, OCH), 2.73–2.67 (m, 1H, CH), 2.62–2.52 (m, 1H, CH), 2.30–2.23 (m, 1H, CH), 1.92–1.85 (m, 1H, CH). 13C{1H}: 146.9 (s, aromatic), 146.7 (s, aromatic), 143.5 (s, aromatic), 143.5 (s, aromatic), 128.5 (s, aromatic), 128.40 (s, aromatic), 128.35 (s, aromatic), 128.28 (s, aromatic), 128.2 (s, aromatic), 127.32 (s, aromatic), 127.30 (s, aromatic), 126.81 (s, aromatic), 126.79 (s, aromatic), 126.22 (s, aromatic), 126.20 (s, aromatic). 126.1 (s, aromatic), 126.0 (s, aromatic), 125.9 (s, aromatic), 88.8 (s, CO), 80.5 (s, CO), 38.5 (s, CH2), 34.7 (s, CH2). IR (ATR, neat): ν 3055 (w), 3022 (w), 2969 (w), 2869 (w) cm−1. HRMS ESI [M + H]+ calcd for C22H21O+ 301.1592; found 301.1572.
2,2-dimethyl-5-phenyltetrahydrofuran (8):
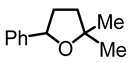
In a 7 mL vial with Teflon screw cup, the diol 4 (0.077 g, 0.396 mmol) and [FeCp2]BF4 (0.011 g, 0.04 mmol) were combined and the mixture was stirred at 45 °C for 3 days. The crude product was purified by filtration over 1.5 × 5 cm Al2O3, using CH2Cl2/hexanes 1:1 to pack/elute. The solvent was removed under high vacuum to obtain the product (8) as a yellow, viscous oil (0.057 g, 0.323 mmol, 82%). NMR (δ, CDCl3) 1H: 7.29–7.21 (m, 4H, aromatic), 7.17–7.15 (m, 1H, aromatic), 4.91–4.85 (m, 1H, OCH), 2.28–2.17 (m, 1H, CH), 1.86–1.75 (m, 3H, CH+CH2), 1.31 (s, 3H, CH3), 1.31 (s, 3H, CH3′).13C{1H}: 143.7 (s, aromatic), 128.3 (s, aromatic), 127.2 (s, aromatic), 125.9 (s, aromatic), 81.3 (s, CO), 80.6 (s, CO), 39.1 (s, CH2), 35.8 (s, CH2), 29.2 (s, CH3), 28.5 (s, CH3′). IR (ATR, neat): ν 3030 (w), 3020 (w), 2964 (m), 2900 (w) cm−1.
2,2-dibutyl-5-phenyltetrahydrofuran (9):
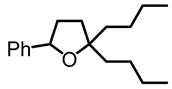
In a 7 mL vial with Teflon screw cup, the diol 5 (0.100 g, 0.359 mmol) and [FeCp2]BF4 (0.010 g, 0.036 mmol) were combined and kept at 70 °C for 48 h. The crude product was purified by filtration over 1.5 × 5 cm Al2O3, using CH2Cl2/hexanes 9:1 to pack/elute. The solvent was removed under high vacuum to obtain the product (9) as a yellow, viscous oil (0.068 g, 0.261 mmol, 73%). NMR (δ, CDCl3) 1H: 7.29–7.12 (m, 5H, aromatic), 4.83–4.79 (m, 1H, OCH), 2.18–2.15 (m, 1H, CH), 1.79–1.71 (m, 2H, CH2), 1.59–1.42 (m, 4H, 2CH2), 1.34–1.18 (m, 9H, CH+4CH2), 0.85–0.82 (m, 6H, 2CH3).13C{1H}: 143.5 (s, aromatic), 128.3 (s, aromatic), 127. 2 (s, aromatic), 125.9 (s, aromatic), 85.8 (s, CO), 80.5 (CO), 39.3 (s, CH2), 38.8 (s, CH2), 35.9 (s, CH2), 35.9 (s, CH2), 26.7 (s, CH2), 26.6 (s, CH2), 23.5 (s, CH3), 14.3 (s, CH3). IR (ATR, neat): ν 3052 (vw), 3026 (vw), 2952 (s), 2929 (s), 2858 (s) cm−1.
2,2-dibutyl-5-methyltetrahydrofuran (10):
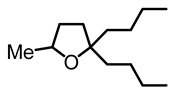
In a 7 mL vial with Teflon screw cup, the diol 6 (0.100 g, 0.500 mmol) and [FeCp2]BF4 (0.013 g, 0.005 mmol) were combined and kept at 70 °C for 64 h. The crude product was purified by filtration over 1.5 × 5 cm Al2O3, using CH2Cl2/hexanes 9:1 to pack/elute. The solvent was removed under high vacuum to obtain the product (10) as a yellow, viscous oil (0.083 g, 0.418 mmol, 84%). NMR (δ, CDCl3) 1H: 3.98–3.88 (m, 1H, OCH), 1.90–1.82 (m, 1H, CHH), 1.67–1.59 (m, 2H, CH2), 1.43–1.30 (m, 4H, 2CH2), 1.21–1.18 (m, 9H, CHH, 4CH2), 1.14 (t, 3H, JHH = 6 Hz, CH3), 0.83 (t, JHH = 6 Hz, 6H, 2CH3).13C{1H}: 85.1 (s, CO), 74.4 (s, CO), 39.5 (s, CH2), 39.2 (s, CH2), 35.5 (s, CH2), 34.1 (s, CH2), 26.6 (s, CH2), 26.5 (s, CH2), 23.43 (s, CH2), 23.39 (s, CH2), 21.5 (s, CH3), 14.2 (s, CH3). IR (ATR, neat): ν 2980 (s), 2960 (s), 2910 (s) cm−1.
4. Conclusions
In summary, we demonstrated for the first time that ferrocenium cations cyclize substituted 1,4-butanediols to afford 2,2,5-trisubstituted tetrahydrofuran derivatives. The isolation of the products is straightforward by filtration through aluminum oxide, as the polarity of the diols is much higher than that of the products. The protocol allows for access to the trisubstituted tetrahydrofurans in a two-step procedure from commercial or easily accessible starting materials. We propose a reaction mechanism involving either a carbocation intermediate or a species in which the ferrocenium cation polarizes the C–OH bond. This method aligns with the principles of green chemistry, as it utilizes an iron-based catalyst and produces only water as a byproduct.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/inorganics13020059/s1, 1H, 13C{1H} NMR, and IR spectra of the diols and the catalysis products in Scheme 1 and Scheme 2 as well as 1H NMR spectra of some reactions in Scheme 3 catalyzed by FeCl3 and KPF6.
Author Contributions
Conceptualization, E.B.B.; Methodology, E.B.B.; Investigation, C.D.A. and K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Banavali Green and Sustainable Chemistry Fund in Arts and Science at the University of Missouri—Saint Louis.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fernandes, R.A.; Pathare, R.S.; Gorve, D.A. Advances in Total Synthesis of Some 2,3,5-Trisubstituted Tetrahydrofuran Natural Products. Chem. Asian J. 2020, 15, 2815–2837. [Google Scholar] [CrossRef] [PubMed]
- De La Torre, A.; Cuyamendous, C.; Bultel-Poncé, V.; Durand, T.; Galano, J.M.; Oger, C. Recent advances in the synthesis of tetrahydrofurans and applications in total synthesis. Tetrahedron 2016, 72, 5003–5025. [Google Scholar] [CrossRef]
- Fernandes, R.A.; Gorve, D.A.; Pathare, R.S. Emergence of 2,3,5-trisubstituted tetrahydrofuran natural products and their synthesis. Org. Biomol. Chem. 2020, 18, 7002–7025. [Google Scholar] [CrossRef] [PubMed]
- Lorente, A.; Lamariano-Merketegi, J.; Albericio, F.; Álvarez, M. Tetrahydrofuran-Containing Macrolides: A Fascinating Gift from the Deep Sea. Chem. Rev. 2013, 113, 4567–4610. [Google Scholar] [CrossRef] [PubMed]
- Nakata, T. Total synthesis of marine polycyclic ethers. Chem. Rev. 2005, 105, 4314–4347. [Google Scholar] [CrossRef]
- González-Andrés, P.; Fernández-Peña, L.; Díez-Poza, C.; Barbero, A. The Tetrahydrofuran Motif in Marine Lipids and Terpenes. Mar. Drugs 2022, 20, 642. [Google Scholar] [CrossRef]
- Kang, E.J.; Lee, E. Total Synthesis of Oxacyclic Macrodiolide Natural Products. Chem. Rev. 2005, 105, 4348–4378. [Google Scholar] [CrossRef]
- Jung, J.H.; Lee, E. Expedient Synthesis of (−)-Amphidinolide X. Angew. Chem. Int. Ed. 2009, 48, 5698–5700. [Google Scholar] [CrossRef]
- Wolfe, J.P. Palladium-Catalyzed Carboetherification and Carboamination Reactions of γ-Hydroxy-and γ-Aminoalkenes for the Synthesis of Tetrahydrofurans and Pyrrolidines. Eur. J. Org. Chem. 2007, 571–582. [Google Scholar] [CrossRef]
- Fernandez de la Pradilla, R.; Montero, C.; Priego, J.; Martínez-Cruz, L.A. A Novel Sulfoxide-Directed Route to Enantiopure Tetrahydrofurans: Application to the Expedient Formal Synthesis of (+)-trans-Kumausyne and (+)-Kumausallene. J. Org. Chem. 1998, 63, 9612–9613. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, X.; Sun, H.; Tao, G.; Chang, X.Y.; Xing, X.; Chen, B.; Xu, C. Development of highly efficient platinum catalysts for hydroalkoxylation and hydroamination of unactivated alkenes. Nat. Commun. 2021, 12, 1953. [Google Scholar] [CrossRef] [PubMed]
- Jalce, G.; Franck, X.; Figadère, B. Diastereoselective synthesis of 2,5-disubstituted tetrahydrofurans. Tetrahedron Asym. 2009, 20, 2537–2581. [Google Scholar] [CrossRef]
- Čorić, I.; Kim, J.H.; Vlaar, T.; Patil, M.; Thiel, W.; List, B. Brønsted acid catalyzed asymmetric SN2-type O-alkylations. Angew. Chem. Int. Ed. 2013, 52, 3490–3493. [Google Scholar] [CrossRef] [PubMed]
- Olah, G.A.; Fung, A.P.; Malhotra, R. Synthetic Methods and Reactions; 99. Preparation of Cyclic Ethers over Superacidic Perfluorinated Resinsulfonic Acid (Nafion-H) Catalyst. Synthesis 1981, 474–476. [Google Scholar] [CrossRef]
- McCullough, L.R.; Childers, D.J.; Watson, R.A.; Kilos, B.A.; Barton, D.G.; Weitz, E.; Kung, H.H.; Notestein, J.M. Acceptorless dehydrogenative coupling of neat alcohols using group VI sulfide catalysts. ACS Sustain. Chem. Eng. 2017, 5, 4890–4896. [Google Scholar] [CrossRef]
- Nalesnik, T.E.; Holy, N.L. Homogeneous catalytic cyclization and oxidation of diols. J. Org. Chem. 1977, 42, 372–374. [Google Scholar] [CrossRef]
- Aricò, F.; Tundo, P.; Maranzana, A.; Tonachini, G. Synthesis of five-membered cyclic ethers by reaction of 1,4-diols with dimethyl carbonate. Chem. Sustain. Chem. 2012, 5, 578–1586. [Google Scholar] [CrossRef]
- Mihailović, M.L.; Gojković, S.; Čeković, Ž. Stereochemistry of cyclic ether formation. Part I. Stereoselective intramolecular cyclisation of aliphatic disecondary 1,4-diols and their sulphonate esters to tetrahydrofurans. Perkin Transactions. J. Chem. Soc. 1972, 2460–2464. [Google Scholar] [CrossRef]
- Jacobus, J. Mechanism and stereochemistry of 1,4-diol ring closure to tetrahydrofuran. J. Org. Chem. 1973, 38, 402–404. [Google Scholar] [CrossRef]
- Sutro, J.L.; Fürstner, A. Total Synthesis of the Allenic Macrolide (+)-Archangiumide. J. Am. Chem. Soc. 2024, 146, 2345–2350. [Google Scholar] [CrossRef]
- Mihelich, E.D. Stereoselective synthesis of highly substituted tetrahydrofurans through acid-catalyzed ring closure of selenyl diols. J. Am. Chem. Soc. 1990, 112, 8995–8997. [Google Scholar] [CrossRef]
- Alvarez-Manzaneda, E.J.; Chaboun, R.; Alvarez, E.; Cabrera, E.; Alvarez-Manzaneda, R.; Haidour, A.; Ramos, J.M. Cerium (IV) ammonium nitrate (CAN): A very efficient reagent for the synthesis of tertiary ethers. Synlett 2006, 1829–1834. [Google Scholar] [CrossRef]
- Sharma, G.V.M.; Kumar, K.R.; Sreenivas, P.; Krishna, P.R.; Chorghade, M.S. Catalytic FeCl3-or Yb(OTf)3-mediated synthesis of substituted tetrahydrofurans and C-aryl glycosides from 1, 4-diols. Tetrahedron Asym. 2002, 13, 687–690. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green chemistry: Principles and practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- Bauer, I.; Knölker, H.-J. Iron catalysis in organic synthesis. Chem. Rev. 2015, 115, 3170–3387. [Google Scholar]
- Rana, S.; Biswas, J.P.; Paul, S.; Paik, A.; Maiti, D. Organic synthesis with the most abundant transition metal–iron: From rust to multitasking catalysts. Chem. Soc. Rev. 2021, 50, 243–472. [Google Scholar]
- Wei, D.; Darcel, C. Iron catalysis in reduction and hydrometalation reactions. Chem. Rev. 2018, 119, 2550–2610. [Google Scholar] [CrossRef]
- Bauer, E.B. Recent Catalytic Applications of Ferrocene and Ferrocenium Cations in the Syntheses of Organic Compounds. Molecules 2024, 29, 5544. [Google Scholar] [CrossRef]
- Toma, Š.; Šebesta, R. Applications of ferrocenium salts in organic synthesis. Synthesis 2015, 47, 1683–1695. [Google Scholar] [CrossRef]
- Fiolek, T.J.; Keel, K.L.; Tepe, J.J. Fluspirilene analogs activate the 20S proteasome and overcome proteasome impairment by intrinsically disordered protein oligomers. ACS Chem. Neurosci. 2021, 12, 1438–1448. [Google Scholar] [CrossRef]
- Too, P.C.; Tnay, Y.L.; Chiba, S. Copper-catalyzed aerobic aliphatic C–H oxygenation with hydroperoxides. Beilstein J. Org. Chem. 2013, 9, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Hirose, D.; Taniguchi, T. Direct Synthesis of 1,4-Diols from Alkenes by Iron-Catalyzed Aerobic Hydration and C-H Hydroxylation. Angew. Chem. Int. Ed. 2014, 53, 2730–2734. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Yasoshima, T.; Momochi, H. A new approach to the synthesis of 1-oxaspiro [4. n] alkanes and tetrahydrofurans by the 1,5-CH insertion reaction of magnesium carbenoids. Tetrahedron Lett. 2012, 53, 2074–2077. [Google Scholar] [CrossRef]
- Rivero, A.R.; Fodran, P.; Ondrejková, A.; Wallentin, C.J. Alcohol etherification via alkoxy radicals generated by visible-light photoredox catalysis. Org. Lett. 2020, 22, 8436–8440. [Google Scholar] [CrossRef] [PubMed]
- Mo, X.; Yakiwchuk, J.; Dansereau, J.; McCubbin, J.A.; Hall, D.G. Unsymmetrical diarylmethanes by ferroceniumboronic acid catalyzed direct Friedel–Crafts reactions with deactivated benzylic alcohols: Enhanced reactivity due to ion-pairing effects. J. Am. Chem. Soc. 2015, 137, 9694–9703. [Google Scholar] [CrossRef]
- Zaragoza, J.P.T.; Cummins, D.C.; Mubarak, M.Q.E.; Siegler, M.A.; de Visser, S.P.; Goldberg, D.P. Hydrogen Atom Abstraction by High-Valent Fe(OH) versus Mn(OH) Porphyrinoid Complexes: Mechanistic Insights from Experimental and Computational Studies. Inorg. Chem. 2019, 58, 16761–16770. [Google Scholar] [CrossRef]
- Babin, V.N.; Belousov, Y.A.; Belousova, T.A.; Borisov, Y.A.; Gumenyuk, V.V.; Nekrasov, Y.S. Reactions of ferricinium salts with Lewis bases. Russ. Chem. Bull. Int. Ed. 2011, 60, 2081–2087. [Google Scholar] [CrossRef]
- Duarte, F.; Paton, R.S. Molecular Recognition in Asymmetric Counteranion Catalysis: Understanding Chiral Phosphate-Mediated Desymmetrization. J. Am. Chem. Soc. 2017, 139, 8886–8896. [Google Scholar] [CrossRef]
- Mahlau, M.; List, B. Asymmetric Counteranion-Directed Catalysis (ACDC): A Remarkably General Approach to Enantioselective Synthesis. Isr. J. Chem. 2012, 52, 630–638. [Google Scholar] [CrossRef]
- Raskatov, J.A.; Thompson, A.L.; Cowley, A.R.; Claridge, T.D.W.; Brown, J.M. Chiral recognition in contact ion-pairs; observation, characterization and analysis. Chem. Sci. 2013, 4, 3140–3147. [Google Scholar] [CrossRef]
- Burchat, A.F.; Chong, J.M.; Nielsen, N. Titration of alkyllithiums with a simple reagent to a blue endpoint. J. Organomet. Chem. 1997, 542, 281–283. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).