
The Role of Methyl Substitution in Spin Crossover of Fe(III) Complexes with Pentadentate Schiff Base Ligands


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
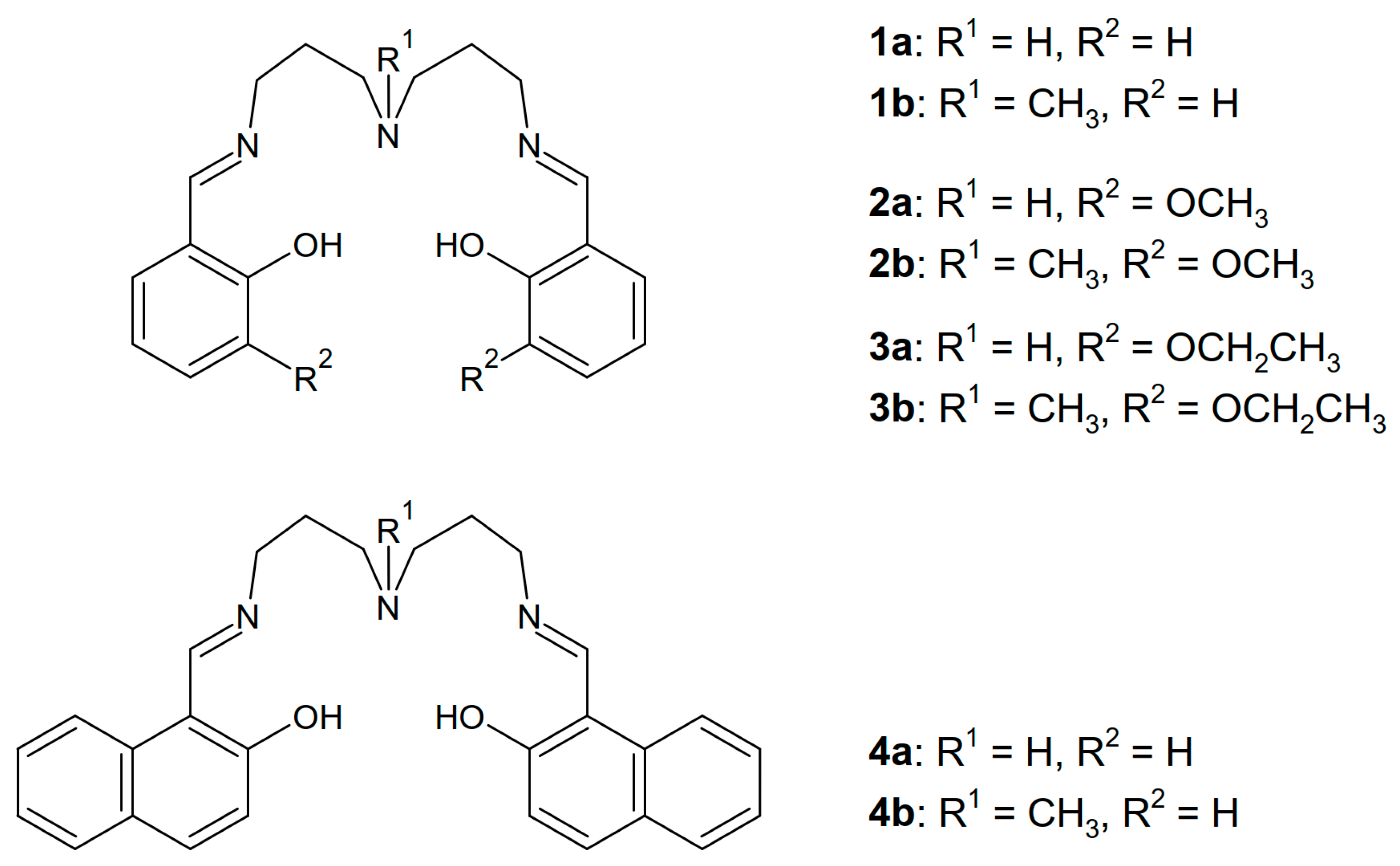
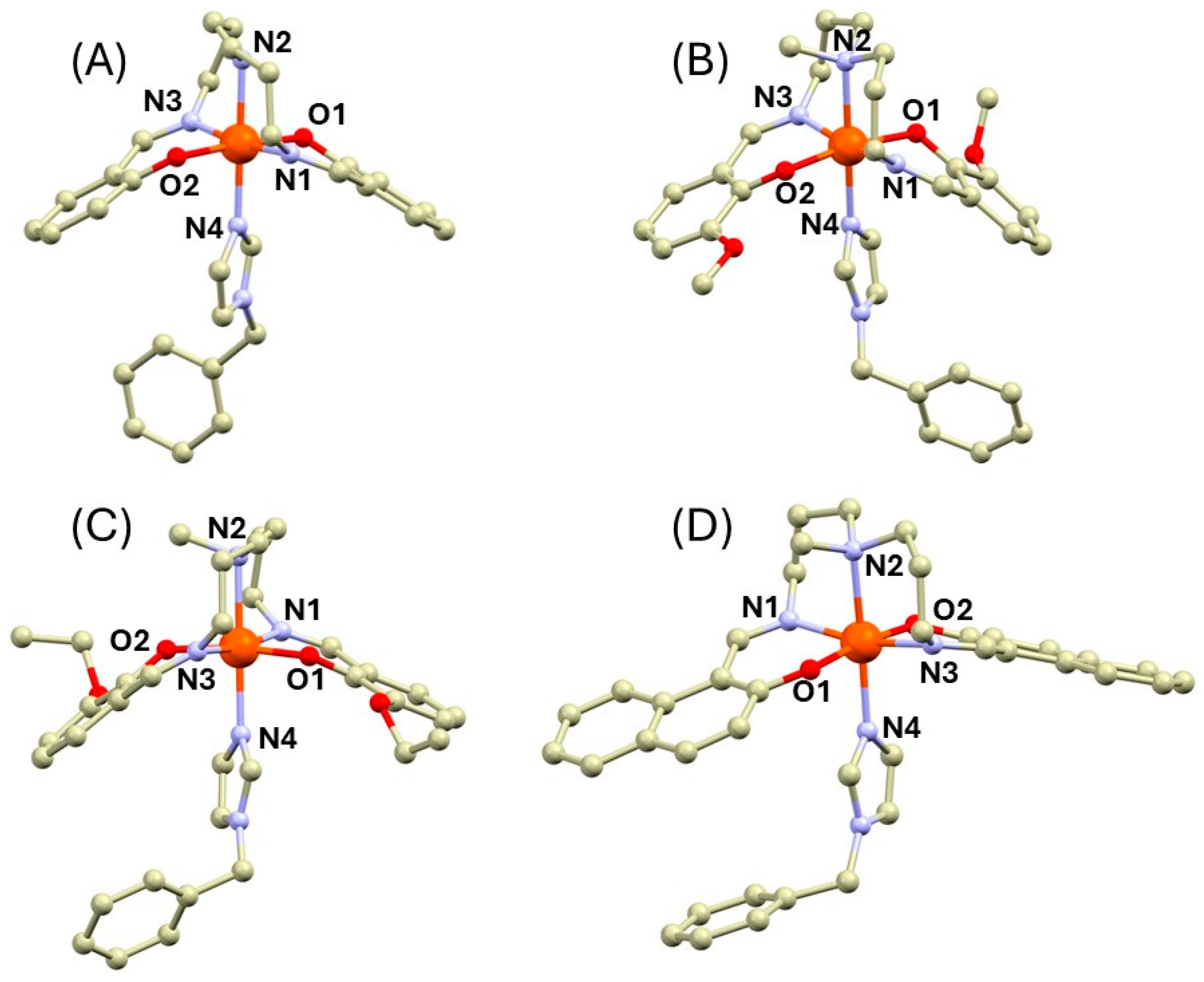
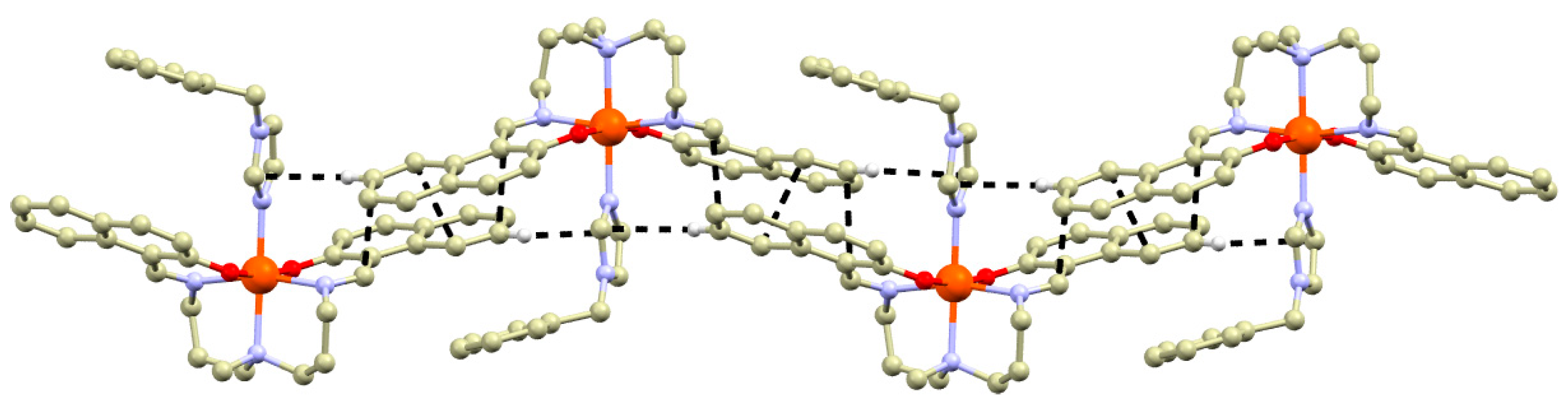
2.1. Synthesis and Crystal Structures
2.2. Magnetic Properties
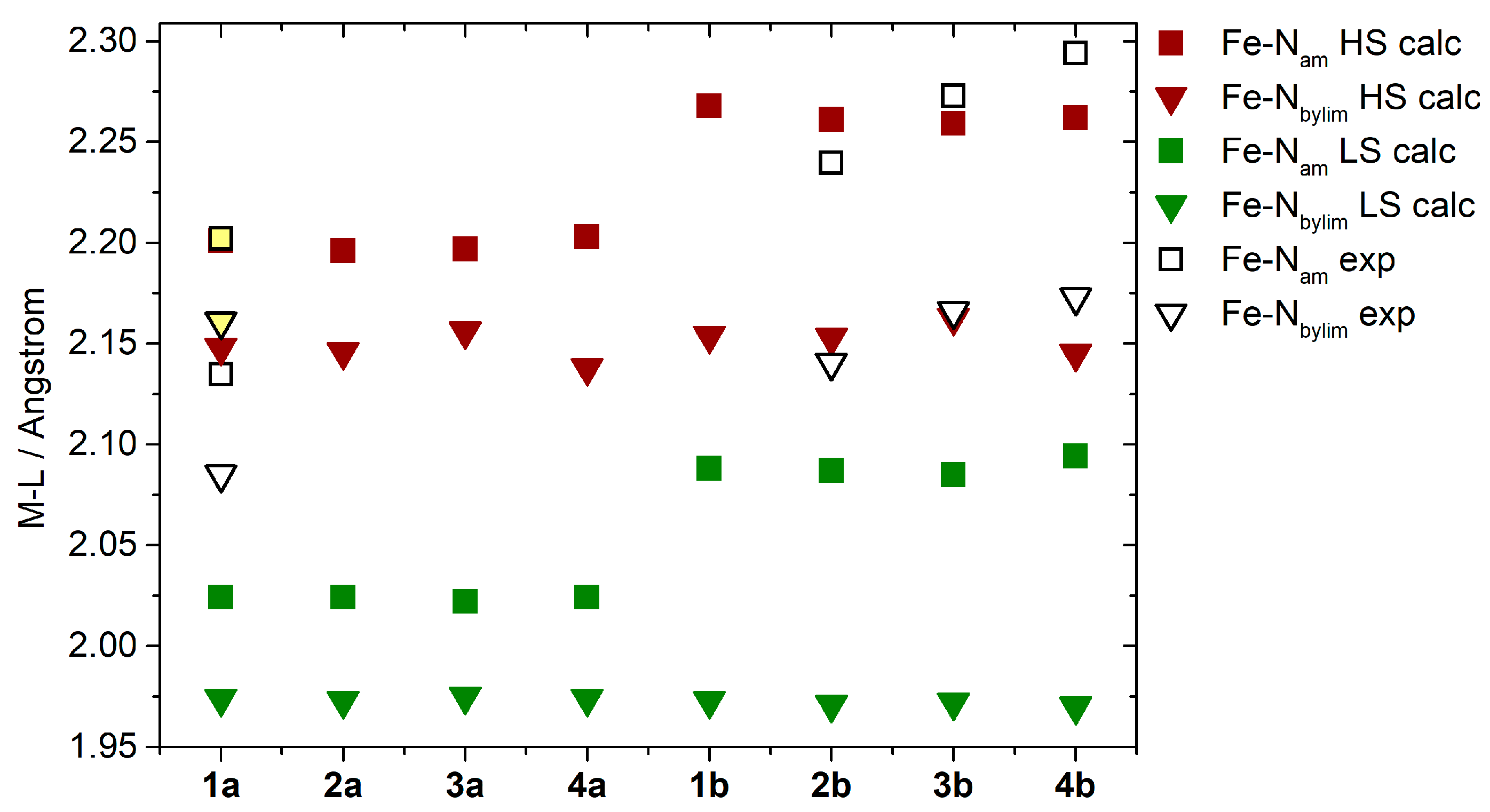
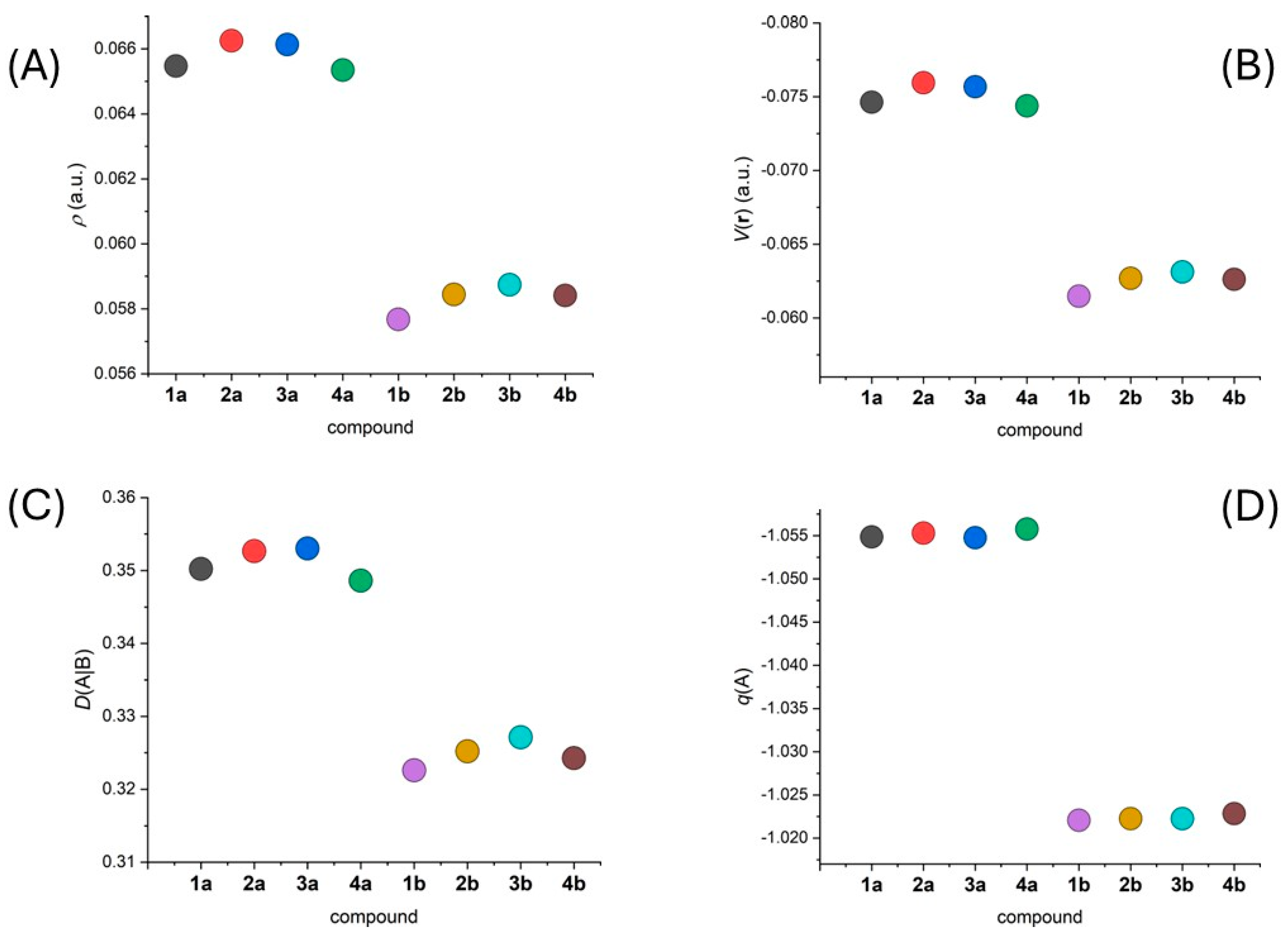
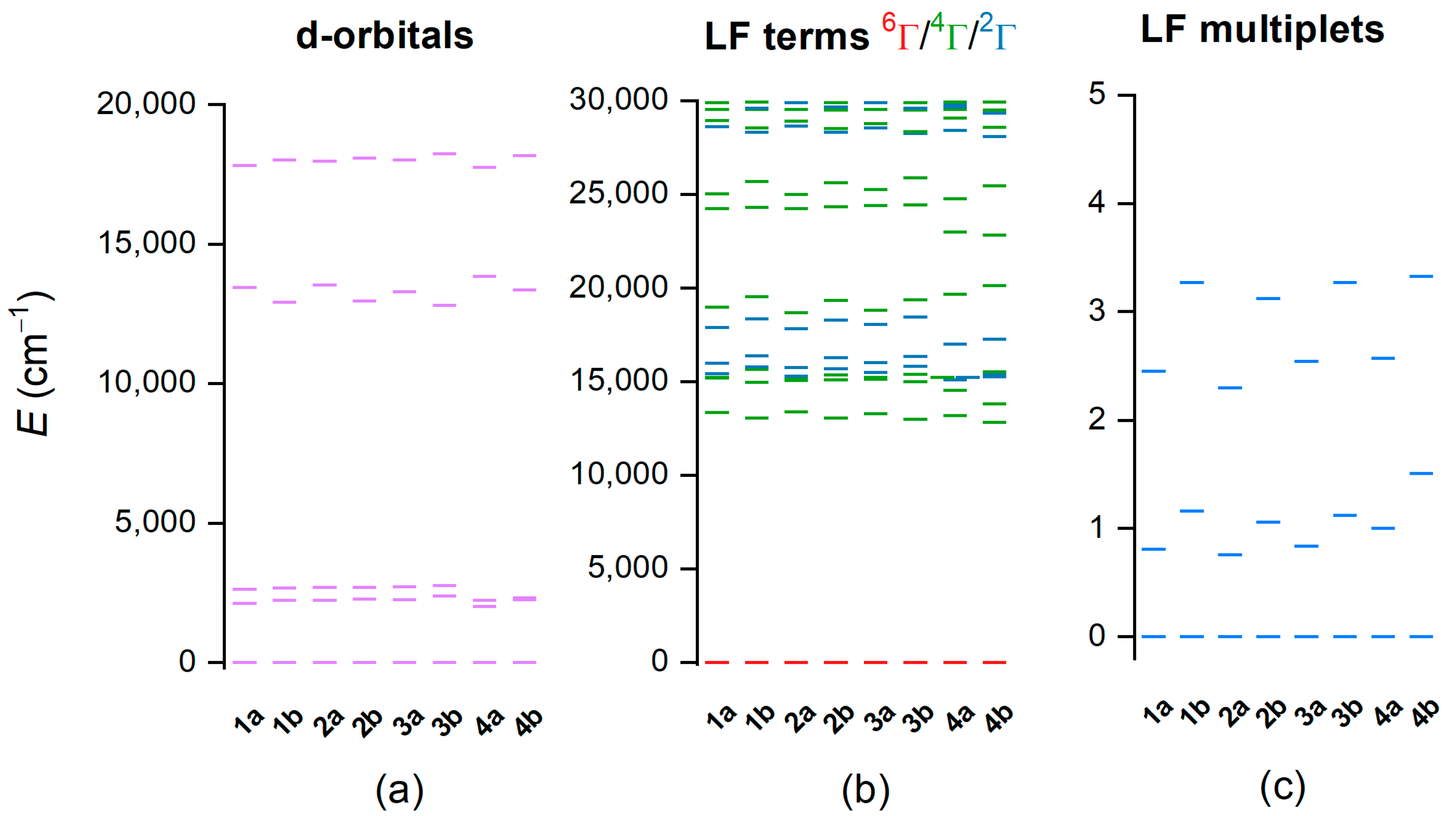
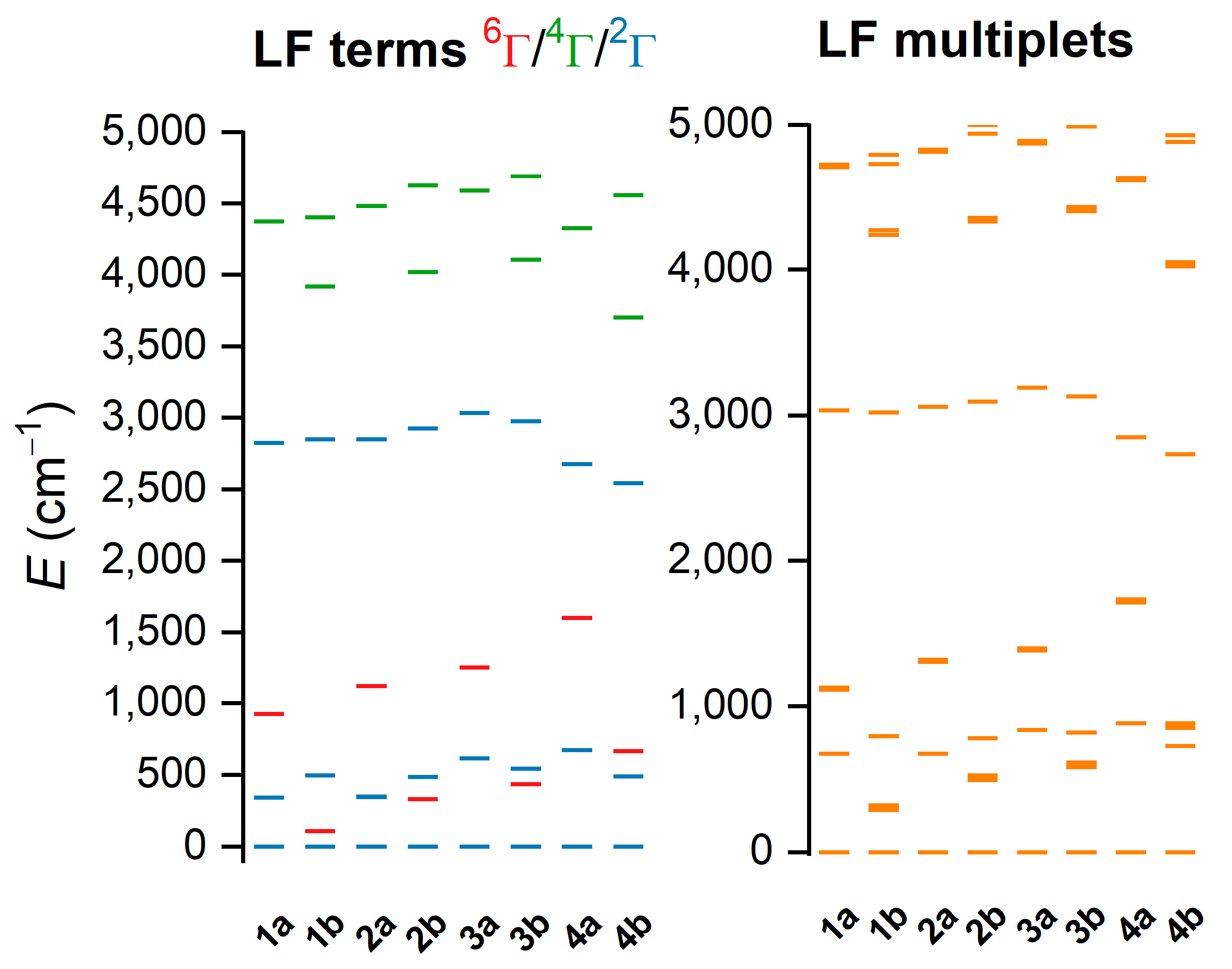
2.3. Theoretical Calculations
3. Materials and Methods
3.1. General Considerations and Instrumentation
3.2. X-Ray Diffraction Analysis
3.3. Synthesis
3.4. Theoretical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Koningsbruggen, P.J.; Maeda, Y.; Oshio, H. Iron(III) spin crossover compounds. In Spin Crossover in Transition Metal Compounds I; Gütlich, P., Goodwin, H.A., Eds.; Springer: Berlin, Germany, 2004; pp. 259–324. [Google Scholar]
- Boonprab, T.; Thammasangwan, W.; Chastanet, G.; Gonidec, M.; Harding, P.; Harding, D.J. Halide Anion Effects and Magnetostructural Relationships in Iron(III) Spin Crossover Complexes. Cryst. Growth Des. 2024, 24, 8145–8152. [Google Scholar] [CrossRef]
- Díaz-Torres, R.; Gómez-Coca, S.; Ruiz, E.; Harding, P.; Harding, D.J. Improving spin crossover characteristics in heteroleptic [FeIII(qsal-5-I)(qsal-5-OMe)]A complexes. Dalton Trans. 2023, 52, 18148–18157. [Google Scholar] [CrossRef]
- Harding, D.J.; Harding, P.; Phonsri, W. Spin crossover in iron(III) complexes. Coord. Chem. Rev. 2016, 313, 38–61. [Google Scholar] [CrossRef]
- Matsumoto, N.; Ohta, S.; Yoshimura, C.; Ohyoshi, A.; Kohata, S.; Okawa, H.; Maeda, Y. Studies on spin-equilibrium iron(III) complexes. Part 1. Syntheses and magnetic properties of a new family of spin cross-over iron(III) complexes with a unidentate ligand over a wide range of the spectrochemical series and a quinquedentate ligand derived. J. Chem. Soc. Dalton Trans. 1985, 12, 2575. [Google Scholar] [CrossRef]
- Boča, R.; Fukuda, Y.; Gembický, M.; Herchel, R.; Jaroščiak, R.; Linert, W.; Renz, F.; Yuzurihara, J. Spin crossover in mononuclear and binuclear iron(III) complexes with pentadentate Schiff-base ligands. Chem. Phys. Lett. 2000, 325, 411–419. [Google Scholar] [CrossRef]
- Tanimura, K.; Kitashima, R.; Bréfuel, N.; Nakamura, M.; Matsumoto, N.; Shova, S.; Tuchagues, J.-P. Infinite chain structure and steep spin crossover of a Fe III Complex with a N3O2 pentadentate schiff-base ligand and 4-aminopyridine. Bull. Chem. Soc. Jpn. 2005, 78, 1279–1282. [Google Scholar] [CrossRef]
- Nemec, I.; Svoboda, I.; Herchel, R. Spin Crossover in Three Mononuclear Iron (III) Schiff Base Complexes. Metals 2019, 9, 849. [Google Scholar] [CrossRef]
- Faulmann, C.; Dorbes, S.; De Bonneval, B.G.; Molnár, G.; Bousseksou, A.; Gomez-Garcia, C.J.; Coronado, E.; Valade, L. Towards molecular conductors with a spin-crossover phenomenon: Crystal structures, magnetic properties and Mössbauer spectra of [Fe(salten)mepepy][M(dmit)2] complexes. Eur. J. Inorg. Chem. 2005, 2005, 3261–3270. [Google Scholar] [CrossRef]
- Bannwarth, A.; Schmidt, S.O.; Peters, G.; Sönnichsen, F.D.; Thimm, W.; Herges, R.; Tuczek, F. FeIII spin-crossover complexes with photoisomerizable ligands: Experimental and theoretical studies on the ligand-driven light-induced spin change effect. Eur. J. Inorg. Chem. 2012, 2012, 2776–2783. [Google Scholar] [CrossRef]
- Nemec, I.; Boča, R.; Herchel, R.; Trávníček, Z.; Gembický, M.; Linert, W. Dinuclear Fe(III) complexes with spin crossover. Monatshefte Chem. Chem. Mon. 2009, 140, 815–828. [Google Scholar] [CrossRef]
- Hayami, S.; Hiki, K.; Kawahara, T.; Maeda, Y.; Urakami, D.; Inoue, K.; Ohama, M.; Kawata, S.; Sato, O. Photo-Induced Spin Transition of Iron(III) Compounds with π-π Intermolecular Interactions. Chem.-A Eur. J. 2009, 15, 3497–3508. [Google Scholar] [CrossRef] [PubMed]
- Si-Guo, W.; Najbul, H.M.; Jie-Yu, Z.; Guo-Zhang, H.; Vu, H.A.N.; Liviu, U.; Wei-Xiong, Z.; Zhao-Ping, N.; Ming-Liang, T. Multiresponsive Spin Crossover Driven by Rotation of Tetraphenylborate Anion in an Iron(III) Complex. CCS Chem. 2020, 3, 453–459. [Google Scholar] [CrossRef]
- Pavlik, J.; Masárová, P.; Nemec, I.; Fuhr, O.; Ruben, M.; Šalitroš, I. Heteronuclear Iron(III)-Schiff Base Complexes with the Hexacyanidocobaltate(III) Anion: On the Quest to Understand the Governing Factors of Spin Crossover. Inorg. Chem. 2020, 59, 2747–2757. [Google Scholar] [CrossRef] [PubMed]
- Herchel, R.; Boča, R.; Gembický, M.; Kožísek, J.; Renz, F. Spin Crossover in a Tetranuclear Cr(III)−Fe(III)3 Complex. Inorg. Chem. 2004, 43, 4103–4105. [Google Scholar] [CrossRef]
- Nemec, I.; Herchel, R.; Boča, R.; Trávníček, Z.; Svoboda, I.; Fuess, H.; Linert, W. Tuning of spin crossover behaviour in iron(III) complexes involving pentadentate Schiff bases and pseudohalides. Dalton Trans. 2011, 40, 10090–10099. [Google Scholar] [CrossRef]
- Nemec, I.; Herchel, R.; Trávníček, Z. The relationship between the strength of hydrogen bonding and spin crossover behaviour in a series of iron(iii) Schiff base complexes. Dalton Trans. 2015, 44, 4474–4484. [Google Scholar] [CrossRef]
- Vela, S.; Paulsen, H. Cooperativity in Spin Crossover Systems. An Atomistic Perspective on the Devil’s Staircase. Inorg. Chem. 2018, 57, 9478–9488. [Google Scholar] [CrossRef] [PubMed]
- Nemec, I.; Boča, R.; Gembický, M.; Dlháň, L.; Herchel, R.; Renz, F. High-spin Schiff-base dinuclear iron(III) complexes bridged by N-oxide ligands. Inorg. Chim. Acta 2009, 362, 4754–4759. [Google Scholar] [CrossRef]
- Pogány, L.; Brachňaková, B.; Masárová, P.; Moncol, J.; Pavlik, J.; Gál, M.; Mazúr, M.; Herchel, R.; Nemec, I.; Šalitroš, I. Impact of the Schiff base ligand substituents on the solid state and solution properties of eleven iron(III) complexes. New J. Chem. 2019, 43, 13916–13928. [Google Scholar] [CrossRef]
- Milocco, F.; De Vries, F.; Siebe, H.S.; Engbers, S.; Demeshko, S.; Meyer, F.; Otten, E. Widening the Window of Spin-Crossover Temperatures in Bis(formazanate)iron(II) Complexes via Steric and Noncovalent Interactions. Inorg. Chem. 2021, 60, 2045–2055. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Najibi, A.; Goerigk, L. DFT-D4 counterparts of leading meta-generalized-gradient approximation and hybrid density functionals for energetics and geometries. J. Comput. Chem. 2020, 41, 2562–2572. [Google Scholar] [CrossRef]
- Bursch, M.; Neugebauer, H.; Ehlert, S.; Grimme, S. Dispersion corrected r2SCAN based global hybrid functionals: r2SCANh, r2SCAN0, and r2SCAN50. J. Chem. Phys. 2022, 156, 134105. [Google Scholar] [CrossRef] [PubMed]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.; Perdew, J.P. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J. Chem. Phys. 2003, 119, 12129–12137. [Google Scholar] [CrossRef]
- Rajchel, Ł.; Żuchowski, P.S.; Hapka, M.; Modrzejewski, M.; Szczęśniak, M.M.; Chałasiński, G. A density functional theory approach to noncovalent interactions via interacting monomer densities. Phys. Chem. Chem. Phys. 2010, 12, 14686–14692. [Google Scholar] [CrossRef]
- Friede, M.; Ehlert, S.; Grimme, S.; Mewes, J.-M. Do Optimally Tuned Range-Separated Hybrid Functionals Require a Reparametrization of the Dispersion Correction? It Depends. J. Chem. Theory Comput. 2023, 19, 8097–8107. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Calculate Root-Mean-Square Deviation (RMSD) of Two Molecules Using Rotation, GitHub. Version rmsd-1.5.1. Available online: https://github.com/charnley/rmsd (accessed on 11 November 2024).
- Li, W.; Li, X.; Robeyns, K.; Wolff, M.; Kfoury, J.; Oláh, J.; Herchel, R.; Demeshko, S.; Meyer, F.; Garcia, Y. Spin-state versatility in FeII4L6 supramolecular cages with a pyridyl-hydrazone ligand scaffold modulated by solvents and counter anions. Dalton Trans. 2024, 53, 1449–1459. [Google Scholar] [CrossRef]
- Figgis, B.N.; Hitchman, M.A. Ligand Field Theory and Its Applications; Wiley: New York, NY, USA, 1999; ISBN 978-0-471-31776-0. [Google Scholar]
- Bader, R. Atoms in Molecules: A Quantum Theory; Oxford University Press: New York, NY, USA, 1994; ISBN 978-0-19-855865-1. [Google Scholar]
- Atanasov, M.; Ganyushin, D.; Sivalingam, K.; Neese, F. A Modern First-Principles View on Ligand Field Theory Through the Eyes of Correlated Multireference Wavefunctions; Springer: Berlin/Heidelberg, Germany, 2011; pp. 149–220. ISBN 978-3-642-27378-0. [Google Scholar]
- Singh, S.K.; Eng, J.; Atanasov, M.; Neese, F. Covalency and chemical bonding in transition metal complexes: An ab initio based ligand field perspective. Coord. Chem. Rev. 2017, 344, 2–25. [Google Scholar] [CrossRef]
- CrysAlisPro, Version 1.171.40.82a, Rigaku Oxford Diffraction: Oxford, UK, 2020.
- Bruker AXS Inc. APEX2, SMART-Plus and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2004. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Shedrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Neese, F. A perspective on the future of quantum chemical software: The example of the ORCA program package. Faraday Discuss. 2024, 254, 295–314. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree-Fock and hybrid DFT calculations. A “chain-of-spheres” algorithm for the Hartree-Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Izsák, R.; Neese, F. An overlap fitted chain of spheres exchange method. J. Chem. Phys. 2011, 135, 144105. [Google Scholar] [CrossRef] [PubMed]
- Helmich-Paris, B.; de Souza, B.; Neese, F.; Izsák, R. An improved chain of spheres for exchange algorithm. J. Chem. Phys. 2021, 155, 104109. [Google Scholar] [CrossRef] [PubMed]
- Caldeweyher, E.; Ehlert, S.; Hansen, A.; Neugebauer, H.; Spicher, S.; Bannwarth, C.; Grimme, S. A generally applicable atomic-charge dependent London dispersion correction. J. Chem. Phys. 2019, 150, 154122. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ratés, M.; Neese, F. Effect of the Solute Cavity on the Solvation Energy and its Derivatives within the Framework of the Gaussian Charge Scheme. J. Comput. Chem. 2020, 41, 922–939. [Google Scholar] [CrossRef]
- AIMAll (Version 19.10.12), Todd A. Keith, TK Gristmill Software: Overland Park, KS, USA, 2019. Available online: https://aim.tkgristmill.com (accessed on 16 March 2023).
- Malmqvist, P.-Å.; Roos, B.O. The CASSCF state interaction method. Chem. Phys. Lett. 1989, 155, 189–194. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Malrieu, J.-P. N-electron valence state perturbation theory: A fast implementation of the strongly contracted variant. Chem. Phys. Lett. 2001, 350, 297–305. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Evangelisti, S.; Leininger, T.; Malrieu, J.-P. Introduction of n -electron valence states for multireference perturbation theory. J. Chem. Phys. 2001, 114, 10252–10264. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nemec, I.; Herchel, R. The Role of Methyl Substitution in Spin Crossover of Fe(III) Complexes with Pentadentate Schiff Base Ligands. Inorganics 2025, 13, 57. https://doi.org/10.3390/inorganics13020057
Nemec I, Herchel R. The Role of Methyl Substitution in Spin Crossover of Fe(III) Complexes with Pentadentate Schiff Base Ligands. Inorganics. 2025; 13(2):57. https://doi.org/10.3390/inorganics13020057
Chicago/Turabian StyleNemec, Ivan, and Radovan Herchel. 2025. "The Role of Methyl Substitution in Spin Crossover of Fe(III) Complexes with Pentadentate Schiff Base Ligands" Inorganics 13, no. 2: 57. https://doi.org/10.3390/inorganics13020057
APA StyleNemec, I., & Herchel, R. (2025). The Role of Methyl Substitution in Spin Crossover of Fe(III) Complexes with Pentadentate Schiff Base Ligands. Inorganics, 13(2), 57. https://doi.org/10.3390/inorganics13020057